Cell Culture Models for Hepatitis E Virus


Abstract
1. Introduction
1.1. Genome Organization
1.2. HEV Life Cycle and Transmission
2. Viruses
2.1. HEV Patient Isolates
2.2. HEV cDNA Clones
3. Cell Models for HEV Infection
3.1. Hepatoma Cell Lines
3.2. Extra-Hepatic Manifestations and Non-Hepatoma Cell Lines
3.3. Primary Cells
3.4. Stem Cell-Derived Models
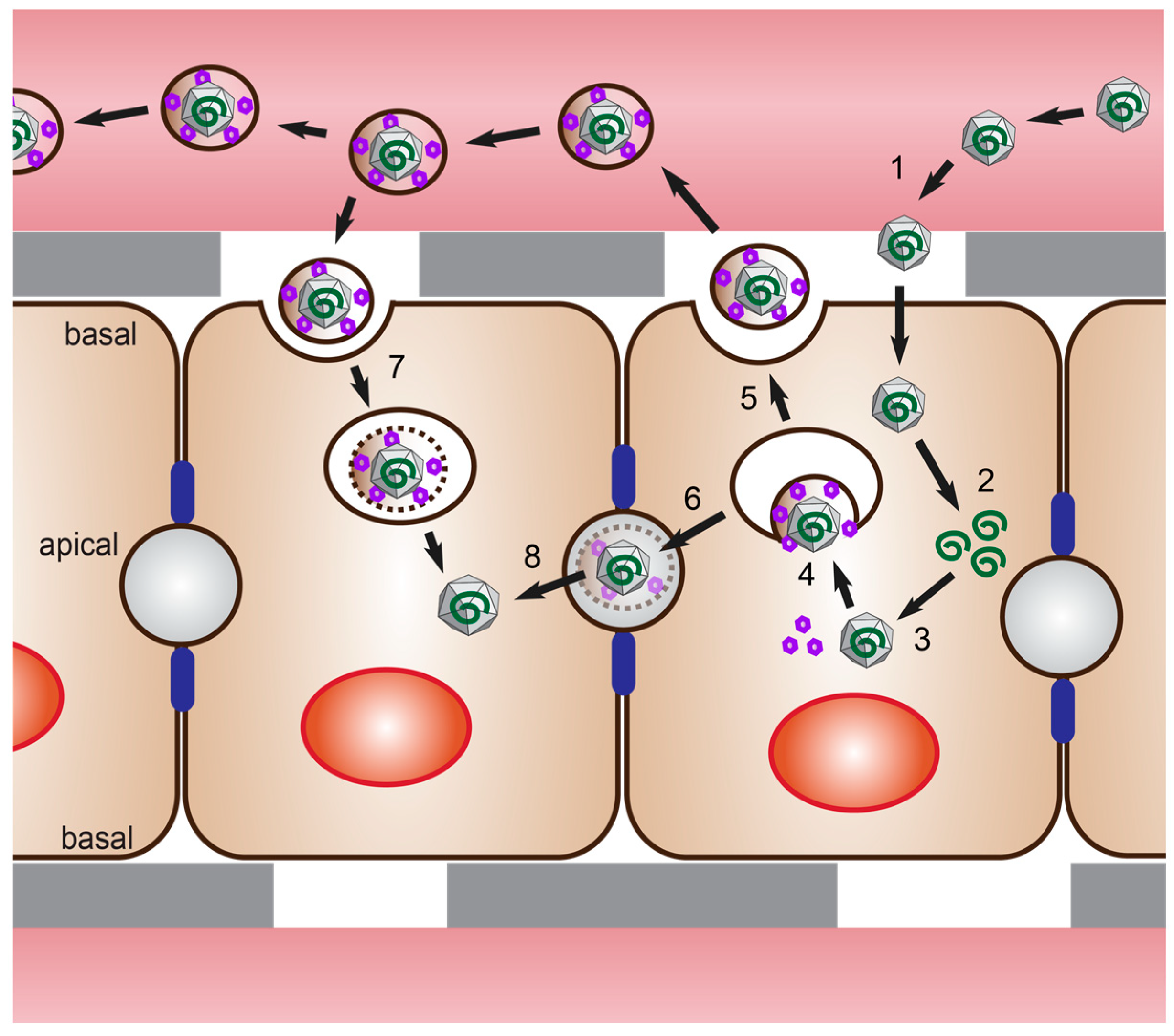
4. Polarized Cell Models for HEV Infection Studies
4.1. Polarized Hepatocytes without Access to Both Domains
4.2. Polarized Hepatocytes with Access to Both Domains
5. Conclusions and Remaining Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kamar, N.; Izopet, J.; Pavio, N.; Aggarwal, R.; Labrique, A.; Wedemeyer, H.; Dalton, H.R. Hepatitis E virus infection. Nat. Rev. Dis. Primers 2017, 3, 17086. [Google Scholar] [CrossRef] [PubMed]
- Pischke, S.; Hiller, J.; Lutgehetmann, M.; Polywka, S.; Rybczynski, M.; Ayuk, F.; Lohse, A.W. Blood-borne Hepatitis E Virus Transmission: A Relevant Risk for Immunosuppressed Patients. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2016, 63, 569–570. [Google Scholar] [CrossRef] [PubMed]
- Webb, G.W.; Dalton, H.R. Hepatitis E: An underestimated emerging threat. Adv. Infect. Dis. 2019, 6, 2049936119837162. [Google Scholar] [CrossRef] [PubMed]
- Khuroo, M.S.; Khuroo, M.S.; Khuroo, N.S. Transmission of Hepatitis E Virus in Developing Countries. Viruses 2016, 8, 253. [Google Scholar] [CrossRef] [PubMed]
- Balayan, M.S.; Andjaparidze, A.G.; Savinskaya, S.S.; Ketiladze, E.S.; Braginsky, D.M.; Savinov, A.P.; Poleschuk, V.F. Evidence for a virus in non-A, non-B hepatitis transmitted via the fecal-oral route. Intervirology 1983, 20, 23–31. [Google Scholar] [CrossRef]
- Nan, Y.; Wu, C.; Zhao, Q.; Zhou, E.M. Zoonotic Hepatitis E Virus: An Ignored Risk for Public Health. Front Microbiol. 2017, 8, 2396. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.H.; Tan, B.H.; Chi-Yuan Teo, E.; Lim, S.G.; Dan, Y.Y.; Wee, A.; Aw, P.P.; Zhu, Y.; Hibberd, M.L.; Tan, C.K.; et al. Chronic Infection With Camelid Hepatitis E Virus in a Liver Transplant Recipient Who Regularly Consumes Camel Meat and Milk. Gastroenterology 2016, 150, 355–357.e3. [Google Scholar] [CrossRef] [PubMed]
- Andonov, A.; Robbins, M.; Borlang, J.; Cao, J.; Hattchete, T.; Stueck, A.; Deschaumbault, Y.; Murnaghan, K.; Varga, J.; Johnston, B. Rat hepatitis E virus linked to severe acute hepatitis in an immunocompetent patient. J. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Sridhar, S.; Yip, C.C.Y.; Wu, S.S.; Cai, J.P.; Zhang, A.J.X.; Leung, K.H.; Chung, T.W.H.; Chan, J.F.W.; Chan, W.M.; Teng, J.L.L.; et al. Rat Hepatitis E Virus as Cause of Persistent Hepatitis after Liver Transplant. Emerg. Infect. Dis. 2018, 24, 2241–2250. [Google Scholar] [CrossRef]
- Dalton, H.R.; Kamar, N. Treatment of hepatitis E virus. Curr. Opin. Infect. Dis. 2016. [Google Scholar] [CrossRef]
- Kinast, V.; Burkard, T.L.; Todt, D.; Steinmann, E. Hepatitis E Virus Drug Development. Viruses 2019, 11, 485. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.W.; Smith, M.M.; Guerra, M.E.; Huang, C.C.; Bradley, D.W.; Fry, K.E.; Reyes, G.R. Hepatitis E virus (HEV): Molecular cloning and sequencing of the full-length viral genome. Virology 1991, 185, 120–131. [Google Scholar] [CrossRef]
- Nimgaonkar, I.; Ding, Q.; Schwartz, R.E.; Ploss, A. Hepatitis E virus: Advances and challenges. Nat. Rev. Gastroenterol. Hepatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Moradpour, D.; Neyts, J.; Gouttenoire, J. Update on hepatitis E virology: Implications for clinical practice. J. Hepatol. 2016, 65, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Takahashi, M.; Tanaka, T.; Yamada, K.; Nishizawa, T.; Okamoto, H. A PSAP motif in the ORF3 protein of hepatitis E virus is necessary for virion release from infected cells. J. Gen. Virol. 2011, 92, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.P.; Anang, S.; Subramani, C.; Madhvi, A.; Bakshi, K.; Srivastava, A.; Nayak, B.; Ct, R.K.; Surjit, M. Endoplasmic Reticulum Stress Induced Synthesis of a Novel Viral Factor Mediates Efficient Replication of Genotype-1 Hepatitis E Virus. PLoS Pathog. 2016, 12, e1005521. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Takahashi, M.; Kobayashi, T.; Nishizawa, T.; Nishiyama, T.; Primadharsini, P.P.; Okamoto, H. Characterization of the Quasi-Enveloped Hepatitis E Virus Particles Released by the Cellular Exosomal Pathway. J. Virol. 2017, 91, e00822-17. [Google Scholar] [CrossRef]
- Nagashima, S.; Takahashi, M.; Jirintai, S.; Kobayashi, T.; Nishizawa, T.; Okamoto, H. The membrane on the surface of hepatitis E virus particles is derived from the intracellular membrane and contains trans-Golgi network protein 2. Arch. Virol. 2014, 159, 979–991. [Google Scholar] [CrossRef]
- Feng, Z.; Hirai-Yuki, A.; McKnight, K.L.; Lemon, S.M. Naked Viruses That Aren’t Always Naked: Quasi-Enveloped Agents of Acute Hepatitis. Annu. Rev. Virol. 2014, 1, 539–560. [Google Scholar] [CrossRef]
- Yin, X.; Li, X.; Feng, Z. Role of Envelopment in the HEV Life Cycle. Viruses 2016, 8, 229. [Google Scholar] [CrossRef]
- Capelli, N.; Marion, O.; Dubois, M.; Allart, S.; Bertrand-Michel, J.; Lhomme, S.; Abravanel, F.; Izopet, J.; Chapuy-Regaud, S. Vectorial Release of Hepatitis E Virus in Polarized Human Hepatocytes. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Dao Thi, V.L.; Wu, X.; Rice, C.M. Stem Cell-Derived Culture Models of Hepatitis E Virus Infection. Cold Spring Harb. Perspect. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H. Culture systems for hepatitis E virus. J. Gastroenterol. 2013, 48, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Chapuy-Regaud, S.; Dubois, M.; Plisson-Chastang, C.; Bonnefois, T.; Lhomme, S.; Bertrand-Michel, J.; You, B.; Simoneau, S.; Gleizes, P.E.; Flan, B.; et al. Characterization of the lipid envelope of exosome encapsulated HEV particles protected from the immune response. Biochimie 2017, 141, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Ambardekar, C.; Lu, Y.; Feng, Z. Distinct Entry Mechanisms for Nonenveloped and Quasi-Enveloped Hepatitis E Viruses. J. Virol. 2016, 90, 4232–4242. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Gass, S.; Giersch, K.; Groth, A.; Kah, J.; Volz, T.; Rapp, G.; Schobel, A.; Lohse, A.W.; Polywka, S.; et al. Human liver chimeric mice as a new model of chronic hepatitis E virus infection and preclinical drug evaluation. J. Hepatol. 2016, 64, 1033–1040. [Google Scholar] [CrossRef]
- Takahashi, M.; Tanaka, T.; Takahashi, H.; Hoshino, Y.; Nagashima, S.; Mizuo, H.; Yazaki, Y.; Takagi, T.; Azuma, M.; et al. Hepatitis E Virus (HEV) strains in serum samples can replicate efficiently in cultured cells despite the coexistence of HEV antibodies: Characterization of HEV virions in blood circulation. J. Clin. Microbiol. 2010, 48, 1112–1125. [Google Scholar] [CrossRef]
- Emerson, S.U.; Nguyen, H.T.; Torian, U.; Burke, D.; Engle, R.; Purcell, R.H. Release of genotype 1 hepatitis E virus from cultured hepatoma and polarized intestinal cells depends on open reading frame 3 protein and requires an intact PXXP motif. J. Virol. 2010, 84, 9059–9069. [Google Scholar] [CrossRef] [PubMed]
- Pillot, J.; Sharma, M.D.; Lazizi, Y.; Budkowska, A.; Dauguet, C.; Galimand, M.; Sarthou, J.L. Immunological Characterization of a Viral Agent Involved in Epidemic and Sporadic Non-a,Non-B Hepatitis. Ann. Inst. Pasteur Virol. 1987, 138, 145–158. [Google Scholar] [CrossRef]
- Tsarev, S.A.; Emerson, S.U.; Reyes, G.R.; Tsareva, T.S.; Legters, L.J.; Malik, I.A.; Iqbal, M.; Purcell, R.H. Characterization of a prototype strain of hepatitis E virus. Proc. Natl. Acad. Sci. USA 1992, 89, 559–563. [Google Scholar] [CrossRef]
- Huang, C.C.; Nguyen, D.; Fernandez, J.; Yun, K.Y.; Fry, K.E.; Bradley, D.W.; Tam, A.W.; Reyes, G.R. Molecular cloning and sequencing of the Mexico isolate of hepatitis E virus (HEV). Virology 1992, 191, 550–558. [Google Scholar] [CrossRef]
- Bradley, D.; Andjaparidze, A.; Cook, E.H.; McCaustland, K.; Balayan, M.; Stetler, H.; Velazquez, O.; Robertson, B.; Humphrey, C.; Kane, M.; et al. Aetiological agent of enterically transmitted non-A, non-B hepatitis. J. Gen. Virol. 1988, 69 Pt 3, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.; Ahmed, A.; Qamar, A.; Dixon, K.; Duncan, J.F.; Islam, N.U.; Rauf, A.; Bryan, J.P.; Malik, I.A.; Legters, L.J. An outbreak of enterically transmitted non-A, non-B hepatitis in Pakistan. Am. J. Trop. Med. Hyg. 1989, 40, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.T.; Li, D.R.; Wei, J.; Huang, X.R.; Yuan, X.T.; Tian, X. Isolation and identification of hepatitis E virus in Xinjiang, China. J. Gen. Virol. 1992, 73 Pt 5, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Emerson, S.U.; Nguyen, H.; Graff, J.; Stephany, D.A.; Brockington, A.; Purcell, R.H. In vitro replication of hepatitis E virus (HEV) genomes and of an HEV replicon expressing green fluorescent protein. J. Virol. 2004, 78, 4838–4846. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dao Thi, V.L.; Liu, P.; Takacs, C.N.; Xiang, K.; Andrus, L.; Gouttenoire, J.; Moradpour, D.; Rice, C.M. Pan-Genotype Hepatitis E Virus Replication in Stem Cell-derived Hepatocellular Systems. Gastroenterology 2017. [Google Scholar] [CrossRef] [PubMed]
- Knegendorf, L.; Drave, S.A.; Dao Thi, V.L.; Debing, Y.; Brown, R.J.P.; Vondran, F.W.R.; Resner, K.; Friesland, M.; Khera, T.; Engelmann, M.; et al. Hepatitis E virus replication and interferon responses in human placental cells. Hepatol. Commun. 2018, 2, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Drave, S.A.; Debing, Y.; Walter, S.; Todt, D.; Engelmann, M.; Friesland, M.; Wedemeyer, H.; Neyts, J.; Behrendt, P.; Steinmann, E. Extra-hepatic replication and infection of hepatitis E virus in neuronal-derived cells. J. Viral Hepat. 2016, 23, 512–521. [Google Scholar] [CrossRef]
- Meng, J.; Pillot, J.; Dai, X.; Fields, H.A.; Khudyakov, Y.E. Neutralization of different geographic strains of the hepatitis E virus with anti-hepatitis E virus-positive serum samples obtained from different sources. Virology 1998, 249, 316–324. [Google Scholar] [CrossRef]
- Huang, R.T.; Li, D.R.; Wei, S.J.; Li, Q.H.; Yuan, X.T.; Geng, L.Q.; Li, X.Y.; Liu, M.X. Cell culture of sporadic hepatitis E virus in China. Clin. Diagn. Lab. Immunol. 1999, 6, 729–733. [Google Scholar]
- Huang, R.; Nakazono, N.; Ishii, K.; Li, D.; Kawamata, O.; Kawaguchi, R.; Tsukada, Y. Hepatitis E virus (87A strain) propagated in A549 cells. J. Med. Virol. 1995, 47, 299–302. [Google Scholar] [CrossRef]
- Shiota, T.; Li, T.C.; Yoshizaki, S.; Kato, T.; Wakita, T.; Ishii, K. The Hepatitis E Virus Capsid C-Terminal Region Is Essential for the Viral Life Cycle: Implication for Viral Genome Encapsidation and Particle Stabilization. J. Virol. 2013, 87, 6031–6036. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Torian, U.; Faulk, K.; Mather, K.; Engle, R.E.; Thompson, E.; Bonkovsky, H.L.; Emerson, S.U. A naturally occurring human/hepatitis E recombinant virus predominates in serum but not in faeces of a chronic hepatitis E patient and has a growth advantage in cell culture. J. Gen. Virol. 2012, 93, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Takahashi, M.; Kusano, E.; Okamoto, H. Development and evaluation of an efficient cell-culture system for Hepatitis E virus. J. Gen. Virol. 2007, 88, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Takahashi, M.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Tanaka, T.; Okamoto, H. Construction of an infectious cDNA clone of hepatitis E virus strain JE03-1760F that can propagate efficiently in cultured cells. J. Gen. Virol. 2009, 90, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.; Nguyen, H.T.; Torian, U.; Engle, R.E.; Faulk, K.; Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Purcell, R.H.; Emerson, S.U. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus-host recombinant. Proc. Natl. Acad. Sci. USA 2011, 108, 2438–2443. [Google Scholar] [CrossRef]
- Yin, X.; Li, X.L.; Ambardekar, C.; Hu, Z.M.; Lhomme, B.; Feng, Z.D. Hepatitis E virus persists in the presence of a type III interferon response. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef]
- Berto, A.; Van der Poel, W.H.M.; Hakze-van der Honing, R.; Martelli, F.; La Ragione, R.M.; Inglese, N.; Collins, J.; Grierson, S.; Johne, R.; Reetz, J.; et al. Replication of hepatitis E virus in three-dimensional cell culture. J. Virol. Methods 2013, 187, 327–332. [Google Scholar] [CrossRef]
- Oshiro, Y.; Yasue, H.; Takahashi, K.; Hattori, S.; Ideno, S.; Urayama, T.; Chiba, M.; Osari, S.; Naito, T.; Takeuchi, K.; et al. Mode of swine hepatitis E virus infection and replication in primary human hepatocytes. J. Gen. Virol. 2014, 95, 2677–2682. [Google Scholar] [CrossRef]
- Johne, R.; Reetz, J.; Ulrich, R.G.; Machnowska, P.; Sachsenroder, J.; Nickel, P.; Hofmann, J. An ORF1-rearranged hepatitis E virus derived from a chronically infected patient efficiently replicates in cell culture. J. Viral. Hepat 2014, 21, 447–456. [Google Scholar] [CrossRef]
- Schemmerer, M.; Johne, R.; Erl, M.; Jilg, W.; Wenzel, J.J. Isolation of Subtype 3c, 3e and 3f-Like Hepatitis E Virus Strains Stably Replicating to High Viral Loads in an Optimized Cell Culture System. Viruses 2019, 11, 483. [Google Scholar] [CrossRef]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Mansuy, J.M.; Rostaing, L.; Kamar, N.; Izopet, J. Characterization of the polyproline region of the hepatitis E virus in immunocompromised patients. J. Virol. 2014, 88, 12017–12025. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Takahashi, M.; Takahashi, H.; Ichiyama, K.; Hoshino, Y.; Nagashima, S.; Mizuo, H.; Okamoto, H. Development and characterization of a genotype 4 hepatitis E virus cell culture system using a HE-JF5/15F strain recovered from a fulminant hepatitis patient. J. Clin. Microbiol. 2009, 47, 1906–1910. [Google Scholar] [CrossRef] [PubMed]
- Cordoba, L.; Feagins, A.R.; Opriessnig, T.; Cossaboom, C.M.; Dryman, B.A.; Huang, Y.W.; Meng, X.J. Rescue of a genotype 4 human hepatitis E virus from cloned cDNA and characterization of intergenotypic chimeric viruses in cultured human liver cells and in pigs. J. Gen. Virol. 2012, 93, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Li, T.C.; Yang, T.T.; Yoshizaki, S.; Ami, Y.; Suzaki, Y.; Ishii, K.; Haga, K.; Nakamura, T.; Ochiai, S.; Takaji, W.; et al. Construction and characterization of an infectious cDNA clone of rat hepatitis E virus. J. Gen. Virol. 2015, 96, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H. Hepatitis E virus cell culture models. Virus Res. 2011, 161, 65–77. [Google Scholar] [CrossRef]
- Lorenzo, F.R.; Tanaka, T.; Takahashi, H.; Ichiyama, K.; Hoshino, Y.; Yamada, K.; Inoue, J.; Takahashi, M.; Okamoto, H. Mutational events during the primary propagation and consecutive passages of hepatitis E virus strain JE03-1760F in cell culture. Virus Res. 2008, 137, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.; Nguyen, H.T.; Faulk, K.; Mather, K.; Torian, U.; Engle, R.E.; Emerson, S.U. Adaptation of a genotype 3 hepatitis E virus to efficient growth in cell culture depends on an inserted human gene segment acquired by recombination. J. Virol. 2012, 86, 5697–5707. [Google Scholar] [CrossRef]
- Debing, Y.; Gisa, A.; Dallmeier, K.; Pischke, S.; Bremer, B.; Manns, M.; Wedemeyer, H.; Suneetha, P.V.; Neyts, J. A mutation in the hepatitis E virus RNA polymerase promotes its replication and associates with ribavirin treatment failure in organ transplant recipients. Gastroenterology 2014, 147, 1008–1011. [Google Scholar] [CrossRef]
- Debing, Y.; Ramiere, C.; Dallmeier, K.; Piorkowski, G.; Trabaud, M.A.; Lebosse, F.; Scholtes, C.; Roche, M.; Legras-Lachuer, C.; de Lamballerie, X.; et al. Hepatitis E virus mutations associated with ribavirin treatment failure result in altered viral fitness and ribavirin sensitivity. J. Hepatol. 2016. [Google Scholar] [CrossRef]
- Reyes, G.R.; Purdy, M.A.; Kim, J.P.; Luk, K.C.; Young, L.M.; Fry, K.E.; Bradley, D.W. Isolation of a cDNA from the virus responsible for enterically transmitted non-A, non-B hepatitis. Science 1990, 247, 1335–1339. [Google Scholar] [CrossRef]
- Panda, S.K.; Ansari, I.H.; Durgapal, H.; Agrawal, S.; Jameel, S. The in vitro-synthesized RNA from a cDNA clone of hepatitis E virus is infectious. J. Virol. 2000, 74, 2430–2437. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.W.; Haqshenas, G.; Kasorndorkbua, C.; Halbur, P.G.; Emerson, S.U.; Meng, X.J. Capped RNA transcripts of full-length cDNA clones of swine hepatitis e virus are replication competent when transfected into Huh7 cells and infectious when intrahepatically inoculated into pigs. J. Virol. 2005, 79, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Montpellier, C.; Wychowski, C.; Sayed, I.M.; Meunier, J.C.; Saliou, J.M.; Ankavay, M.; Bull, A.; Pillez, A.; Abravanel, F.; Helle, F.; et al. Hepatitis E Virus Lifecycle and Identification of 3 Forms of the ORF2 Capsid Protein. Gastroenterology 2018, 154, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Mishra, N.; Verbeken, E.; Ramaekers, K.; Dallmeier, K.; Neyts, J. A rat model for hepatitis E virus. Dis. Model Mech. 2016, 9, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.F.; Pierson, F.W.; Toth, T.E.; Meng, X.J. Construction and characterization of infectious cDNA clones of a chicken strain of hepatitis E virus (HEV), avian HEV. J. Gen. Virol. 2005, 86, 2585–2593. [Google Scholar] [CrossRef]
- Cao, D.; Huang, Y.W.; Meng, X.J. The nucleotides on the stem-loop RNA structure in the junction region of the hepatitis E virus genome are critical for virus replication. J. Virol. 2010, 84, 13040–13044. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Ying, D.; Lhomme, S.; Tang, Z.; Walker, C.M.; Xia, N.; Zheng, Z.; Feng, Z. Origin, antigenicity, and function of a secreted form of ORF2 in hepatitis E virus infection. Proc. Natl. Acad. Sci. USA 2018, 115, 4773–4778. [Google Scholar] [CrossRef]
- Ding, Q.; Heller, B.; Capuccino, J.M.V.; Song, B.K.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles (vol 114, pg 1147, 2017). Proc. Natl. Acad. Sci. USA 2017, 114, E4897. [Google Scholar] [CrossRef]
- Gouttenoire, J.; Pollan, A.; Abrami, L.; Oechslin, N.; Mauron, J.; Matter, M.; Oppliger, J.; Szkolnicka, D.; Thi, V.L.D.; van der Goot, F.G.; et al. Palmitoylation mediates membrane association of hepatitis E virus ORF3 protein and is required for infectious particle secretion. PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef]
- Meister, T.L.; Bruening, J.; Todt, D.; Steinmann, E. Cell culture systems for the study of hepatitis E virus. Antivir. Res. 2019, 163, 34–49. [Google Scholar] [CrossRef]
- Meng, J.; Dubreuil, P.; Pillot, J. A new PCR-based seroneutralization assay in cell culture for diagnosis of hepatitis E. J. Clin. Microbiol. 1997, 35, 1373–1377. [Google Scholar] [PubMed]
- Shiota, T.; Li, T.C.; Yoshizaki, S.; Kato, T.; Wakita, T.; Ishii, K. Establishment of hepatitis E virus infection-permissive and -non-permissive human hepatoma PLC/PRF/5 subclones. Microbiol. Immunol. 2015, 59, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Bendall, R.; Legrand-Abravanel, F.; Xia, N.S.; Ijaz, S.; Izopet, J.; Dalton, H.R. Hepatitis E. Lancet 2012, 379, 2477–2488. [Google Scholar] [CrossRef]
- McLean, B.N.; Gulliver, J.; Dalton, H.R. Hepatitis E virus and neurological disorders. Pract. Neurol. 2017, 17, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.P.; Kasorndorkbua, C.; Halbur, P.G.; Haqshenas, G.; Guenette, D.K.; Toth, T.E.; Meng, X.J. Evidence of extrahepatic sites of replication of the hepatitis E virus in a swine model. J. Clin. Microbiol. 2001, 39, 3040–3046. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.D.; Lemon, S.M. Peek-a-boo: Membrane hijacking and the pathogenesis of viral hepatitis. Trends Microbiol. 2014, 22, 59–64. [Google Scholar] [CrossRef]
- Navaneethan, U.; Al Mohajer, M.; Shata, M.T. Hepatitis E and pregnancy: Understanding the pathogenesis. Liver Int. 2008, 28, 1190–1199. [Google Scholar] [CrossRef]
- Zhou, X.; Huang, F.; Xu, L.; Lin, Z.M.; de Vrij, F.M.S.; Ayo-Martin, A.C.; van der Kroeg, M.; Zhao, M.Z.; Yin, Y.B.; Wang, W.S.; et al. Hepatitis E Virus Infects Neurons and Brains. J. Infect. Dis. 2017, 215, 1197–1206. [Google Scholar] [CrossRef]
- Helsen, N.; Debing, Y.; Paeshuyse, J.; Dallmeier, K.; Boon, R.; Coll, M.; Sancho-Bru, P.; Claes, C.; Neyts, J.; Verfaillie, C.M. Stem cell-derived hepatocytes: A novel model for hepatitis E virus replication. J. Hepatol. 2015. [Google Scholar] [CrossRef]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Bottger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef]
- Elaut, G.; Henkens, T.; Papeleu, P.; Snykers, S.; Vinken, M.; Vanhaecke, T.; Rogiers, V. Molecular mechanisms underlying the dedifferentiation process of isolated hepatocytes and their cultures. Curr. Drug Metab. 2006, 7, 629–660. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.W.; White, R.; Reed, E.; Short, M.; Zhang, Y.F.; Fuerst, T.R.; Lanford, R.E. In vitro propagation and production of hepatitis E virus from in vivo-infected primary macaque hepatocytes. Virology 1996, 215, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.W.; White, R.; Yarbough, P.O.; Murphy, B.J.; McAtee, C.P.; Lanford, R.E.; Fuerst, T.R. In vitro infection and replication of hepatitis E virus in primary cynomolgus macaque hepatocytes. Virology 1997, 238, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Gouilly, J.; Chen, Q.; Siewiera, J.; Cartron, G.; Levy, C.; Dubois, M.; Al-Daccak, R.; Izopet, J.; Jabrane-Ferrat, N.; El Costa, H. Genotype specific pathogenicity of hepatitis E virus at the human maternal-fetal interface. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Y.; Xu, L.; Wang, Y.J.; Wang, W.S.; Sprengers, D.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q.W. Requirement of the eukaryotic translation initiation factor 4F complex in hepatitis E virus replication. Antivir. Res. 2015, 124, 11–19. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Qu, C.; Wang, S.; Zhou, J.; Cao, W.; Xu, L.; Ma, B.; Hakim, M.S.; Yin, Y.; et al. The RNA genome of hepatitis E virus robustly triggers an antiviral interferon response. Hepatology 2018, 67, 2096–2112. [Google Scholar] [CrossRef]
- Schwartz, R.E.; Trehan, K.; Andrus, L.; Sheahan, T.P.; Ploss, A.; Duncan, S.A.; Rice, C.M.; Bhatia, S.N. Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2544–2548. [Google Scholar] [CrossRef]
- Szkolnicka, D.; Zhou, W.L.; Lucendo-Villarin, B.; Hay, D.C. Pluripotent Stem Cell-Derived Hepatocytes: Potential and Challenges in Pharmacology. Annu. Rev. Pharm. 2013, 53, 147–159. [Google Scholar] [CrossRef]
- Wang, J.; Qu, B.; Zhang, F.; Zhang, C.; Deng, W.; Dao Thi, V.L.; Xia, Y. Stem Cell-Derived Hepatocyte-Like Cells as Model for Viral Hepatitis Research. Stem Cells Int. 2019, 2019. [Google Scholar] [CrossRef]
- Xia, Y.; Carpentier, A.; Cheng, X.M.; Block, P.D.; Zhao, Y.; Zhang, Z.S.; Protzer, U.; Liang, T.J. Human stem cell-derived hepatocytes as a model for hepatitis B virus infection, spreading and virus-host interactions. J. Hepatol. 2017, 66, 494–503. [Google Scholar] [CrossRef]
- Holmgren, G.; Sjogren, A.K.; Barragan, I.; Sabirsh, A.; Sartipy, P.; Synnergren, J.; Bjorquist, P.; Ingelman-Sundberg, M.; Andersson, T.B.; Edsbagge, J. Long-Term Chronic Toxicity Testing Using Human Pluripotent Stem Cell-Derived Hepatocytes. Drug Metab. Dispos. 2014, 42, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.H.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Zare, M.; Soleimani, M.; Mohammadian, M.; Akbarzadeh, A.; Havasi, P.; Zarghami, N. Efficient biotechnological approach for lentiviral transduction of induced pluripotent stem cells. Artif. Cell Nanomed. B 2016, 44, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Dao Thi, V.L.; Wu, X.F.; Belote, R.L.; Andreo, U.; Takacs, C.N.; Fernandez, J.P.; Vale Silva, L.A.; Prallet, S.; Decker, C.C.; Fu, R.M.; et al. Stem cell-derived polarized hepatocytes. 2019; submitted work. [Google Scholar]
- Shlomai, A.; Schwartz, R.E.; Ramanan, V.; Bhatta, A.; de Jong, Y.P.; Bhatia, S.N.; Rice, C.M. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc. Natl. Acad. Sci. USA 2014, 111, 12193–12198. [Google Scholar] [CrossRef]
- Paganelli, M.; Dallmeier, K.; Nyabi, O.; Scheers, I.; Kabamba, B.; Neyts, J.; Goubau, P.; Najimi, M.; Sokal, E.M. Differentiated umbilical cord matrix stem cells as a new in vitro model to study early events during hepatitis B virus infection. Hepatology 2013, 57, 59–69. [Google Scholar] [CrossRef]
- Wu, X.; Robotham, J.M.; Lee, E.; Dalton, S.; Kneteman, N.M.; Gilbert, D.M.; Tang, H. Productive hepatitis C virus infection of stem cell-derived hepatocytes reveals a critical transition to viral permissiveness during differentiation. PLoS Pathog. 2012, 8, e1002617. [Google Scholar] [CrossRef]
- Roelandt, P.; Obeid, S.; Paeshuyse, J.; Vanhove, J.; Van Lommel, A.; Nahmias, Y.; Nevens, F.; Neyts, J.; Verfaillie, C.M. Human pluripotent stem cell-derived hepatocytes support complete replication of hepatitis C virus. J. Hepatol. 2012, 57, 246–251. [Google Scholar] [CrossRef]
- Yoshida, T.; Takayama, K.; Kondoh, M.; Sakurai, F.; Tani, H.; Sakamoto, N.; Matsuura, Y.; Mizuguchi, H.; Yagi, K. Use of human hepatocyte-like cells derived from induced pluripotent stem cells as a model for hepatocytes in hepatitis C virus infection. Biochem. Biophys. Res. Commun. 2011, 416, 119–124. [Google Scholar] [CrossRef]
- Lang, J.S.; Vera, D.; Cheng, Y.C.; Tang, H.L. Modeling Dengue Virus-Hepatic Cell Interactions Using Human Pluripotent Stem Cell-Derived Hepatocyte-like Cells. Stem Cell Rep. 2016, 7, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.F.; Thi, V.L.D.; Huang, Y.M.; Billerbeck, E.; Saha, D.; Hoffmann, H.H.; Wang, Y.M.; Silva, L.A.V.; Sarbanes, S.; Sun, T.; et al. Intrinsic Immunity Shapes Viral Resistance of Stem Cells. Cell 2018, 172, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Tricot, T.; Helsen, N.; Kaptein, S.J.F.; Neyts, J.; Verfaillie, C.M. Human stem cell-derived hepatocyte-like cells support Zika virus replication and provide a relevant model to assess the efficacy of potential antivirals. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; March, S.; Galstian, A.; Hanson, K.; Carvalho, T.; Mota, M.M.; Bhatia, S.N. Hypoxia promotes liver-stage malaria infection in primary human hepatocytes in vitro. Dis. Model. Mech. 2014, 7, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Dao Thi, V.L.; Debing, Y.; Wu, X.; Rice, C.M.; Neyts, J.; Moradpour, D.; Gouttenoire, J. Sofosbuvir Inhibits Hepatitis E Virus Replication In Vitro and Results in an Additive Effect When Combined With Ribavirin. Gastroenterology 2016, 150, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, X.; Debing, Y.; Chen, K.; Van Der Laan, L.J.; Neyts, J.; Janssen, H.L.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. Calcineurin inhibitors stimulate and mycophenolic acid inhibits replication of hepatitis E virus. Gastroenterology 2014, 146, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Lhomme, S.; Abravanel, F.; Cointault, O.; Esposito, L.; Cardeau-Desangles, I.; Del Bello, A.; Dorr, G.; Lavayssiere, L.; Nogier, M.B.; et al. An Early Viral Response Predicts the Virological Response to Ribavirin in Hepatitis E Virus Organ Transplant Patients. Transplantation 2015, 99, 2124–2131. [Google Scholar] [CrossRef]
- Zhou, X.Y.; Wang, Y.J.; Metselaar, H.J.; Janssen, H.L.A.; Peppelenbosch, M.P.; Pan, Q.W. Rapamycin and everolimus facilitate hepatitis E virus replication: Revealing a basal defense mechanism of PI3K-PKB-mTOR pathway. J. Hepatol. 2014, 61, 746–754. [Google Scholar] [CrossRef]
- Treyer, A.; Musch, A. Hepatocyte Polarity. Compr. Physiol. 2013, 3, 243–287. [Google Scholar] [CrossRef]
- Lenggenhager, D.; Gouttenoire, J.; Malehmir, M.; Bawohl, M.; Honcharova-Biletska, H.; Kreutzer, S.; Semela, D.; Neuweiler, J.; Hurlimann, S.; Aepli, P.; et al. Visualization of hepatitis E virus RNA and proteins in the human liver. J. Hepatol. 2017, 67, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Gural, N.; Mancio-Silva, L.; He, J.; Bhatia, S.N. Engineered Livers for Infectious Diseases. Cell Mol. Gastroenterol. Hepatol. 2018, 5, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Tascher, G.; Burban, A.; Camus, S.; Plumel, M.; Chanon, S.; Le Guevel, R.; Shevchenko, V.; Van Dorsselaer, A.; Lefai, E.; Guguen-Guillouzo, C.; et al. In-Depth Proteome Analysis Highlights HepaRG Cells as a Versatile Cell System Surrogate for Primary Human Hepatocytes. Cells-Basel 2018, 8, 192. [Google Scholar] [CrossRef] [PubMed]
- Rogee, S.; Talbot, N.; Caperna, T.; Bouquet, J.; Barnaud, E.; Pavio, N. New models of hepatitis E virus replication in human and porcine hepatocyte cell lines. J. Gen. Virol. 2013, 94, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhou, X.; Wang, W.; Wang, Y.; Yin, Y.; Laan, L.J.; Sprengers, D.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. IFN regulatory factor 1 restricts hepatitis E virus replication by activating STAT1 to induce antiviral IFN-stimulated genes. Faseb J. 2016, 30, 3352–3367. [Google Scholar] [CrossRef] [PubMed]
- Kelm, J.M.; Timmins, N.E.; Brown, C.J.; Fussenegger, M.; Nielsen, L.K. Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol. Bioeng. 2003, 83, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Molina-Jimenez, F.; Benedicto, I.; Viet, L.D.T.; Gondar, V.; Lavillette, D.; Marin, J.J.; Briz, O.; Moreno-Otero, R.; Aldabe, R.; Baumert, T.F.; et al. Matrigel-embedded 3D culture of Huh-7 cells as a hepatocyte-like polarized system to study hepatitis C virus cycle. Virology 2012, 425, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Ananthanarayanan, A.; Nugraha, B.; Triyatni, M.; Hart, S.; Sankuratri, S.; Yu, H. Scalable Spheroid Model of Human Hepatocytes for Hepatitis C Infection and Replication. Mol. Pharm. 2014, 11, 2106–2114. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.J.; Elazar, M.; Xiong, A.M.; Lee, W.; Chiao, E.; Baker, J.; Frank, C.W.; Glenn, J.S. Viral infection of human progenitor and liver-derived cells encapsulated in three-dimensional PEG-based hydrogel. Biomed. Mater. 2009, 4. [Google Scholar] [CrossRef]
- Aly, H.H.; Shimotohno, K.; Hijikata, M. 3D cultured immortalized human hepatocytes useful to develop drugs for blood-borne HCV. Biochem. Biophys. Res. Commun. 2009, 379, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Aizaki, H.; Nagamori, S.; Matsuda, M.; Kawakami, H.; Hashimoto, O.; Ishiko, H.; Kawada, M.; Matsuura, T.; Hasumura, S.; Matsuura, Y.; et al. Production and release of infectious hepatitis C virus from human liver cell cultures in the three-dimensional radial-flow bioreactor. Virology 2003, 314, 16–25. [Google Scholar] [CrossRef][Green Version]
- Bouwknegt, M.; Frankena, K.; Rutjes, S.A.; Wellenberg, G.J.; Husman, A.M.D.R.; van der Poel, W.H.M.; de Jong, M.C.M. Estimation of hepatitis E virus transmission among pigs due to contact-exposure. Vet. Res. 2008, 39. [Google Scholar] [CrossRef] [PubMed]
- Khetani, S.R.; Bhatia, S.N. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 2008, 26, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Khetani, S.R.; Jones, C.T.; Syder, A.J.; Trehan, K.; Gaysinskaya, V.A.; Mu, K.; Ritola, K.; Rice, C.M.; Bhatia, S.N. Persistent hepatitis C virus infection in microscale primary human hepatocyte cultures. Proc. Natl. Acad. Sci. USA 2010, 107, 3141–3145. [Google Scholar] [CrossRef] [PubMed]
- March, S.; Ng, S.; Velmurugan, S.; Galstian, A.; Shan, J.; Logan, D.J.; Carpenter, A.E.; Thomas, D.; Sim, B.K.L.; Mota, M.M.; et al. A Microscale Human Liver Platform that Supports the Hepatic Stages of Plasmodium falciparum and vivax. Cell Host Microbe 2013, 14, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Berger, D.R.; Ware, B.R.; Davidson, M.D.; Allsup, S.R.; Khetani, S.R. Enhancing the Functional Maturity of Induced Pluripotent Stem Cell-Derived Human Hepatocytes by Controlled Presentation of Cell-Cell Interactions In Vitro. Hepatology 2015, 61, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, K.; Owens, D.J.; Raju, R.; Firpo, M.; O’Brien, T.D.; Verfaillie, C.M.; Hu, W.S. Spheroid Culture for Enhanced Differentiation of Human Embryonic Stem Cells to Hepatocyte-Like Cells. Stem Cells Dev. 2014, 23, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.R.; Takebe, T.; Miyazaki, L.; Takayama, M.; Koike, H.; Kimura, M.; Enomura, M.; Zheng, Y.W.; Sekine, K.; Taniguchi, H. Efficient Hepatic Differentiation of Human Induced Pluripotent Stem Cells in a Three-Dimensional Microscale Culture. Stem Cells Tissue Repair Methods Protoc. 2014, 1210, 131–141. [Google Scholar] [CrossRef]
- Luo, Y.; Lou, C.; Zhang, S.; Zhu, Z.Y.; Xing, Q.Z.; Wang, P.; Liu, T.; Liu, H.; Li, C.L.; Shi, W.X.; et al. Three-dimensional hydrogel culture conditions promote the differentiation of human induced pluripotent stem cells into hepatocytes. Cytotherapy 2018, 20, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.Z.; Zheng, Y.W.; Ogawa, M.; Miyagi, E.; Taniguchi, H. Human liver organoids generated with single donor-derived multiple cells rescue mice from acute liver failure. Stem Cell Res. 2018, 9. [Google Scholar] [CrossRef]
- Nie, Y.Z.; Zheng, Y.W.; Miyakawa, K.; Murata, S.; Zhang, R.R.; Sekine, K.; Ueno, Y.; Takebe, T.; Wakita, T.; Ryo, A.; et al. Recapitulation of hepatitis B virus-host interactions in liver organoids from human induced pluripotent stem cells. Ebiomedicine 2018, 35, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, I.J.; Raub, T.J.; Borchardt, R.T. Characterization of the Human-Colon Carcinoma Cell-Line (Caco-2) as a Model System for Intestinal Epithelial Permeability. Gastroenterology 1989, 96, 736–749. [Google Scholar] [CrossRef]
- Hirai-Yuki, A.; Hensley, L.; Whitmire, J.K.; Lemon, S.M. Biliary Secretion of Quasi-Enveloped Human Hepatitis A Virus. Mbio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Snooks, M.J.; Bhat, P.; Mackenzie, J.; Counihan, N.A.; Vaughan, N.; Anderson, D.A. Vectorial entry and release of hepatitis a virus in polarized human hepatocytes. J. Virol. 2008, 82, 8733–8742. [Google Scholar] [CrossRef] [PubMed]
- Bhat, P.; Snooks, M.J.; Anderson, D.A. Hepatocytes traffic and export hepatitis B virus basolaterally by polarity-dependent mechanisms. J. Virol. 2011, 85, 12474–12481. [Google Scholar] [CrossRef]
- Belouzard, S.; Danneels, A.; Feneant, L.; Seron, K.; Rouille, Y.; Dubuisson, J. Entry and Release of Hepatitis C Virus in Polarized Human Hepatocytes. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Everson, G.T.; Polokoff, M.A. Hepg2—A Human Hepatoblastoma Cell-Line Exhibiting Defects in Bile-Acid Synthesis and Conjugation. J. Biol. Chem. 1986, 261, 2197–2201. [Google Scholar]



{kind=link}
{kind=link}
| (a) | |||||
| Genotype | Strain | cDNA Clone | Adaptation in Cell Culture | Susceptible Cells | Ref |
| ENANB | Yes | PLC/PRF/5 | [29] | ||
| 1 | Sar-55 | Yes | Huh 7 | [28] | |
| PLC/PRF/5 | [35] | ||||
| Caco-2 | [28] | ||||
| HLCs | [36] | ||||
| Sar55/S17 | Yes | BeWo | [37] | ||
| Introduced S17 sequence from Kernow-C1/p6 | JEG-1 | [37] | |||
| M03.13 | [38] | ||||
| F23 | PLC/PRF/5 | [39] | |||
| 87A | 2BS | [40,41] | |||
| A549 | [40,41] | ||||
| 2 | MEX-14 | HLCs | [36] | ||
| 3 | HEV83-2-27 | Yes | PLC/PRF/5 | [42] | |
| LBPR-0379 | Yes | Insertion of 39 amino acids from S19 ribosomal protein in wt virus, selected in cell culture | HepG2 | [43] | |
| JE03-1706 | Yes | Gained 13 mutations after 10 passages | PLC/PRF/5 | [44] | |
| A549 | [45] | ||||
| Kernow-C1 | Yes | S17 insertion and 54 synonymous mutations in wt virus, selected after six passages in cell culture | Hep G2 | [46] | |
| Huh-7 | [46] | ||||
| SH-SH5Y | [38] | ||||
| SK-N-MC | [38] | ||||
| U97 | [38] | ||||
| U343 | [38] | ||||
| M03.13 | [38] | ||||
| BeWo | [37] | ||||
| JEG-3 | [37] | ||||
| LLC-PK1 (swine) | [46] | ||||
| OHH1.Li (deer) | [46] | ||||
| MDCK (dog) | [46] | ||||
| CRFK (cat) | [46] | ||||
| LLC-RK1 (rabbit) | [46] | ||||
| CMH (chicken) | [46] | ||||
| Hepa 1-6 (mouse) | [46] | ||||
| PHH | [47] | ||||
| HLCs | [36] | ||||
| NLSWIE3 | PLC/PRF/5 | [48] | |||
| swJR-P5 | PHH | [49] | |||
| swJB-E10 | PHH | [49] | |||
| swJB-M8 | PHH | [49] | |||
| US-2 | HLCs | [36] | |||
| 47832 | Insertion in the hypervariable region of ORF1 in wt virus, gained 25 point mutations after two passages | A549 PLC/PRF/5 HepG2/C3A Huh-7 Lunet BLR MRC-5 | [50] | ||
| [51] | |||||
| [51] | |||||
| [51] | |||||
| [51] | |||||
| 14-16753 | A549 PLC/PRF/5 HepG2/C3A Huh-7 Lunet BLR | [51] | |||
| [51] | |||||
| [51] | |||||
| [51] | |||||
| 14-22707 | A549 PLC/PRF/5 Huh-7 Lunet BLR | [51] | |||
| [51] | |||||
| [51] | |||||
| 15-22016 | PLC/PRF/5 HepG2/C3A Huh-7 Lunet BLR | [51] | |||
| [51] | |||||
| [51] | |||||
| TLS 09/M0 | A 75-nt insertion in the polyproline region after 48 months of infection in vivo | F2 (subclone of HepG2/C3A) | [52] | ||
| 4 | HE-JF5 | Gained 10 mutations after six passages | PLC/PRF/5 A549 | [53] | |
| TW6196 | Yes | HepG2 | [54] | ||
| swJB-H7 | PHH | [49] | |||
| Rat | R63/DEU/2009 8 | Yes | Gained 9 mutations after two passages | PLC/PRF/5 | [55] |
| (b) | |||||
| Patient Isolates | cDNA Clones | ||||
| Replication in cell culture | + | ++ | |||
| Replication in animal models | ++ | + | |||
| Physiological relevance | +++ | + | |||
| Pan-genotype | +++ | - | |||
| Reproducibility | - | +++ | |||
| Genetic manipulation | - | +++ | |||
| Cancer Cells | Stem Cell-Derived Cells | Primary Cells | |
|---|---|---|---|
| Availability | +++ | ++ | - |
| Reproducibility | +++ | ++ | - |
| Genetic modification | +++ | ++ | - |
| Physiologically relevant | - | ++ | +++ |
| Pan-genotype | - | ++ | +++ |
| Cellular polarity | + | ++ | ++ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, R.M.; Decker, C.C.; Dao Thi, V.L. Cell Culture Models for Hepatitis E Virus. Viruses 2019, 11, 608. https://doi.org/10.3390/v11070608
Fu RM, Decker CC, Dao Thi VL. Cell Culture Models for Hepatitis E Virus. Viruses. 2019; 11(7):608. https://doi.org/10.3390/v11070608
Chicago/Turabian StyleFu, Rebecca Menhua, Charlotte Caroline Decker, and Viet Loan Dao Thi. 2019. "Cell Culture Models for Hepatitis E Virus" Viruses 11, no. 7: 608. https://doi.org/10.3390/v11070608
APA StyleFu, R. M., Decker, C. C., & Dao Thi, V. L. (2019). Cell Culture Models for Hepatitis E Virus. Viruses, 11(7), 608. https://doi.org/10.3390/v11070608