Advances in the Development of Antiviral Strategies against Parvovirus B19


Abstract
1. Introduction
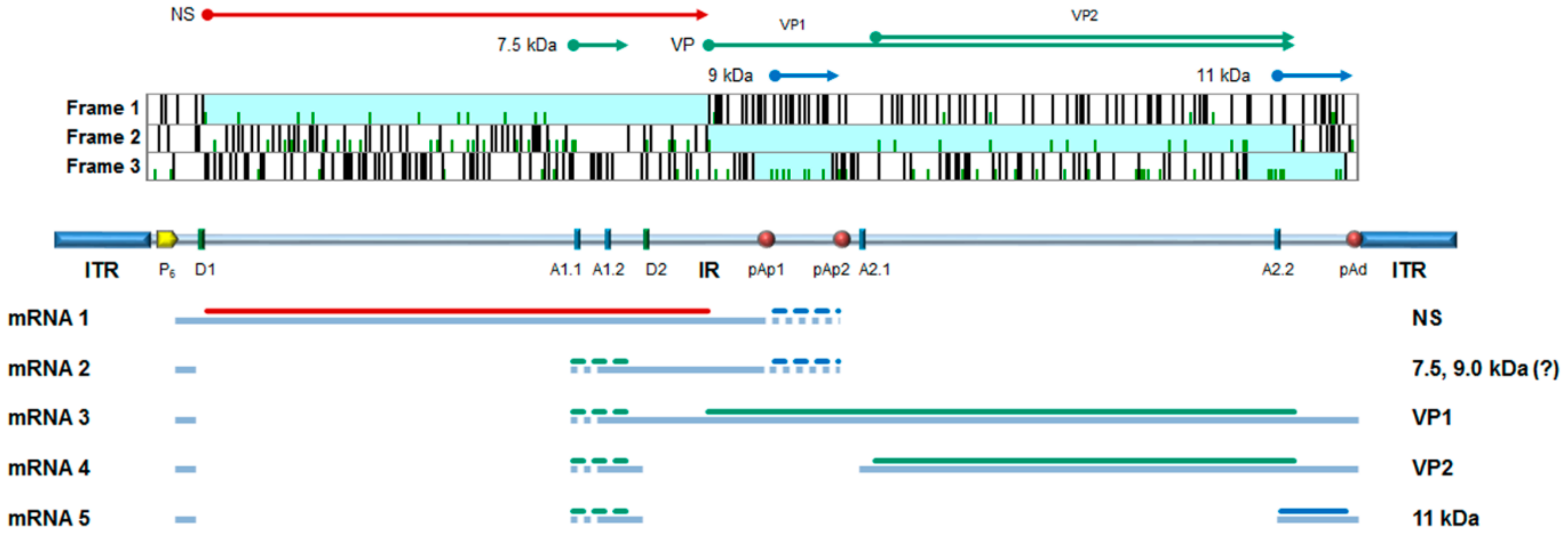
2. B19V Structure
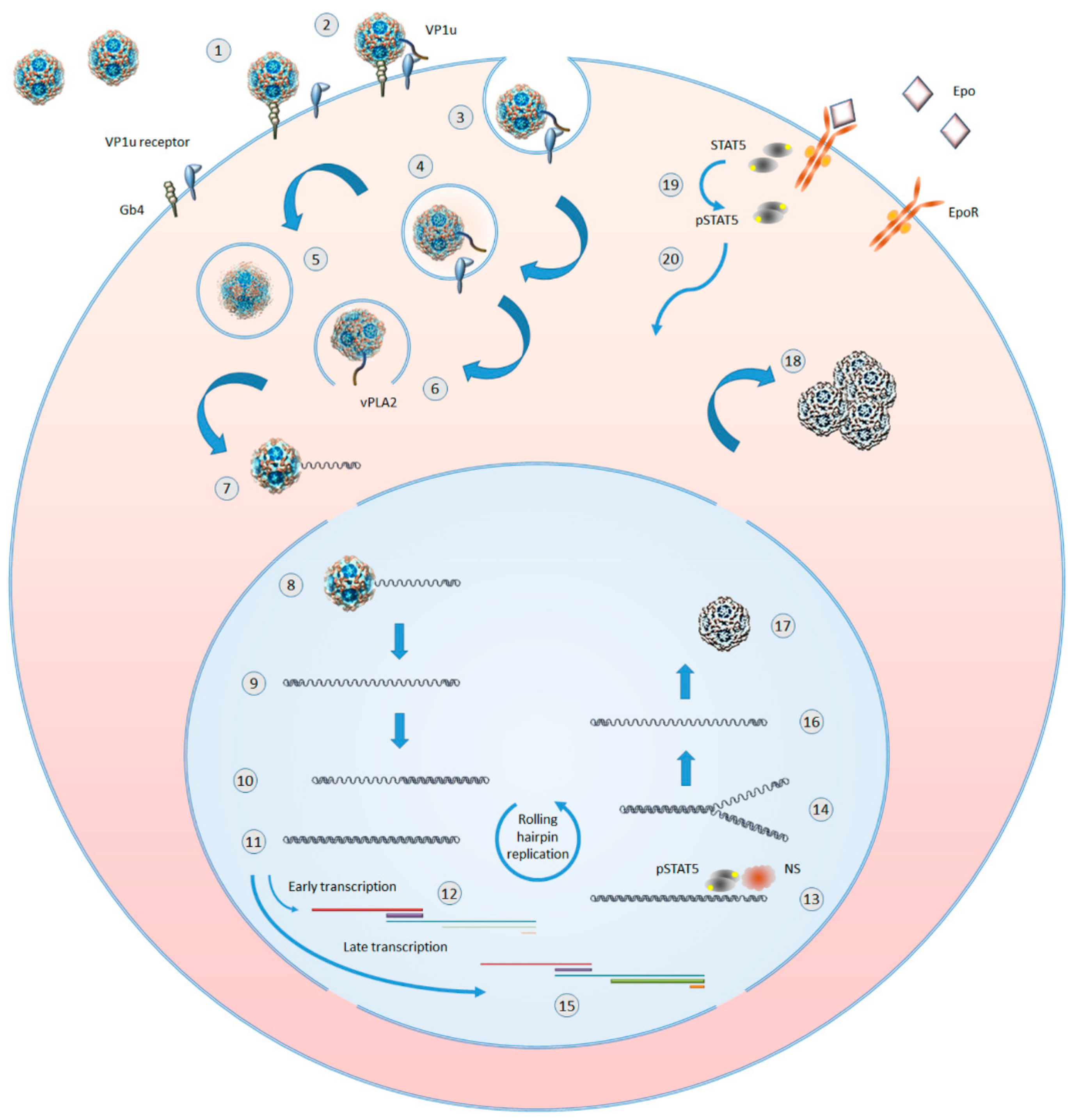
3. The Lifecycle
4. The Pathologies
5. Need for Treatment and Current Options
6. Passive Immunization
7. The Quest for Antiviral Agents
7.1. Hydroxyurea
7.2. Nucleotide Analogues: Cidofovir (CDV) and Brincidofovir (BCV)
7.3. Serendipity Approach: Coumarin Derivatives
7.4. Direct Antiviral Agents
7.5. Other Compounds
8. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family Parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Gallinella, G. Parvovirus B19 Achievements and Challenges. ISRN Virol. 2013. [Google Scholar] [CrossRef]
- Qiu, J.; Soderlund-Venermo, M.; Young, N.S. Human Parvoviruses. Clin. Microbiol. Rev. 2017, 30, 43–113. [Google Scholar] [CrossRef] [PubMed]
- Mietzsch, M.; Penzes, J.J.; Agbandje-McKenna, M. Twenty-Five Years of Structural Parvovirology. Viruses 2019, 11, 362. [Google Scholar] [CrossRef] [PubMed]
- Gallinella, G. The clinical use of parvovirus B19 assays: Recent advances. Expert Rev. Mol. Diagn. 2018, 18, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, R.; Buczkowska, A.; Mikolajewicz, K.; Krotkiewski, H.; Czerwinski, M. P1PK, GLOB, and FORS blood group systems and GLOB collection: Biochemical and clinical aspects. Do we understand it all yet? Transfus. Med. Rev. 2014, 28, 126–136. [Google Scholar] [CrossRef]
- Brown, K.E.; Anderson, S.M.; Young, N.S. Erythrocyte P antigen: Cellular receptor for B19 parvovirus. Science 1993, 262, 114–117. [Google Scholar] [CrossRef]
- Brown, K.E.; Hibbs, J.R.; Gallinella, G.; Anderson, S.M.; Lehman, E.D.; McCarthy, P.; Young, N.S. Resistance to parvovirus B19 infection due to lack of virus receptor (erythrocyte P antigen). N. Engl. J. Med. 1994, 330, 1192–1196. [Google Scholar] [CrossRef]
- Chipman, P.R.; Agbandje-McKenna, M.; Kajigaya, S.; Brown, K.E.; Young, N.S.; Baker, T.S.; Rossmann, M.G. Cryo-electron microscopy studies of empty capsids of human parvovirus B19 complexed with its cellular receptor. Proc. Natl. Acad. Sci. USA 1996, 93, 7502–7506. [Google Scholar] [CrossRef]
- Kaufmann, B.; Baxa, U.; Chipman, P.R.; Rossmann, M.G.; Modrow, S.; Seckler, R. Parvovirus B19 does not bind to membrane-associated globoside in vitro. Virology 2005, 332, 189–198. [Google Scholar] [CrossRef]
- Nasir, W.; Nilsson, J.; Olofsson, S.; Bally, M.; Rydell, G.E. Parvovirus B19 VLP recognizes globoside in supported lipid bilayers. Virology 2014, 456–457, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Bonsch, C.; Zuercher, C.; Lieby, P.; Kempf, C.; Ros, C. The globoside receptor triggers structural changes in the B19 virus capsid that facilitate virus internalization. J. Virol. 2010, 84, 11737–11746. [Google Scholar] [CrossRef] [PubMed]
- Leisi, R.; Ruprecht, N.; Kempf, C.; Ros, C. Parvovirus B19 uptake is a highly selective process controlled by VP1u, a novel determinant of viral tropism. J. Virol. 2013, 87, 13161–13167. [Google Scholar] [CrossRef] [PubMed]
- Leisi, R.; Di Tommaso, C.; Kempf, C.; Ros, C. The Receptor-Binding Domain in the VP1u Region of Parvovirus B19. Viruses 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Leisi, R.; Von Nordheim, M.; Ros, C.; Kempf, C. The VP1u Receptor Restricts Parvovirus B19 Uptake to Permissive Erythroid Cells. Viruses 2016, 8, 265. [Google Scholar] [CrossRef] [PubMed]
- Quattrocchi, S.; Ruprecht, N.; Bonsch, C.; Bieli, S.; Zurcher, C.; Boller, K.; Kempf, C.; Ros, C. Characterization of the early steps of human parvovirus B19 infection. J. Virol. 2012, 86, 9274–9284. [Google Scholar] [CrossRef] [PubMed]
- Caliaro, O.; Marti, A.; Ruprecht, N.; Leisi, R.; Subramanian, S.; Hafenstein, S.; Ros, C. Parvovirus B19 Uncoating Occurs in the Cytoplasm without Capsid Disassembly and It Is Facilitated by Depletion of Capsid-Associated Divalent Cations. Viruses 2019, 11, 430. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Qiu, J. Human parvovirus B19: A mechanistic overview of infection and DNA replication. Future Virol. 2015, 10, 155–167. [Google Scholar] [CrossRef]
- Ganaie, S.S.; Qiu, J. Recent Advances in Replication and Infection of Human Parvovirus B19. Front. Cell. Infect. Microbiol. 2018, 8, 166. [Google Scholar] [CrossRef]
- Bua, G.; Manaresi, E.; Bonvicini, F.; Gallinella, G. Parvovirus B19 Replication and Expression in Differentiating Erythroid Progenitor Cells. PLoS ONE 2016, 11, e0148547. [Google Scholar] [CrossRef]
- Chen, A.Y.; Guan, W.; Lou, S.; Liu, Z.; Kleiboeker, S.; Qiu, J. Role of erythropoietin receptor signaling in parvovirus B19 replication in human erythroid progenitor cells. J. Virol. 2010, 84, 12385–12396. [Google Scholar] [CrossRef]
- Chen, A.Y.; Kleiboeker, S.; Qiu, J. Productive parvovirus B19 infection of primary human erythroid progenitor cells at hypoxia is regulated by STAT5A and MEK signaling but not HIFalpha. PLoS Pathog. 2011, 7, e1002088. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zou, W.; Wang, Z.; Xiong, M.; Chen, A.Y.; Xu, P.; Ganaie, S.S.; Badawi, Y.; Kleiboeker, S.; Nishimune, H.; Ye, S.Q.; et al. Human Parvovirus B19 Utilizes Cellular DNA Replication Machinery for Viral DNA Replication. J. Virol. 2018, 92, e01881-17. [Google Scholar] [CrossRef]
- Ganaie, S.S.; Zou, W.; Xu, P.; Deng, X.; Kleiboeker, S.; Qiu, J. Phosphorylated STAT5 directly facilitates parvovirus B19 DNA replication in human erythroid progenitors through interaction with the MCM complex. PLoS Pathog. 2017, 13, e1006370. [Google Scholar] [CrossRef]
- Ganaie, S.S.; Chen, A.Y.; Huang, C.; Xu, P.; Kleiboeker, S.; Du, A.; Qiu, J. RNA Binding Protein RBM38 Regulates Expression of the 11-Kilodalton Protein of Parvovirus B19, Which Facilitates Viral DNA Replication. J. Virol. 2018, 92, e02050-17. [Google Scholar] [CrossRef]
- Xu, P.; Chen, A.Y.; Ganaie, S.S.; Cheng, F.; Shen, W.; Wang, X.; Kleiboeker, S.; Li, Y.; Qiu, J. The 11-Kilodalton Nonstructural Protein of Human Parvovirus B19 Facilitates Viral DNA Replication by Interacting with Grb2 through Its Proline-Rich Motifs. J. Virol. 2019, 93, e01464-18. [Google Scholar] [CrossRef]
- Chen, A.Y.; Qiu, J. Parvovirus infection-induced cell death and cell cycle arrest. Future Virol. 2010, 5, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Qiu, J. Parvovirus infection-induced DNA damage response. Future Virol. 2013, 8, 245–257. [Google Scholar] [CrossRef]
- Pyoria, L.; Toppinen, M.; Mantyla, E.; Hedman, L.; Aaltonen, L.M.; Vihinen-Ranta, M.; Ilmarinen, T.; Soderlund-Venermo, M.; Hedman, K.; Perdomo, M.F. Extinct type of human parvovirus B19 persists in tonsillar B cells. Nat. Commun. 2017, 8, 14930. [Google Scholar] [CrossRef]
- Von Kietzell, K.; Pozzuto, T.; Heilbronn, R.; Grossl, T.; Fechner, H.; Weger, S. Antibody-mediated enhancement of parvovirus B19 uptake into endothelial cells mediated by a receptor for complement factor C1q. J. Virol. 2014, 88, 8102–8115. [Google Scholar] [CrossRef] [PubMed]
- Adamson-Small, L.A.; Ignatovich, I.V.; Laemmerhirt, M.G.; Hobbs, J.A. Persistent parvovirus B19 infection in non-erythroid tissues: Possible role in the inflammatory and disease process. Virus Res. 2014, 190, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Bua, G.; Gallinella, G. How does parvovirus B19 DNA achieve lifelong persistence in human cells? Future Virol. 2017, 12, 549–553. [Google Scholar] [CrossRef]
- Janovitz, T.; Wong, S.; Young, N.S.; Oliveira, T.; Falck-Pedersen, E. Parvovirus B19 integration into human CD36+ erythroid progenitor cells. Virology 2017, 511, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Norja, P.; Hokynar, K.; Aaltonen, L.M.; Chen, R.; Ranki, A.; Partio, E.K.; Kiviluoto, O.; Davidkin, I.; Leivo, T.; Eis-Hubinger, A.M.; et al. Bioportfolio: Lifelong persistence of variant and prototypic erythrovirus DNA genomes in human tissue. Proc. Natl. Acad. Sci. USA 2006, 103, 7450–7453. [Google Scholar] [CrossRef] [PubMed]
- Toppinen, M.; Perdomo, M.F.; Palo, J.U.; Simmonds, P.; Lycett, S.J.; Soderlund-Venermo, M.; Sajantila, A.; Hedman, K. Bones hold the key to DNA virus history and epidemiology. Sci. Rep. 2015, 5, 17226. [Google Scholar] [CrossRef] [PubMed]
- Muhlemann, B.; Margaryan, A.; Damgaard, P.B.; Allentoft, M.E.; Vinner, L.; Hansen, A.J.; Weber, A.; Bazaliiskii, V.I.; Molak, M.; Arneborg, J.; et al. Ancient human parvovirus B19 in Eurasia reveals its long-term association with humans. Proc. Natl. Acad. Sci. USA 2018, 115, 7557–7562. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.R. A review of blood diseases and cytopenias associated with human parvovirus B19 infection. Rev. Med. Virol. 2015, 25, 224–240. [Google Scholar] [CrossRef]
- Tsitsikas, D.A.; Gallinella, G.; Patel, S.; Seligman, H.; Greaves, P.; Amos, R.J. Bone marrow necrosis and fat embolism syndrome in sickle cell disease: Increased susceptibility of patients with non-SS genotypes and a possible association with human parvovirus B19 infection. Blood Rev. 2014, 28, 23–30. [Google Scholar] [CrossRef]
- Verdonschot, J.; Hazebroek, M.; Merken, J.; Debing, Y.; Dennert, R.; Brunner-La Rocca, H.P.; Heymans, S. Relevance of cardiac parvovirus B19 in myocarditis and dilated cardiomyopathy: Review of the literature. Eur. J. Heart Fail. 2016, 18, 1430–1441. [Google Scholar] [CrossRef]
- Kerr, J.R. The role of parvovirus B19 in the pathogenesis of autoimmunity and autoimmune disease. J. Clin. Pathol. 2016, 69, 279–291. [Google Scholar] [CrossRef]
- Lunardi, C.; Tinazzi, E.; Bason, C.; Dolcino, M.; Corrocher, R.; Puccetti, A. Human parvovirus B19 infection and autoimmunity. Autoimmun. Rev. 2008, 8, 116–120. [Google Scholar] [CrossRef]
- Thammasri, K.; Rauhamaki, S.; Wang, L.; Filippou, A.; Kivovich, V.; Marjomaki, V.; Naides, S.J.; Gilbert, L. Human parvovirus B19 induced apoptotic bodies contain altered self-antigens that are phagocytosed by antigen presenting cells. PLoS ONE 2013, 8, e67179. [Google Scholar] [CrossRef]
- Puttaraksa, K.; Pirttinen, H.; Karvonen, K.; Nykky, J.; Naides, S.J.; Gilbert, L. Parvovirus B19V Non-structural Protein NS1 Induces dsDNA Autoantibodies and End Organ Damage in Non-autoimmune Mice. J. Infect. Dis. 2018, 219, 1418–1429. [Google Scholar] [CrossRef]
- Bonvicini, F.; Bua, G.; Gallinella, G. Parvovirus B19 infection in pregnancy-awareness and opportunities. Curr. Opin. Virol. 2017, 27, 8–14. [Google Scholar] [CrossRef]
- Pasquinelli, G.; Bonvicini, F.; Foroni, L.; Salfi, N.; Gallinella, G. Placental endothelial cells can be productively infected by Parvovirus B19. J. Clin. Virol. 2009, 44, 33–38. [Google Scholar] [CrossRef]
- Bonvicini, F.; Puccetti, C.; Salfi, N.C.; Guerra, B.; Gallinella, G.; Rizzo, N.; Zerbini, M. Gestational and fetal outcomes in B19 maternal infection: A problem of diagnosis. J. Clin. Microbiol. 2011, 49, 3514–3518. [Google Scholar] [CrossRef]
- Bascietto, F.; Liberati, M.; Murgano, D.; Buca, D.; Iacovelli, A.; Flacco, M.E.; Manzoli, L.; Familiari, A.; Scambia, G.; D’Antonio, F. Outcome of fetuses with congenital parvovirus B19 infection: Systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2018, 52, 569–576. [Google Scholar] [CrossRef]
- Xiong, Y.Q.; Tan, J.; Liu, Y.M.; He, Q.; Li, L.; Zou, K.; Sun, X. The risk of maternal parvovirus B19 infection during pregnancy on fetal loss and fetal hydrops: A systematic review and meta-analysis. J. Clin. Virol. 2019, 114, 12–20. [Google Scholar] [CrossRef]
- Mage, V.; Lipsker, D.; Barbarot, S.; Bessis, D.; Chosidow, O.; Del Giudice, P.; Aractingi, S.; Avouac, J.; Bernier, C.; Descamps, V.; et al. Different patterns of skin manifestations associated with parvovirus B19 primary infection in adults. J. Am. Acad. Dermatol. 2014, 71, 62–69. [Google Scholar] [CrossRef]
- Dijkmans, A.C.; de Jong, E.P.; Dijkmans, B.A.; Lopriore, E.; Vossen, A.; Walther, F.J.; Oepkes, D. Parvovirus B19 in pregnancy: Prenatal diagnosis and management of fetal complications. Curr. Opin. Obstet. Gynecol. 2012, 24, 95–101. [Google Scholar] [CrossRef]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef]
- Bansal, G.P.; Hatfield, J.A.; Dunn, F.E.; Kramer, A.A.; Brady, F.; Riggin, C.H.; Collett, M.S.; Yoshimoto, K.; Kajigaya, S.; Young, N.S. Candidate recombinant vaccine for human B19 parvovirus. J. Infect. Dis. 1993, 167, 1034–1044. [Google Scholar] [CrossRef]
- Ballou, W.R.; Reed, J.L.; Noble, W.; Young, N.S.; Koenig, S. Safety and immunogenicity of a recombinant parvovirus B19 vaccine formulated with MF59C.1. J. Infect. Dis. 2003, 187, 675–678. [Google Scholar] [CrossRef]
- Bernstein, D.I.; El Sahly, H.M.; Keitel, W.A.; Wolff, M.; Simone, G.; Segawa, C.; Wong, S.; Shelly, D.; Young, N.S.; Dempsey, W. Safety and immunogenicity of a candidate parvovirus B19 vaccine. Vaccine 2011, 29, 7357–7363. [Google Scholar] [CrossRef]
- Chandramouli, S.; Medina-Selby, A.; Coit, D.; Schaefer, M.; Spencer, T.; Brito, L.A.; Zhang, P.; Otten, G.; Mandl, C.W.; Mason, P.W.; et al. Generation of a parvovirus B19 vaccine candidate. Vaccine 2013, 31, 3872–3878. [Google Scholar] [CrossRef]
- Penkert, R.R.; Young, N.S.; Surman, S.L.; Sealy, R.E.; Rosch, J.; Dormitzer, P.R.; Settembre, E.C.; Chandramouli, S.; Wong, S.; Hankins, J.S.; et al. Saccharomyces cerevisiae-derived virus-like particle parvovirus B19 vaccine elicits binding and neutralizing antibodies in a mouse model for sickle cell disease. Vaccine 2017, 35, 3615–3620. [Google Scholar] [CrossRef]
- Mouthon, L.; Lortholary, O. Intravenous immunoglobulins in infectious diseases: Where do we stand? Clin. Microbiol. Infect. 2003, 9, 333–338. [Google Scholar] [CrossRef]
- Mouthon, L.; Guillevin, L.; Tellier, Z. Intravenous immunoglobulins in autoimmune- or parvovirus B19-mediated pure red-cell aplasia. Autoimmun. Rev. 2005, 4, 264–269. [Google Scholar] [CrossRef]
- Mouthon, L.; Michel, M.; Gandre, C.; Montagnier-Petrissans, C.; Chevreul, K. Costs of intravenous immunoglobulin therapy in patients with unconfirmed parvovirus b19 pure red cell aplasia. Clin. Infect. Dis. 2015, 60, 488. [Google Scholar] [CrossRef][Green Version]
- Perez, E.E.; Orange, J.S.; Bonilla, F.; Chinen, J.; Chinn, I.K.; Dorsey, M.; El-Gamal, Y.; Harville, T.O.; Hossny, E.; Mazer, B.; et al. Update on the use of immunoglobulin in human disease: A review of evidence. J. Allergy Clin. Immunol. 2017, 139, S1–S46. [Google Scholar] [CrossRef]
- Crabol, Y.; Terrier, B.; Rozenberg, F.; Pestre, V.; Legendre, C.; Hermine, O.; Montagnier-Petrissans, C.; Guillevin, L.; Mouthon, L.; Groupe d’experts de l’Assistance Publique-Hôpitaux de Paris. Intravenous immunoglobulin therapy for pure red cell aplasia related to human parvovirus b19 infection: A retrospective study of 10 patients and review of the literature. Clin. Infect. Dis. 2013, 56, 968–977. [Google Scholar] [CrossRef]
- Modrof, J.; Berting, A.; Tille, B.; Klotz, A.; Forstner, C.; Rieger, S.; Aberham, C.; Gessner, M.; Kreil, T.R. Neutralization of human parvovirus B19 by plasma and intravenous immunoglobulins. Transfusion 2008, 48, 178–186. [Google Scholar] [CrossRef]
- Chaigne, B.; Mouthon, L. Mechanisms of action of intravenous immunoglobulin. Transfus. Apher. Sci. 2017, 56, 45–49. [Google Scholar] [CrossRef]
- Kerr, J.R.; Cunniffe, V.S.; Kelleher, P.; Bernstein, R.M.; Bruce, I.N. Successful intravenous immunoglobulin therapy in 3 cases of parvovirus B19-associated chronic fatigue syndrome. Clin. Infect. Dis. 2003, 36, e100–e106. [Google Scholar] [CrossRef]
- Attard, L.; Bonvicini, F.; Gelsomino, F.; Manfredi, R.; Cascavilla, A.; Viale, P.; Varani, S.; Gallinella, G. Paradoxical response to intravenous immunoglobulin in a case of Parvovirus B19-associated chronic fatigue syndrome. J. Clin. Virol. 2015, 62, 54–57. [Google Scholar] [CrossRef]
- Gigler, A.; Dorsch, S.; Hemauer, A.; Williams, C.; Kim, S.; Young, N.S.; Zolla-Pazner, S.; Wolf, H.; Gorny, M.K.; Modrow, S. Generation of neutralizing human monoclonal antibodies against parvovirus B19 proteins. J. Virol. 1999, 73, 1974–1979. [Google Scholar]
- Manaresi, E.; Gallinella, G.; Zerbini, M.; Venturoli, S.; Gentilomi, G.; Musiani, M. IgG immune response to B19 parvovirus VP1 and VP2 linear epitopes by immunoblot assay. J. Med. Virol. 1999, 57, 174–178. [Google Scholar] [CrossRef]
- Manaresi, E.; Zuffi, E.; Gallinella, G.; Gentilomi, G.; Zerbini, M.; Musiani, M. Differential IgM response to conformational and linear epitopes of parvovirus B19 VP1 and VP2 structural proteins. J. Med. Virol. 2001, 64, 67–73. [Google Scholar] [CrossRef]
- Sato, H.; Hirata, J.; Furukawa, M.; Kuroda, N.; Shiraki, H.; Maeda, Y.; Okochi, K. Identification of the region including the epitope for a monoclonal antibody which can neutralize human parvovirus B19. J. Virol. 1991, 65, 1667–1672. [Google Scholar]
- Sato, H.; Hirata, J.; Kuroda, N.; Shiraki, H.; Maeda, Y.; Okochi, K. Identification and mapping of neutralizing epitopes of human parvovirus B19 by using human antibodies. J. Virol. 1991, 65, 5485–5490. [Google Scholar]
- Saikawa, T.; Anderson, S.; Momoeda, M.; Kajigaya, S.; Young, N.S. Neutralizing linear epitopes of B19 parvovirus cluster in the VP1 unique and VP1-VP2 junction regions. J. Virol. 1993, 67, 3004–3009. [Google Scholar]
- Sun, Y.; Klose, T.; Liu, Y.; Modrow, S.; Rossmann, M.G. Structure of Parvovirus B19 Decorated by Fabs from a Human Antibody. J. Virol. 2019, 93, e01732-18. [Google Scholar] [CrossRef]
- Zuffi, E.; Manaresi, E.; Gallinella, G.; Gentilomi, G.A.; Venturoli, S.; Zerbini, M.; Musiani, M. Identification of an immunodominant peptide in the parvovirus B19 VP1 unique region able to elicit a long-lasting immune response in humans. Viral Immunol. 2001, 14, 151–158. [Google Scholar] [CrossRef]
- Hattangadi, S.M.; Wong, P.; Zhang, L.; Flygare, J.; Lodish, H.F. From stem cell to red cell: Regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood 2011, 118, 6258–6268. [Google Scholar] [CrossRef]
- Filippone, C.; Franssila, R.; Kumar, A.; Saikko, L.; Kovanen, P.E.; Soderlund-Venermo, M.; Hedman, K. Erythroid progenitor cells expanded from peripheral blood without mobilization or preselection: Molecular characteristics and functional competence. PLoS ONE 2010, 5, e9496. [Google Scholar] [CrossRef]
- Wong, S.; Brown, K.E. Development of an improved method of detection of infectious parvovirus B19. J. Clin. Virol. 2006, 35, 407–413. [Google Scholar] [CrossRef]
- Manaresi, E.; Bua, G.; Bonvicini, F.; Gallinella, G. A flow-FISH assay for the quantitative analysis of parvovirus B19 infected cells. J. Virol. Methods 2015, 223, 50–54. [Google Scholar] [CrossRef]
- Bonvicini, F.; Filippone, C.; Delbarba, S.; Manaresi, E.; Zerbini, M.; Musiani, M.; Gallinella, G. Parvovirus B19 genome as a single, two-state replicative and transcriptional unit. Virology 2006, 347, 447–454. [Google Scholar] [CrossRef]
- Bonvicini, F.; Filippone, C.; Manaresi, E.; Zerbini, M.; Musiani, M.; Gallinella, G. Functional analysis and quantitative determination of the expression profile of human parvovirus B19. Virology 2008, 381, 168–177. [Google Scholar] [CrossRef]
- Zhi, N.; Zadori, Z.; Brown, K.E.; Tijssen, P. Construction and sequencing of an infectious clone of the human parvovirus B19. Virology 2004, 318, 142–152. [Google Scholar] [CrossRef]
- Manaresi, E.; Conti, I.; Bua, G.; Bonvicini, F.; Gallinella, G. A Parvovirus B19 synthetic genome: Sequence features and functional competence. Virology 2017, 508, 54–62. [Google Scholar] [CrossRef]
- Bonvicini, F.; Bua, G.; Conti, I.; Manaresi, E.; Gallinella, G. Hydroxyurea inhibits parvovirus B19 replication in erythroid progenitor cells. Biochem. Pharmacol. 2017, 136, 32–39. [Google Scholar] [CrossRef]
- Bonvicini, F.; Bua, G.; Manaresi, E.; Gallinella, G. Antiviral effect of cidofovir on parvovirus B19 replication. Antivir. Res. 2015, 113, 11–18. [Google Scholar] [CrossRef]
- Bonvicini, F.; Bua, G.; Manaresi, E.; Gallinella, G. Enhanced inhibition of parvovirus B19 replication by cidofovir in extendedly exposed erythroid progenitor cells. Virus Res. 2016, 220, 47–51. [Google Scholar] [CrossRef]
- Bua, G.; Conti, I.; Manaresi, E.; Sethna, P.; Foster, S.; Bonvicini, F.; Gallinella, G. Antiviral activity of brincidofovir on parvovirus B19. Antivir. Res. 2019, 162, 22–29. [Google Scholar] [CrossRef]
- Conti, I.; Morigi, R.; Locatelli, A.; Rambaldi, M.; Bua, G.; Gallinella, G.; Leoni, A. Synthesis of 3-(Imidazo[2,1-b]thiazol-6-yl)-2H-chromen-2-one Derivatives and Study of Their Antiviral Activity against Parvovirus B19. Molecules 2019, 24, 1037. [Google Scholar] [CrossRef]
- Xu, P.; Ganaie, S.S.; Wang, X.; Wang, Z.; Kleiboeker, S.; Horton, N.C.; Heier, R.F.; Meyers, M.J.; Tavis, J.E.; Qiu, J. Endonuclease Activity Inhibition of the NS1 Protein of Parvovirus B19 as a Novel Target for Antiviral Drug Development. Antimicrob. Agents Chemother. 2019, 63, e01879-18. [Google Scholar] [CrossRef]
- Gallicchio, V.S. Ribonucleotide reductase: Target therapy for human disease. Expert Opin. Ther. Pat. 2005, 15, 659–673. [Google Scholar] [CrossRef]
- Neyts, J.; De Clercq, E. Hydroxyurea Potentiates the Antiherpesvirus Activities of Purine and Pyrimidine Nucleoside and Nucleoside Phosphonate Analogs. Antimicrob. Agents Chemother. 1999, 43, 2885–2892. [Google Scholar] [CrossRef][Green Version]
- Lori, F.; Foli, A.; Kelly, L.M.; Lisziewicz, J. Virostatics: A new class of anti-HIV drugs. Curr. Med. Chem. 2007, 14, 233–241. [Google Scholar] [CrossRef]
- Bhave, S.; Elford, H.; McVoy, M.A. Ribonucleotide reductase inhibitors hydroxyurea, didox, and trimidox inhibit human cytomegalovirus replication in vitro and synergize with ganciclovir. Antivir. Res. 2013, 100, 151–158. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Z.; Hou, C.; Wang, M.; Chen, X.; Lin, Q.; Song, R.; Lou, M.; Zhu, L.; Qiu, Y.; et al. Inhibition of hepatitis B virus replication by targeting ribonucleotide reductase M2 protein. Biochem. Pharm. 2016, 103, 118–128. [Google Scholar] [CrossRef]
- Platt, O.S. Hydroxyurea for the treatment of sickle cell anemia. N. Engl. J. Med. 2008, 358, 1362–1369. [Google Scholar] [CrossRef]
- Green, N.S.; Barral, S. Emerging science of hydroxyurea therapy for pediatric sickle cell disease. Pediatric Res. 2013, 75, 196. [Google Scholar] [CrossRef]
- McGann, P.T.; Ware, R.E. Hydroxyurea therapy for sickle cell anemia. Expert Opin. Drug Saf. 2015, 14, 1749–1758. [Google Scholar] [CrossRef]
- Hankins, J.S.; Penkert, R.R.; Lavoie, P.; Tang, L.; Sun, Y.; Hurwitz, J.L. Parvovirus B19 infection in children with sickle cell disease in the hydroxyurea era. Exp. Biol. Med. 2016, 241, 749–754. [Google Scholar] [CrossRef]
- De Clercq, E.; Holy, A. Acyclic nucleoside phosphonates: A key class of antiviral drugs. Nat. Rev. Drug Discov. 2005, 4, 928–940. [Google Scholar] [CrossRef]
- De Clercq, E. Acyclic nucleoside phosphonates: Past, present and future. Bridging chemistry to HIV, HBV, HCV, HPV, adeno-, herpes-, and poxvirus infections: The phosphonate bridge. Biochem. Pharm. 2007, 73, 911–922. [Google Scholar] [CrossRef]
- Aldern, K.A.; Ciesla, S.L.; Winegarden, K.L.; Hostetler, K.Y. Increased antiviral activity of 1-O-hexadecyloxypropyl-[2-(14)C]cidofovir in MRC-5 human lung fibroblasts is explained by unique cellular uptake and metabolism. Mol. Pharmacol. 2003, 63, 678–681. [Google Scholar] [CrossRef]
- Williams-Aziz, S.L.; Hartline, C.B.; Harden, E.A.; Daily, S.L.; Prichard, M.N.; Kushner, N.L.; Beadle, J.R.; Wan, W.B.; Hostetler, K.Y.; Kern, E.R. Comparative activities of lipid esters of cidofovir and cyclic cidofovir against replication of herpesviruses in vitro. Antimicrob. Agents Chemother. 2005, 49, 3724–3733. [Google Scholar] [CrossRef]
- Hostetler, K.Y. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: Current state of the art. Antivir. Res. 2009, 82, A84–A98. [Google Scholar] [CrossRef]
- McMullan, L.K.; Flint, M.; Dyall, J.; Albarino, C.; Olinger, G.G.; Foster, S.; Sethna, P.; Hensley, L.E.; Nichol, S.T.; Lanier, E.R.; et al. The lipid moiety of brincidofovir is required for in vitro antiviral activity against Ebola virus. Antivir. Res. 2016, 125, 71–78. [Google Scholar] [CrossRef]
- Magee, W.C.; Evans, D.H. The antiviral activity and mechanism of action of (S)-[3-hydroxy-2-(phosphonomethoxy)propyl] (HPMP) nucleosides. Antivir. Res. 2012, 96, 169–180. [Google Scholar] [CrossRef]
- Johnson, J.A.; Gangemi, J.D. Selective inhibition of human papillomavirus-induced cell proliferation by (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine. Antimicrob. Agents Chemother. 1999, 43, 1198–1205. [Google Scholar] [CrossRef]
- Randhawa, P.; Farasati, N.A.; Shapiro, R.; Hostetler, K.Y. Ether lipid ester derivatives of cidofovir inhibit polyomavirus BK replication in vitro. Antimicrob. Agents Chemother. 2006, 50, 1564–1566. [Google Scholar] [CrossRef]
- Jiang, Z.G.; Cohen, J.; Marshall, L.J.; Major, E.O. Hexadecyloxypropyl-cidofovir (CMX001) suppresses JC virus replication in human fetal brain SVG cell cultures. Antimicrob. Agents Chemother. 2010, 54, 4723–4732. [Google Scholar] [CrossRef]
- Andrei, G.; Topalis, D.; De Schutter, T.; Snoeck, R. Insights into the mechanism of action of cidofovir and other acyclic nucleoside phosphonates against polyoma- and papillomaviruses and non-viral induced neoplasia. Antivir. Res. 2015, 114, 21–46. [Google Scholar] [CrossRef]
- Tsang, S.H.; Wang, R.; Nakamaru-Ogiso, E.; Knight, S.A.; Buck, C.B.; You, J. The Oncogenic Small Tumor Antigen of Merkel Cell Polyomavirus Is an Iron-Sulfur Cluster Protein That Enhances Viral DNA Replication. J. Virol. 2016, 90, 1544–1556. [Google Scholar] [CrossRef]
- Painter, W.; Robertson, A.; Trost, L.C.; Godkin, S.; Lampert, B.; Painter, G. First pharmacokinetic and safety study in humans of the novel lipid antiviral conjugate CMX001, a broad-spectrum oral drug active against double-stranded DNA viruses. Antimicrob. Agents Chemother. 2012, 56, 2726–2734. [Google Scholar] [CrossRef]
- Tippin, T.K.; Morrison, M.E.; Brundage, T.M.; Mommeja-Marin, H. Brincidofovir Is Not a Substrate for the Human Organic Anion Transporter 1: A Mechanistic Explanation for the Lack of Nephrotoxicity Observed in Clinical Studies. Ther. Drug Monit. 2016, 38, 777–786. [Google Scholar] [CrossRef]
- Chemaly, R.F.; Hill, J.A.; Voigt, S.; Peggs, K.S. In vitro comparison of currently available and investigational antiviral agents against pathogenic human double-stranded DNA viruses: A systematic literature review. Antivir. Res. 2019, 163, 50–58. [Google Scholar] [CrossRef]
- Penta, S. Advances in Structure and Activity Relationship of Coumarin Derivatives; Academic Press: Cambridge, MA, USA, 2015; pp. 1–182. [Google Scholar]
- Hassan, M.Z.; Osman, H.; Ali, M.A.; Ahsan, M.J. Therapeutic potential of coumarins as antiviral agents. Eur. J. Med. Chem. 2016, 123, 236–255. [Google Scholar] [CrossRef]
- Tewary, S.K.; Zhao, H.; Deng, X.; Qiu, J.; Tang, L. The human parvovirus B19 non-structural protein 1 N-terminal domain specifically binds to the origin of replication in the viral DNA. Virology 2014, 449, 297–303. [Google Scholar] [CrossRef]
- Sanchez, J.L.; Romero, Z.; Quinones, A.; Torgeson, K.R.; Horton, N.C. DNA Binding and Cleavage by the Human Parvovirus B19 NS1 Nuclease Domain. Biochemistry 2016, 55, 6577–6593. [Google Scholar] [CrossRef]
- Van Linthout, S.; Elsanhoury, A.; Klein, O.; Sosnowski, M.; Miteva, K.; Lassner, D.; Abou-El-Enein, M.; Pieske, B.; Kuhl, U.; Tschope, C. Telbivudine in chronic lymphocytic myocarditis and human parvovirus B19 transcriptional activity. ESC Heart Fail 2018, 5, 818–829. [Google Scholar] [CrossRef]
- Zobel, T.; Bock, C.T.; Kuhl, U.; Rohde, M.; Lassner, D.; Schultheiss, H.P.; Schmidt-Lucke, C. Telbivudine Reduces Parvovirus B19-Induced Apoptosis in Circulating Angiogenic Cells. Viruses 2019, 11, 227. [Google Scholar] [CrossRef]
- Zimmermann, O.; Rodewald, C.; Radermacher, M.; Vetter, M.; Wiehe, J.M.; Bienek-Ziolkowski, M.; Hombach, V.; Torzewski, J. Interferon beta-1b therapy in chronic viral dilated cardiomyopathy—Is there a role for specific therapy? J. Card. Fail. 2010, 16, 348–356. [Google Scholar] [CrossRef]
- Schmidt-Lucke, C.; Spillmann, F.; Bock, T.; Kuhl, U.; Van Linthout, S.; Schultheiss, H.P.; Tschope, C. Interferon beta modulates endothelial damage in patients with cardiac persistence of human parvovirus b19 infection. J. Infect. Dis. 2010, 201, 936–945. [Google Scholar] [CrossRef]
- Schultheiss, H.P.; Piper, C.; Sowade, O.; Waagstein, F.; Kapp, J.F.; Wegscheider, K.; Groetzbach, G.; Pauschinger, M.; Escher, F.; Arbustini, E.; et al. Betaferon in chronic viral cardiomyopathy (BICC) trial: Effects of interferon-beta treatment in patients with chronic viral cardiomyopathy. Clin. Res. Cardiol. 2016, 105, 763–773. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Compound/Cells | Max Inhibition | EC50 (µM [CI]) | CC50 (µM [CI]) | SI (CC50/EC50) | References and Notes |
|---|---|---|---|---|---|
| HU | Ref [82] | ||||
| UT7/EpoS1 | 100% at >1000 µM | 96.2 [81.5–118.1] | 581.9 [426.5–812.3] | 6.0 | moi in the range 101–104 |
| EPCs | 100% at >1000 µM | 147.1 [121.4–190.4] | 584.8 [478.5–722.3] | 4.0 | moi in the range 101–104 |
| CDV | Ref [83,85] | ||||
| UT7/EpoS1 | 100% at 500 µM | 16.1 [12.9–20.2] | >500 | ND | moi in the range 101–104 |
| EPCs | ND | 320.5 [173.9–590.7] | >500 | ND | moi in the range 101–104 |
| BCV | Ref [85] | ||||
| UT7/EpoS1 | 100% at >10 µM | 0.22 [0.19–0.25] | 66.8 [62.0–72.9] | 303.6 | moi in the range 101–104 |
| EPCs | 100% at >100 µM | 9.35 [8.84–9.89] | 102.9 [87.6–120.8] | 11.0 | moi in the range 101–104 |
| BCV (S) | Ref [85] | ||||
| UT7/EpoS1 | 100% at >10 µM | 0.63 [0.58–0.68] | 59.9 [52.7–68.1] | 95.1 | moi 104 |
| EPCs | 100% at >100 µM | 14.3 [11.8–17.3] | 93.4 [68.9–126.6] | 6.5 | moi 104 |
| BCV (R) | Ref [85] | ||||
| UT7/EpoS1 | 100% at >100 µM | 54.7 [42.7–69.9] | 72.1 [62.5–83.1] | 1.3 | moi 104 |
| EPCs | 100% at >500 µM | 93.0 [77.4–111.8] | 146.2 [106.4–200] | 1.6 | moi 104 |
| Coumarin derivatives | Ref [86] | ||||
| UT7/EpoS1 | 82.1 at 12.5 µM | 6.7 | ND | 4.0 at 12.5 µM | Compound #7; moi 104 |
| EPCs | 65.0 at 12.5 µM | ND | ND | 2.7 at 12.5 µM | Compound #8; moi 104 |
| Flavonoid compounds | Ref [87] | ||||
| UT7/EpoS1 | Following transfection (IIF) | ||||
| Compound #7 | NR | 44.2 [25.6–62.8] | 194.0 [171.0–216.3] | 4.4 | |
| Compound #135 | NR | 61.1 [60.8–61.4] | 227.0 [206.0–248.0] | 3.7 | |
| Compound #201 | NR | 55.1 [47.2–63.0] | 180.9 [160.4–201.4] | 3.3 | |
| EPCs | Following infection (IIF) | ||||
| Compound #7 | NR | 37.6 [34.0–41.2] | 55.9 [53.8–58.0] | 1.5 | |
| Compound #135 | NR | 53.9 [46.8–61.0] | 89.8 [82.0–97.6] | 1.7 | |
| Compound #201 | NR | 33.5 [32.1–34.9] | 60.0 [57.6–62.4] | 1.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manaresi, E.; Gallinella, G. Advances in the Development of Antiviral Strategies against Parvovirus B19. Viruses 2019, 11, 659. https://doi.org/10.3390/v11070659
Manaresi E, Gallinella G. Advances in the Development of Antiviral Strategies against Parvovirus B19. Viruses. 2019; 11(7):659. https://doi.org/10.3390/v11070659
Chicago/Turabian StyleManaresi, Elisabetta, and Giorgio Gallinella. 2019. "Advances in the Development of Antiviral Strategies against Parvovirus B19" Viruses 11, no. 7: 659. https://doi.org/10.3390/v11070659
APA StyleManaresi, E., & Gallinella, G. (2019). Advances in the Development of Antiviral Strategies against Parvovirus B19. Viruses, 11(7), 659. https://doi.org/10.3390/v11070659