Characterization of Biomarker Levels in Crimean–Congo Hemorrhagic Fever and Hantavirus Fever with Renal Syndrome

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Patients
2.3. Cytokines and Chemokines
2.4. Statistical Analyses
3. Results
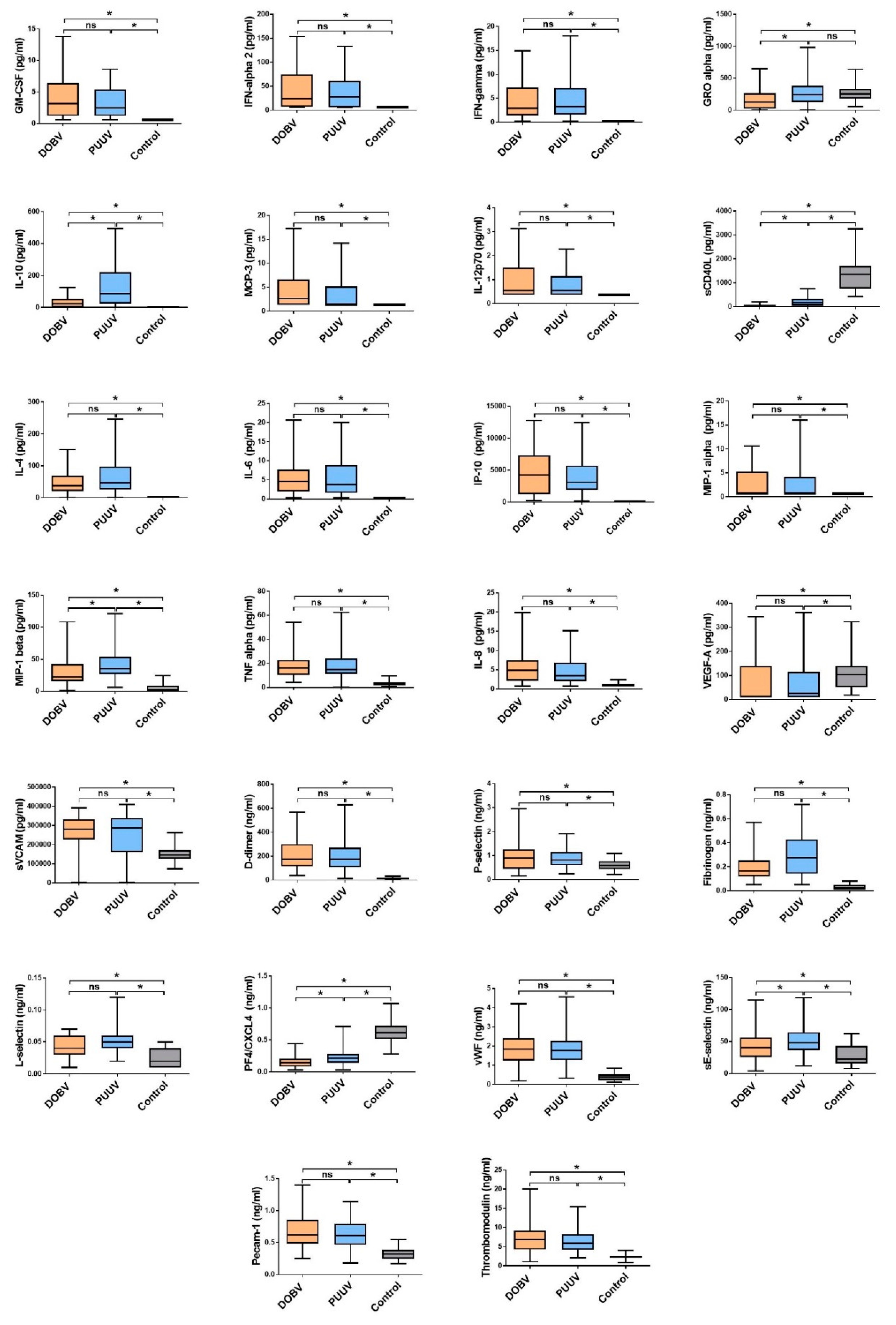
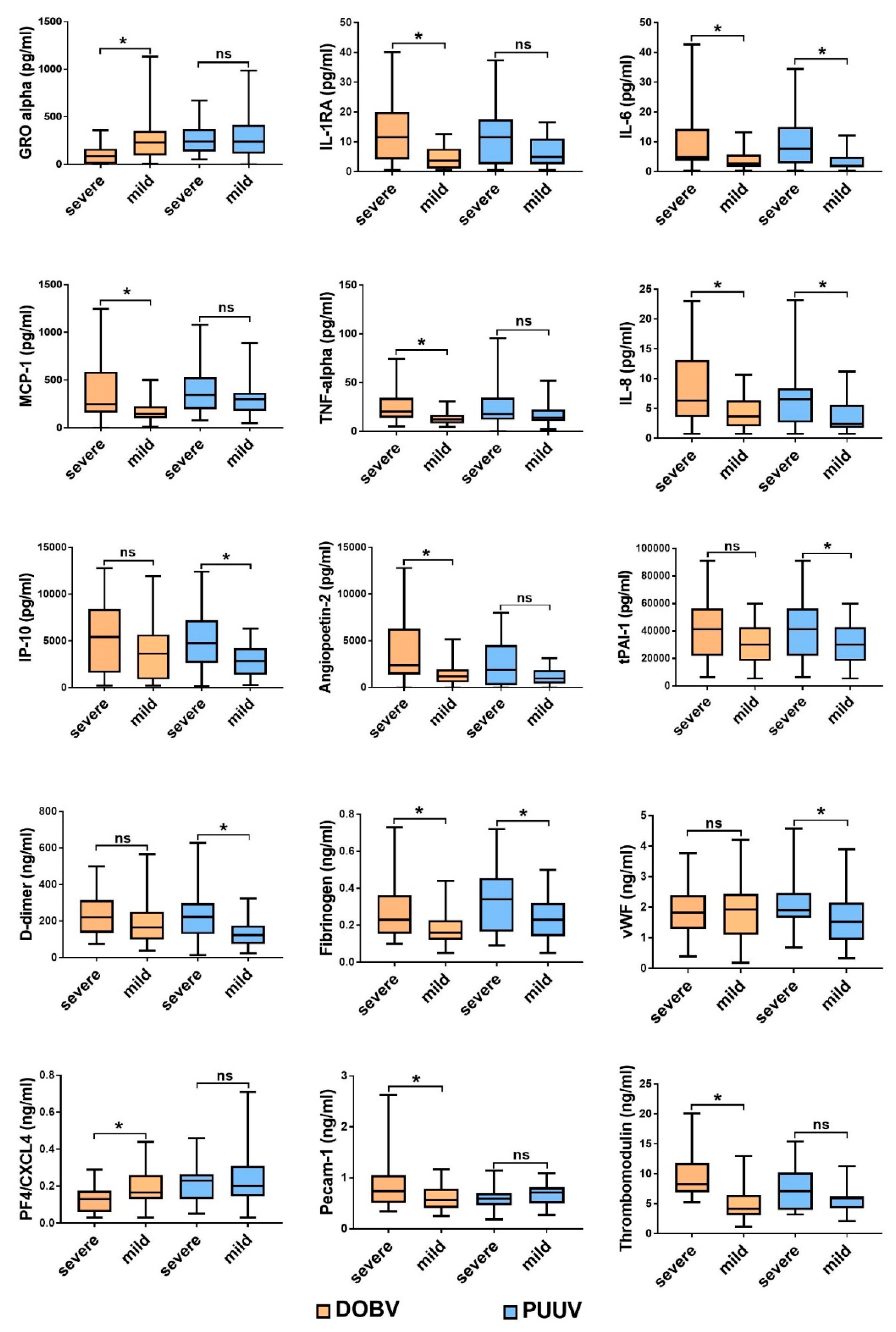
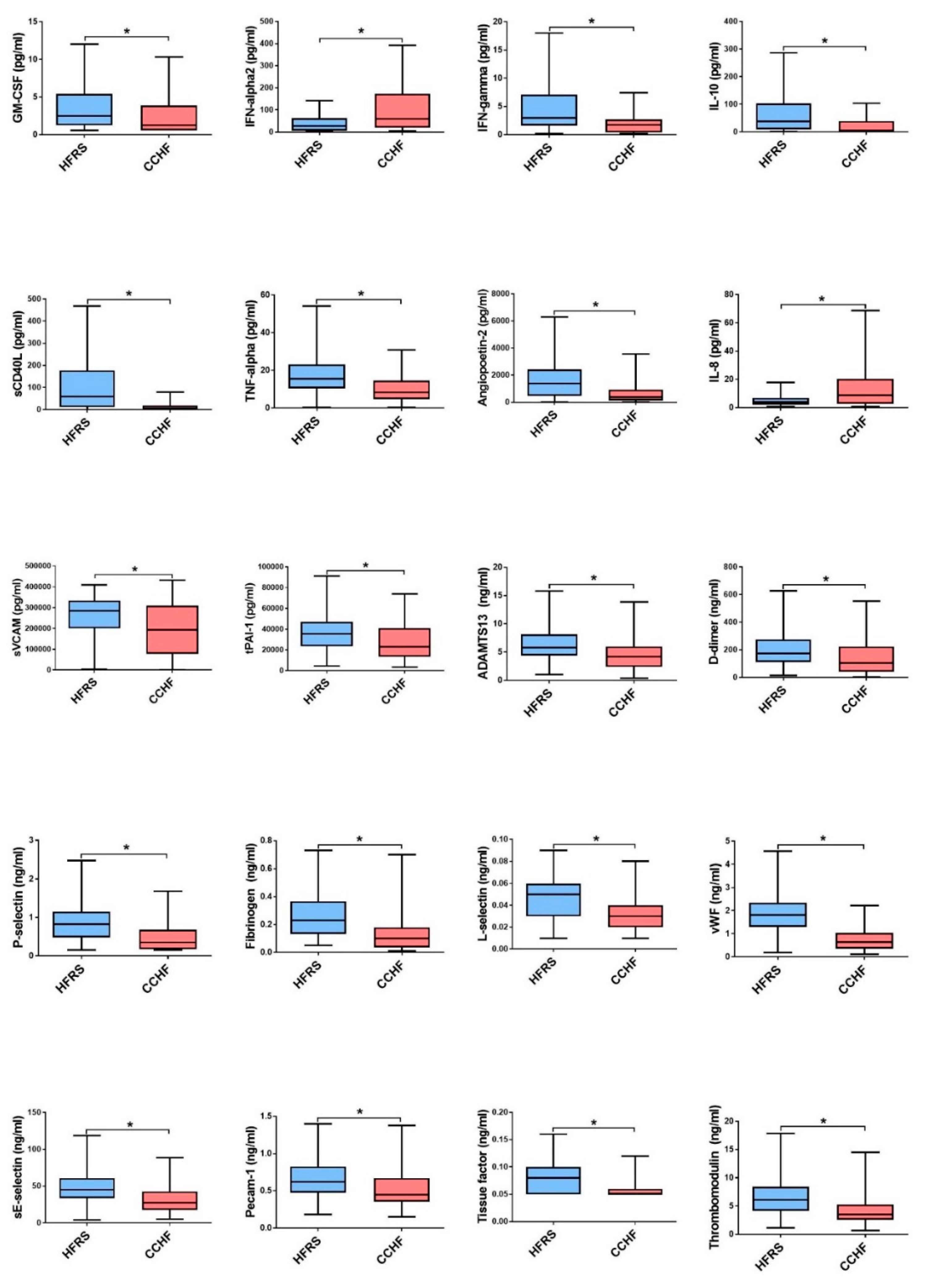
3.1. Comparison of Cytokine and Chemokine Levels in HFRS Patients
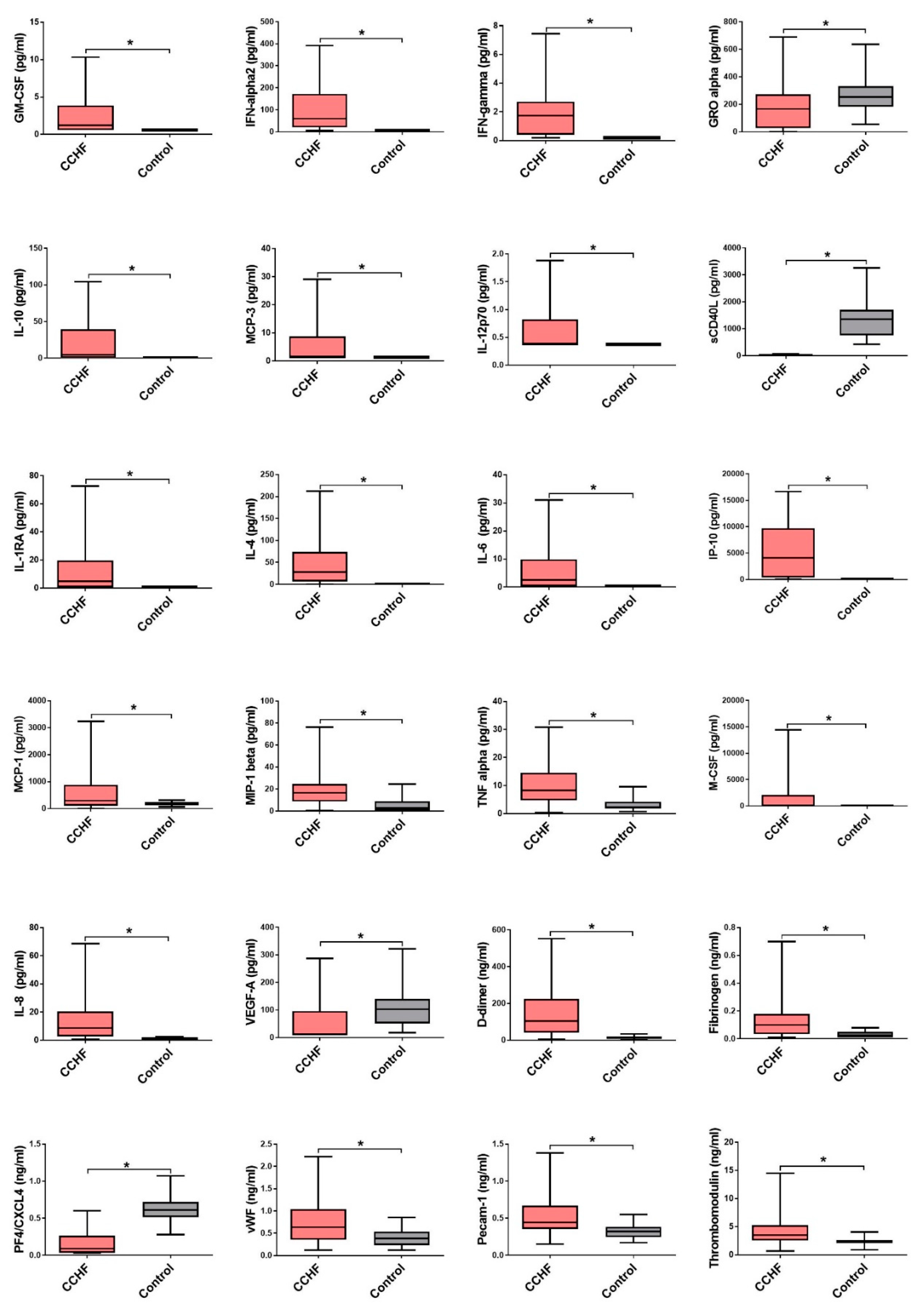
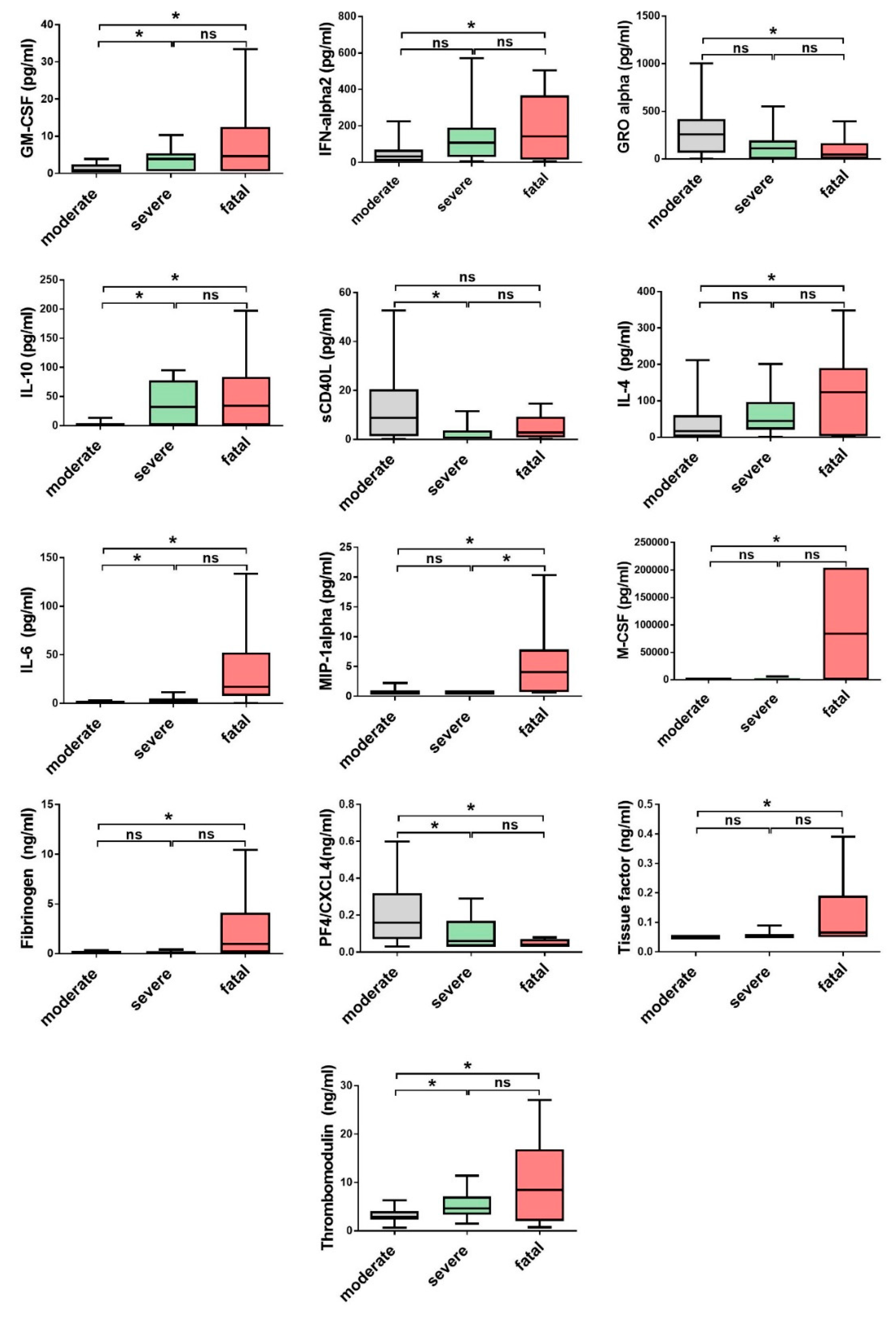
3.2. Comparison of Cytokine and Chemokine Levels in CCHF Patients
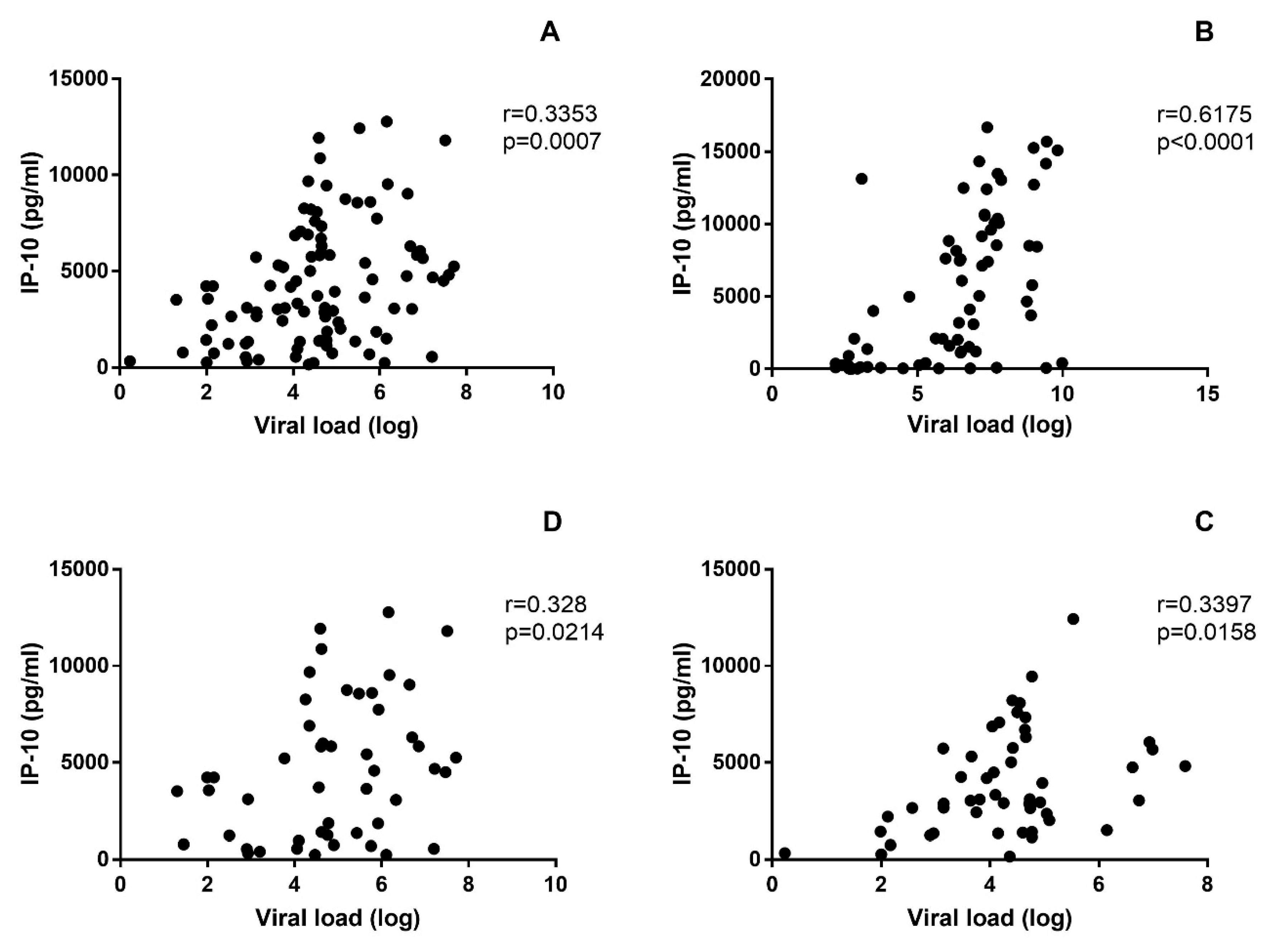
3.3. Soluble Biomarkers Involved in the Pathogenesis of both VHF
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Whitehouse, C.A. Crimean-Congo hemorrhagic fever. Antivir. Res. 2004, 64, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Ergonul, O. Crimean-Congo haemorrhagic fever. Lancet Infect. Dis. 2006, 6, 203–214. [Google Scholar] [CrossRef]
- Weber, F.; Mirazimi, A. Interferon and cytokine responses to Crimean Congo hemorrhagic fever virus; an emerging and neglected viral zonoosis. Cytokine Growth Factor Rev. 2008, 19, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Thomson, G.; Dowall, S.; Bruce, C.; Cook, N.; Easterbrook, L.; O’Donoghue, L.; Summers, S.; Ajazaj, L.; Hewson, R.; et al. Review of Crimean Congo hemorrhagic fever infection in Kosova in 2008 and 2009: Prolonged viremias and virus detected in urine by PCR. Vector Borne Zoonotic Dis. 2012, 12, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Bente, D.A.; Forrester, N.L.; Watts, D.M.; McAuley, A.J.; Whitehouse, C.A.; Bray, M. Crimean-Congo hemorrhagic fever: History, epidemiology, pathogenesis, clinical syndrome and genetic diversity. Antivir. Res. 2013, 100, 159–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaheri, A.; Strandin, T.; Hepojoki, J.; Sironen, T.; Henttonen, H.; Makela, S.; Mustonen, J. Uncovering the mysteries of hantavirus infections. Nat. Rev. Microbiol. 2013, 11, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, E.; Grouls, S.; Hettwer, D.; Rafat, N.; Tonshoff, B.; Zeier, M. Mobilization of circulating endothelial progenitor cells correlates with the clinical course of hantavirus disease. J. Virol. 2014, 88, 483–489. [Google Scholar] [CrossRef]
- Avsic-Zupanc, T.; Petrovec, M.; Furlan, P.; Kaps, R.; Elgh, F.; Lundkvist, A. Hemorrhagic fever with renal syndrome in the Dolenjska region of Slovenia—A 10-year survey. Clin. Infect. Dis. 1999, 28, 860–865. [Google Scholar] [CrossRef]
- Pal, E.; Strle, F.; Avsic-Zupanc, T. Hemorrhagic fever with renal syndrome in the Pomurje region of Slovenia—An 18-year survey. Wien. Klin. Wochenschr. 2005, 117, 398–405. [Google Scholar] [CrossRef]
- Pal, E.; Korva, M.; Rus, K.R.; Kejzar, N.; Bogovic, P.; Kurent, A.; Avsic-Zupanc, T.; Strle, F. Sequential assessment of clinical and laboratory parameters in patients with hemorrhagic fever with renal syndrome. PLoS ONE 2018, 13, e0197661. [Google Scholar] [CrossRef]
- Krautkramer, E.; Grouls, S.; Stein, N.; Reiser, J.; Zeier, M. Pathogenic old world hantaviruses infect renal glomerular and tubular cells and induce disassembling of cell-to-cell contacts. J. Virol. 2011, 85, 9811–9823. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Bino, S.; Velo, E.; Harxhi, A.; Kota, M.; Antoniadis, A. Cytokine levels in Crimean-Congo hemorrhagic fever. J. Clin. Virol. 2006, 36, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Duh, D.; Saksida, A.; Petrovec, M.; Ahmeti, S.; Dedushaj, I.; Panning, M.; Drosten, C.; Avsic-Zupanc, T. Viral load as predictor of Crimean-Congo hemorrhagic fever outcome. Emerg. Infect. Dis. 2007, 13, 1769–1772. [Google Scholar] [CrossRef] [PubMed]
- Saksida, A.; Duh, D.; Wraber, B.; Dedushaj, I.; Ahmeti, S.; Avsic-Zupanc, T. Interacting roles of immune mechanisms and viral load in the pathogenesis of crimean-congo hemorrhagic fever. Clin. Vaccine Immunol. 2010, 17, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Kaya, S.; Elaldi, N.; Kubar, A.; Gursoy, N.; Yilmaz, M.; Karakus, G.; Gunes, T.; Polat, Z.; Gozel, M.G.; Engin, A.; et al. Sequential determination of serum viral titers, virus-specific IgG antibodies, and TNF-alpha, IL-6, IL-10, and IFN-gamma levels in patients with Crimean-Congo hemorrhagic fever. BMC Infect. Dis. 2014, 14, 416. [Google Scholar] [CrossRef]
- Ozsurekci, Y.; Arasli, M.; Karadag Oncel, E.; Caglayik, D.Y.; Kaya, A.; Icagasioglu, F.D.; Engin, A.; Korukluoglu, G.; Elaldi, N.; Ceyhan, M. Can the mild clinical course of crimean-congo hemorrhagic fever in children be explained by cytokine responses? J. Med. Virol. 2013, 85, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- Garanina, E.; Martynova, E.; Davidyuk, Y.; Kabwe, E.; Ivanov, K.; Titova, A.; Markelova, M.; Zhuravleva, M.; Cherepnev, G.; Shakirova, V.G.; et al. Cytokine Storm Combined with Humoral Immune Response Defect in Fatal Hemorrhagic Fever with Renal Syndrome Case, Tatarstan, Russia. Viruses 2019, 11, 601. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Tsergouli, K.; Caglayik, D.Y.; Bino, S.; Como, N.; Uyar, Y.; Korukluoglu, G. Cytokines as biomarkers of Crimean-Congo hemorrhagic fever. J. Med. Virol. 2016, 88, 21–27. [Google Scholar] [CrossRef]
- Korva, M.; Saksida, A.; Kejzar, N.; Schmaljohn, C.; Avsic-Zupanc, T. Viral load and immune response dynamics in patients with haemorrhagic fever with renal syndrome. Clin. Microbiol. Infect. 2013, 19, e358–e366. [Google Scholar] [CrossRef] [Green Version]
- Saksida, A.; Duh, D.; Korva, M.; Avsic-Zupanc, T. Dobrava virus RNA load in patients who have hemorrhagic fever with renal syndrome. J. Infect. Dis. 2008, 197, 681–685. [Google Scholar] [CrossRef]
- Frese, M.; Kochs, G.; Feldmann, H.; Hertkorn, C.; Haller, O. Inhibition of bunyaviruses, phleboviruses, and hantaviruses by human MxA protein. J. Virol. 1996, 70, 915–923. [Google Scholar] [PubMed]
- Kanerva, M.; Melen, K.; Vaheri, A.; Julkunen, I. Inhibition of puumala and tula hantaviruses in Vero cells by MxA protein. Virology 1996, 224, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Andersson, I.; Bladh, L.; Mousavi-Jazi, M.; Magnusson, K.E.; Lundkvist, A.; Haller, O.; Mirazimi, A. Human MxA protein inhibits the replication of Crimean-Congo hemorrhagic fever virus. J. Virol. 2004, 78, 4323–4329. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.R.; Prescott, J.; Brown, K.S.; Best, S.M.; Ebihara, H.; Feldmann, H. Antagonism of type I interferon responses by new world hantaviruses. J. Virol. 2010, 84, 11790–11801. [Google Scholar] [CrossRef] [PubMed]
- Resman Rus, K.; Korva, M.; Bogovic, P.; Pal, E.; Strle, F.; Avsic-Zupanc, T. Delayed Interferon Type 1-Induced Antiviral State Is a Potential Factor for Hemorrhagic Fever With Renal Syndrome Severity. J. Infect. Dis. 2018, 217, 926–932. [Google Scholar] [CrossRef]
- Andersson, I.; Karlberg, H.; Mousavi-Jazi, M.; Martinez-Sobrido, L.; Weber, F.; Mirazimi, A. Crimean-Congo hemorrhagic fever virus delays activation of the innate immune response. J. Med. Virol. 2008, 80, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Fajs, L.; Resman, K.; Avsic-Zupanc, T. Crimean-Congo hemorrhagic fever virus nucleoprotein suppresses IFN-beta-promoter-mediated gene expression. Arch. Virol. 2014, 159, 345–348. [Google Scholar] [CrossRef]
- Fisman, D.N. Hemophagocytic syndromes and infection. Emerg. Infect. Dis. 2000, 6, 601–608. [Google Scholar] [CrossRef]
- Fernandez-Mestre, M.T.; Gendzekhadze, K.; Rivas-Vetencourt, P.; Layrisse, Z. TNF-alpha-308A allele, a possible severity risk factor of hemorrhagic manifestation in dengue fever patients. Tissue Antigens 2004, 64, 469–472. [Google Scholar] [CrossRef]
- Ergonul, O.; Seref, C.; Eren, S.; Celikbas, A.; Baykam, N.; Dokuzoguz, B.; Gonen, M.; Can, F. Cytokine response in crimean-congo hemorrhagic fever virus infection. J. Med Virol. 2017, 89, 1707–1713. [Google Scholar] [CrossRef]
- Strandin, T.; Makela, S.; Mustonen, J.; Vaheri, A. Neutrophil Activation in Acute Hemorrhagic Fever With Renal Syndrome Is Mediated by Hantavirus-Infected Microvascular Endothelial Cells. Front. Immunol. 2018, 9, 2098. [Google Scholar] [CrossRef] [Green Version]
- Linderholm, M.; Ahlm, C.; Settergren, B.; Waage, A.; Tarnvik, A. Elevated plasma levels of tumor necrosis factor (TNF)-alpha, soluble TNF receptors, interleukin (IL)-6, and IL-10 in patients with hemorrhagic fever with renal syndrome. J. Infect. Dis. 1996, 173, 38–43. [Google Scholar] [CrossRef]
- Muranyi, W.; Bahr, U.; Zeier, M.; Van Der Woude, F.J. Hantavirus infection. J. Am. Soc. Nephrol. 2005, 16, 3669–3679. [Google Scholar] [CrossRef]
- Sadeghi, M.; Eckerle, I.; Daniel, V.; Burkhardt, U.; Opelz, G.; Schnitzler, P. Cytokine expression during early and late phase of acute Puumala hantavirus infection. BMC Immunol. 2011, 12, 65. [Google Scholar] [CrossRef]
- Saksida, A.; Wraber, B.; Avsic-Zupanc, T. Serum levels of inflammatory and regulatory cytokines in patients with hemorrhagic fever with renal syndrome. BMC Infect. Dis. 2011, 11, 142. [Google Scholar] [CrossRef]
- Wang, P.Z.; Li, Z.D.; Yu, H.T.; Zhang, Y.; Wang, W.; Jiang, W.; Bai, X.F. Elevated serum concentrations of inflammatory cytokines and chemokines in patients with haemorrhagic fever with renal syndrome. J. Int. Med. Res. 2012, 40, 648–656. [Google Scholar] [CrossRef]
- Tsergouli, K.; Papa, A. Immune response in Dobrava-Belgrade virus infections. Arch. Virol. 2016, 161, 3413–3420. [Google Scholar] [CrossRef]
- Guo, J.; Guo, X.; Wang, Y.; Tian, F.; Luo, W.; Zou, Y. Cytokine response to Hantaan virus infection in patients with hemorrhagic fever with renal syndrome. J. Med. Virol. 2017, 89, 1139–1145. [Google Scholar] [CrossRef]
- Wauquier, N.; Becquart, P.; Padilla, C.; Baize, S.; Leroy, E.M. Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Negl. Trop. Dis. 2010, 4, e837. [Google Scholar] [CrossRef]
- McElroy, A.K.; Erickson, B.R.; Flietstra, T.D.; Rollin, P.E.; Nichol, S.T.; Towner, J.S.; Spiropoulou, C.F. Ebola hemorrhagic Fever: Novel biomarker correlates of clinical outcome. J. Infect. Dis. 2014, 210, 558–566. [Google Scholar] [CrossRef]
- Ferro, T.; Neumann, P.; Gertzberg, N.; Clements, R.; Johnson, A. Protein kinase C-alpha mediates endothelial barrier dysfunction induced by TNF-alpha. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 278, L1107–L1117. [Google Scholar] [CrossRef]
- Desai, T.R.; Leeper, N.J.; Hynes, K.L.; Gewertz, B.L. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J. Surg. Res. 2002, 104, 118–123. [Google Scholar] [CrossRef]
- Baize, S.; Leroy, E.M.; Georges, A.J.; Georges-Courbot, M.C.; Capron, M.; Bedjabaga, I.; Lansoud-Soukate, J.; Mavoungou, E. Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol. 2002, 128, 163–168. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Tikhonov, I.; Rodas, J.D.; Zapata, J.C.; Yang, Y.; Djavani, M.; Salvato, M.S. Arenavirus-mediated liver pathology: Acute lymphocytic choriomeningitis virus infection of rhesus macaques is characterized by high-level interleukin-6 expression and hepatocyte proliferation. J. Virol. 2003, 77, 1727–1737. [Google Scholar] [CrossRef]
- Vernet, M.A.; Reynard, S.; Fizet, A.; Schaeffer, J.; Pannetier, D.; Guedj, J.; Rives, M.; Georges, N.; Garcia-Bonnet, N.; Sylla, A.I.; et al. Clinical, virological, and biological parameters associated with outcomes of Ebola virus infection in Macenta, Guinea. JCI Insight 2017, 2, e88864. [Google Scholar] [CrossRef] [Green Version]
- Baigildina, A.A.; Khaiboullina, S.F.; Martynova, E.V.; Anokhin, V.A.; Lombardi, V.C.; Rizvanov, A.A. Inflammatory cytokines kinetics define the severity and phase of nephropathia epidemica. Biomark. Med. 2015, 9, 99–107. [Google Scholar] [CrossRef]
- Papa, A.; Yagci Caglayik, D.; Christova, I.; Tsergouli, K.; Korukluoglu, G.; Uyar, Y. Crimean-Congo hemorrhagic fever: CXCL10 correlates with the viral load. J. Med. Virol. 2015, 87, 899–903. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, B.; Ma, Y.; Yi, J.; Zhang, C.; Zhang, Y.; Xu, Z.; Wang, J.; Yang, K.; Yang, A.; et al. Hantaan virus infection induces CXCL10 expression through TLR3, RIG-I, and MDA-5 pathways correlated with the disease severity. Mediat. Inflamm. 2014, 2014, 697837. [Google Scholar] [CrossRef]
- Yamada, M.; Kim, S.; Egashira, K.; Takeya, M.; Ikeda, T.; Mimura, O.; Iwao, H. Molecular mechanism and role of endothelial monocyte chemoattractant protein-1 induction by vascular endothelial growth factor. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1996–2001. [Google Scholar] [CrossRef]
- Lee, Y.R.; Liu, M.T.; Lei, H.Y.; Liu, C.C.; Wu, J.M.; Tung, Y.C.; Lin, Y.S.; Yeh, T.M.; Chen, S.H.; Liu, H.S. MCP-1, a highly expressed chemokine in dengue haemorrhagic fever/dengue shock syndrome patients, may cause permeability change, possibly through reduced tight junctions of vascular endothelium cells. J. Gen. Virol. 2006, 87, 3623–3630. [Google Scholar] [CrossRef]
- Perez, A.B.; Garcia, G.; Sierra, B.; Alvarez, M.; Vazquez, S.; Cabrera, M.V.; Rodriguez, R.; Rosario, D.; Martinez, E.; Denny, T.; et al. IL-10 levels in Dengue patients: Some findings from the exceptional epidemiological conditions in Cuba. J. Med. Virol. 2004, 73, 230–234. [Google Scholar] [CrossRef]
- Abhishek, K.S.; Chakravarti, A.; Baveja, C.P.; Kumar, N.; Siddiqui, O.; Kumar, S. Association of interleukin-2, -4 and-10 with dengue severity. Indian J. Pathol. Microbiol. 2017, 60, 66–69. [Google Scholar] [CrossRef]
- Butthep, P.; Chunhakan, S.; Yoksan, S.; Tangnararatchakit, K.; Chuansumrit, A. Alteration of cytokines and chemokines during febrile episodes associated with endothelial cell damage and plasma leakage in dengue hemorrhagic fever. Pediatr. Infect. Dis. J. 2012, 31, e232–e238. [Google Scholar] [CrossRef]
- Rice, G.E.; Munro, J.M.; Corless, C.; Bevilacqua, M.P. Vascular and nonvascular expression of INCAM-110. A target for mononuclear leukocyte adhesion in normal and inflamed human tissues. Am. J. Pathol. 1991, 138, 385–393. [Google Scholar]
- Van Gorp, E.C.; Suharti, C.; Ten Cate, H.; Dolmans, W.M.; Van Der Meer, J.W.; Ten Cate, J.W.; Brandjes, D.P. Review: Infectious diseases and coagulation disorders. J. Infect. Dis. 1999, 180, 176–186. [Google Scholar] [CrossRef]
- Scholz, A.; Plate, K.H.; Reiss, Y. Angiopoietin-2: A multifaceted cytokine that functions in both angiogenesis and inflammation. Ann. N. Y. Acad. Sci. 2015, 1347, 45–51. [Google Scholar] [CrossRef]
- Gavrilovskaya, I.N.; Gorbunova, E.E.; Mackow, N.A.; Mackow, E.R. Hantaviruses direct endothelial cell permeability by sensitizing cells to the vascular permeability factor VEGF, while angiopoietin 1 and sphingosine 1-phosphate inhibit hantavirus-directed permeability. J. Virol. 2008, 82, 5797–5806. [Google Scholar] [CrossRef]
- Kerber, R.; Krumkamp, R.; Korva, M.; Rieger, T.; Wurr, S.; Duraffour, S.; Oestereich, L.; Gabriel, M.; Sissoko, D.; Anglaret, X.; et al. Kinetics of Soluble Mediators of the Host Response in Ebola Virus Disease. J. Infect. Dis. 2018, 218, S496–S503. [Google Scholar] [CrossRef] [Green Version]
- De Sousa Cardozo, F.T.G.; Baimukanova, G.; Lanteri, M.C.; Keating, S.M.; Ferreira, F.M.; Heitman, J.; Pannuti, C.S.; Pati, S.; Romano, C.M.; Sabino, E.C. Serum from dengue virus-infected patients with and without plasma leakage differentially affects endothelial cells barrier function in vitro. PLoS ONE 2017, 12, e0178820. [Google Scholar] [CrossRef]
- Soe, H.J.; Khan, A.M.; Manikam, R.; Samudi Raju, C.; Vanhoutte, P.; Sekaran, S.D. High dengue virus load differentially modulates human microvascular endothelial barrier function during early infection. J. Gen. Virol. 2017, 98, 2993–3007. [Google Scholar] [CrossRef]
- Perdomo-Celis, F.; Salvato, M.S.; Medina-Moreno, S.; Zapata, J.C. T-Cell Response to Viral Hemorrhagic Fevers. Vaccines 2019, 7, 11. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Disease Course | No. of Enrolled Patients | Self-Reported Onset of Symptoms (Median; Min/Max) | Day of Hospitalization (Median; Min/Max) | Viral load (Median *; Min/Max) | IgG Pos/Neg (Min; Max Titer) | IgM Pos/Neg (Min; Max Titer) |
|---|---|---|---|---|---|---|---|
| CCHF | moderate | 25 | 7 (2–10) | 2 (1–7) | 5.6 (2.2–7.9) | 2/25(800; 1600) | 7/25 (800; >6400) |
| severe | 18 | 5 (2–8) | 2 (1–4) | 6.9 (2.7–8.9) | 1/18 (100) | 6/18 (1600; 6400) | |
| fatal | 14 | 4 (2–7) | 2 (1–2) | 8.9 (2.7–10.0) | 0/14 | 9/14 (400; 3200) | |
| ∑ | 70 | 6 (2–10) | 2 (1–7) | 6.6 (2.0–10.0) | 5/70 (100; 1600) | 23/70 (400; >6400) | |
| PUU | mild | 25 | 6 (3–10) | 2 (1–6) | 3.7 (0.2–7.0) | 15/25 (400; >6400) | 25/25 (100; >6400) |
| severe | 25 | 6 (3–9) | 2 (1–8) | 4.6 (2.6–7.6) | 13/25 (100; >6400) | 24/25 (400; >6400) | |
| ∑ | 50 | 6 (3–10) | 2 (1–8) | 4.4 (0.2–7.6) | 27/50 (100; >6400) | 49/50 (100; >6400) | |
| DOB | mild | 25 | 7 (3–11) | 2 (1–8) | 4.6 (1.3–8.3) | 9/25 (800; 3200) | 24/25 (800; >6400) |
| severe | 22 | 7 (3–11) | 2 (1–6) | 5.7 (2.1–7.7) | 9/22 (100; >6400) | 22/22 (1600; >6400) | |
| fatal | 3 | 4 (3–4) | 2 (1–4) | 5.2 (4.3–6.8) | 0/3 | 3/3 (1600; 6400) | |
| ∑ | 50 | 7 (3–11) | 2 (1–8) | 4.8 (1.3–8.3) | 18/50 (100; >6400) | 49/50 (800; >6400) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korva, M.; Resman Rus, K.; Pavletič, M.; Saksida, A.; Knap, N.; Jelovšek, M.; Strašek Smrdel, K.; Jakupi, X.; Humolli, I.; Dedushaj, J.; et al. Characterization of Biomarker Levels in Crimean–Congo Hemorrhagic Fever and Hantavirus Fever with Renal Syndrome. Viruses 2019, 11, 686. https://doi.org/10.3390/v11080686
Korva M, Resman Rus K, Pavletič M, Saksida A, Knap N, Jelovšek M, Strašek Smrdel K, Jakupi X, Humolli I, Dedushaj J, et al. Characterization of Biomarker Levels in Crimean–Congo Hemorrhagic Fever and Hantavirus Fever with Renal Syndrome. Viruses. 2019; 11(8):686. https://doi.org/10.3390/v11080686
Chicago/Turabian StyleKorva, Miša, Katarina Resman Rus, Miša Pavletič, Ana Saksida, Nataša Knap, Mateja Jelovšek, Katja Strašek Smrdel, Xhevat Jakupi, Isme Humolli, Jusuf Dedushaj, and et al. 2019. "Characterization of Biomarker Levels in Crimean–Congo Hemorrhagic Fever and Hantavirus Fever with Renal Syndrome" Viruses 11, no. 8: 686. https://doi.org/10.3390/v11080686
APA StyleKorva, M., Resman Rus, K., Pavletič, M., Saksida, A., Knap, N., Jelovšek, M., Strašek Smrdel, K., Jakupi, X., Humolli, I., Dedushaj, J., Petrovec, M., & Avšič-Županc, T. (2019). Characterization of Biomarker Levels in Crimean–Congo Hemorrhagic Fever and Hantavirus Fever with Renal Syndrome. Viruses, 11(8), 686. https://doi.org/10.3390/v11080686