Strain-Dependent Porcine Circovirus Type 2 (PCV2) Entry and Replication in T-Lymphoblasts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Porcine T-Lymphoblasts In Vitro
2.2. Virus Preparation and Particle Quantification
2.3. PCV2 Peplication Kinetics in T-Lymphoblasts
2.4. PCV2 Attachment to T-Lymphoblasts
2.4.1. Visualization of PCV2 Attachment
2.4.2. Determination of the Expression of GAGs on T-Lymphoblasts
2.4.3. Evaluation of PCV2 Particles Binding to CS-Expressed T-Lymphoblasts
2.4.4. Effect of Enzymatic Treatment on PCV2 Infection in T-Lymphoblasts
2.5. PCV2 Entry and Disassembly in T-Lymphoblasts
2.5.1. Visualization of PCV2 Internalization
2.5.2. Effect of Different Entry Inhibitors on PCV2 Infection in T-Lymphoblasts
2.5.3. Effect of Acidotropic Agents on PCV2 Infection in T-Lymphoblasts
2.5.4. Effect of AEBSF on PCV2 Infection in T-Lymphoblasts
2.6. Toxicity Assays
2.7. Amino Acid and 3D Structural Analyses of PCV2 Cap Sequences from Strains 1121 and Stoon1010
2.8. Statistical Analysis
3. Results
3.1. The In Vitro Generated T-Lymphoblasts Support PCV2 Replication
3.2. PCV2 Attachment to T-Lymphoblasts is Mediated by Chondroitin Sulfate
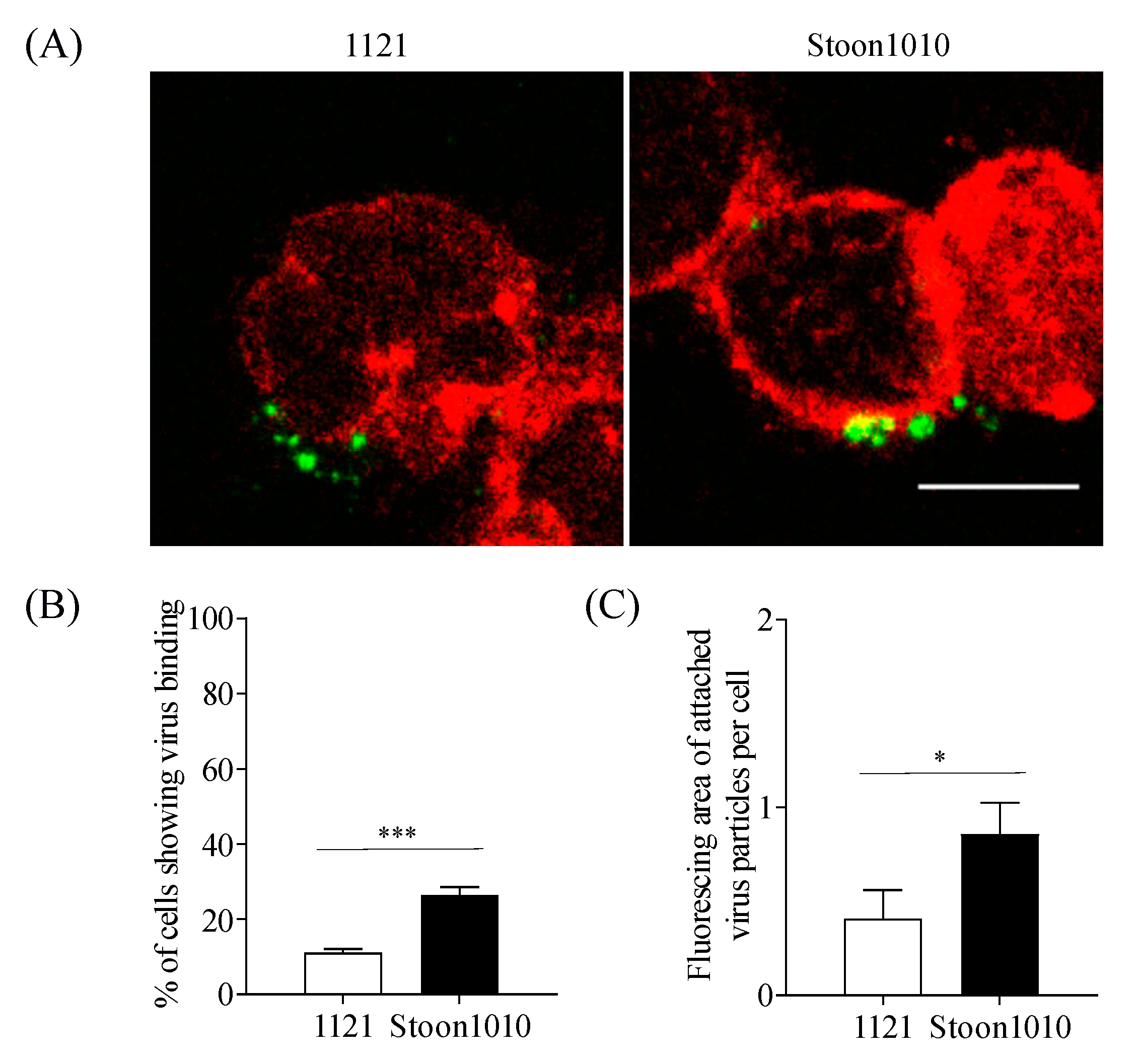
3.2.1. PCV2 Attachment is Restricted to 11–26% of T-Lymphoblasts
3.2.2. CS, but Not Other GAGs, is Expressed on T-Lymphoblasts
3.2.3. PCV2 Particles Were Able to Bind to T-Lymphoblasts With and Without CS Molecules
3.2.4. Enzymatic Removal of CS Decreased PCV2 Infection of T-Lymphoblasts
3.3. PCV2 Entry and Disassembly in T-Lymphoblasts
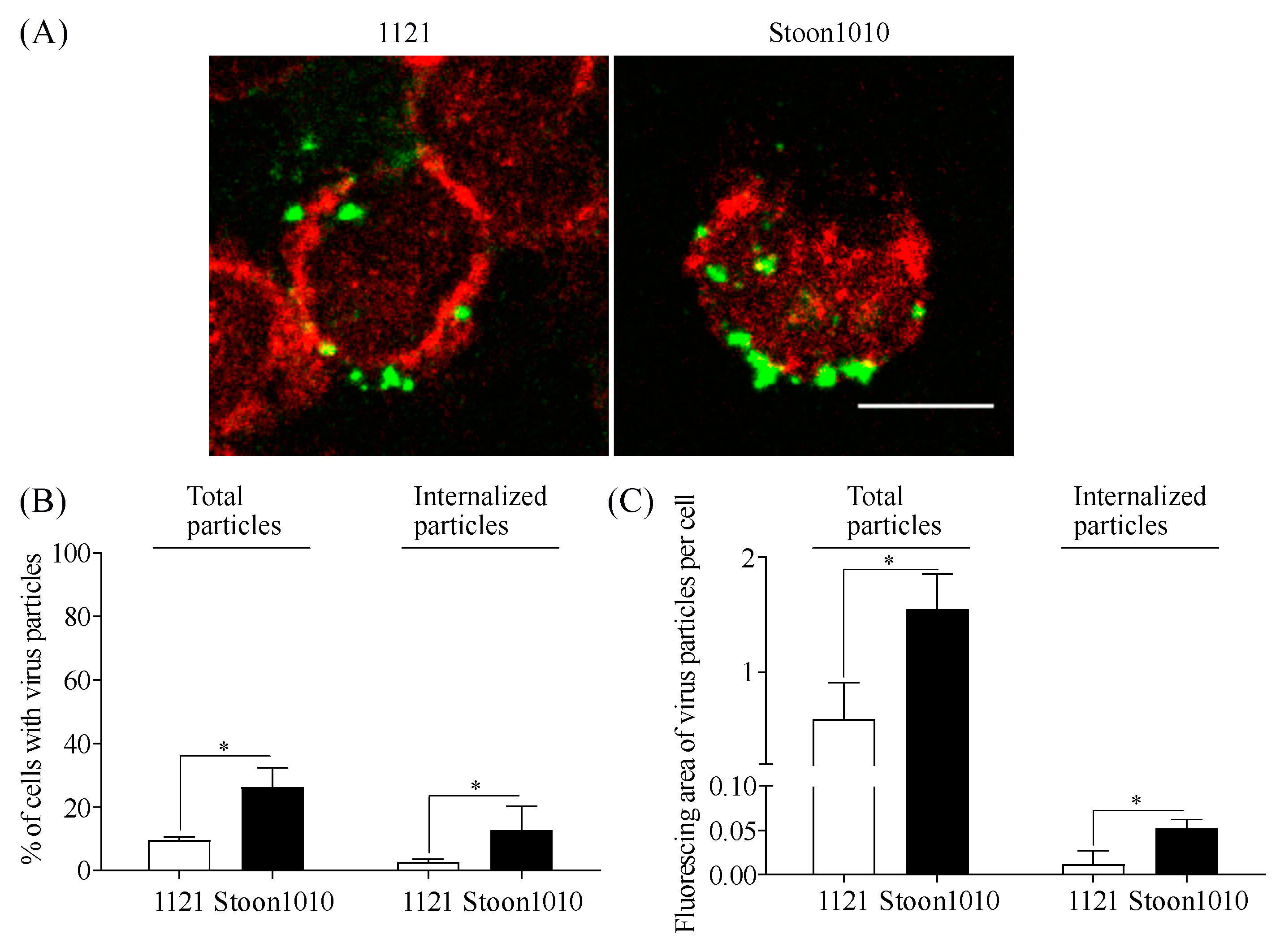
3.3.1. Visualization of PCV2 Internalization
3.3.2. PCV2 Strain 1121, but Not Stoon1010, Exploits Macropinocytosis for Its Entry
3.3.3. PCV2 Entry Occurs via Clathrin-Mediated Endocytosis and is Independent of Caveolae-Mediated Endocytosis
3.3.4. PCV2 Entry Into T-Lymphoblasts Requires Actin
3.3.5. Participation of Small GTPases in PCV2 Entry into T-Lymphoblasts
3.3.6. PCV2 Infection of T-Lymphoblasts Requires a Low-pH Step
3.3.7. PCV2 Disassembly is Mediated by Serine Proteases
3.4. Structural Difference of PCV2 Cap between Strains 1121 and Stoon1010
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AEBSF | 4-(2-aminoethyl) benzenesulfonyl fluoride |
| ATCC | American Type Culture Collection |
| Cap | capsid protein |
| CHO | Chinese hamster ovary |
| ConA | concanavalin A |
| CS | chondroitin sulfate |
| DABCO | 1,4-diazobicyclo-2.2.2-octane |
| DPBS | Dulbecco’s PBS |
| DSHB | the Developmental Studies Hybridoma Bank |
| FCS | fetal calf serum |
| GAG | glycosaminoglycan |
| HA | hyaluronic acid |
| HIV | human immunodeficiency virus |
| HS | heparan sulfate |
| HSV | herpes simplex virus |
| IL-2 | interleukin-2 |
| IPMA | immunoperoxidase monolayer assay |
| I-TASSER | iterative threading assembly refinement |
| JEV | Japanese encephalitis virus |
| KS | keratan sulfate |
| mAb | monoclonal antibody |
| mβCD | methyl-β-cyclodextrin |
| MTT | metabolize 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| ORF | open reading frame |
| PBMC | porcine blood mononuclear cell |
| PCV | porcine circovirus |
| PCVAD | PCV2-associated disease |
| PF | paraformaldehyde |
| PMWS | postweaning multisystemic wasting syndrome |
| Rep | replicase protein |
| RT | room temperature |
| SK | swine kidney |
References
- Todd, D.; Bendinelli, M.; Biagini, P.; Hino, S.; Mankertz, A.; Mishiro, S.; Niel, C.; Okamoto, H.; Raidal, S.; Ritchie, B.W.; et al. Circoviridae. In Virus Taxonomy, VIIIth Report of the International Committee for the Taxonomy of Viruses; Elsevier/Academic Press: London, UK, 2005; pp. 327–334. [Google Scholar]
- Afolabi, K.O.; Iweriebor, B.C.; Okoh, A.I.; Obi, L.C. Global Status of Porcine circovirus Type 2 and Its Associated Diseases in Sub-Saharan Africa. Adv. Virol. 2017, 2017, 6807964. [Google Scholar] [CrossRef] [PubMed]
- Tischer, I.; Gelderblom, H.; Vettermann, W.; Koch, M.A. A very small porcine virus with circular single-stranded DNA. Nature 1982, 295, 64. [Google Scholar] [CrossRef] [PubMed]
- Crowther, R.A.; Berriman, J.A.; Curran, W.L.; Allan, G.M.; Todd, D. Comparison of the structures of three circoviruses: Chicken anemia virus, porcine circovirus type 2, and beak and feather disease virus. J. Virol. 2003, 77, 13036–13041. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.A.; Viancelli, A.; Rigotto, C.; Pilotto, M.R.; Esteves, P.A.; Kunz, A.; Barardi, C.R. Surveillance of human and swine adenovirus, human norovirus and swine circovirus in water samples in Santa Catarina, Brazil. J. Water Health 2012, 10, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verreault, D.; Letourneau, V.; Gendron, L.; Masse, D.; Gagnon, C.A.; Duchaine, C. Airborne porcine circovirus in Canadian swine confinement buildings. Vet. Microbiol. 2010, 141, 224–230. [Google Scholar] [CrossRef]
- Blunt, R.; McOrist, S.; McKillen, J.; McNair, I.; Jiang, T.; Mellits, K. House fly vector for porcine circovirus 2b on commercial pig farms. Vet. Microbiol. 2011, 149, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.L.; Chen, S.N.; Liu, W.; Li, X.P.; Deng, S.F.; Wen, X.H.; Luo, M.L.; Lv, D.H.; Wei, W.K.; Chen, R.A. Molecular detection and genome characterization of porcine circovirus type 2 in rats captured on commercial swine farms. Arch. Virol. 2016, 161, 3237–3244. [Google Scholar] [CrossRef] [PubMed]
- Halami, M.Y.; Müller, H.; Böttcher, J.; Vahlenkamp, T.W. Whole-genome sequences of two strains of porcine circovirus 2 isolated from calves in Germany. Genome Announc. 2014, 2, e01150-13. [Google Scholar] [CrossRef]
- Wang, G.S.; Sun, N.; Tian, F.L.; Wen, Y.J.; Xu, C.; Li, J.; Chen, Q.; Wang, J.B. Genetic analysis of porcine circovirus type 2 from dead minks. J. Gen. Virol. 2016, 97, 2316–2322. [Google Scholar] [CrossRef]
- Song, T.; Zhang, S.; Hao, J.; Xin, S.; Hui, W.; Tang, M.; Li, W.; Tian, R.; Liu, X.; Rui, P.; et al. First detection and genetic analysis of fox-origin porcine circovirus type 2. Transbound. Emerg. Dis. 2018, 66, 1–6. [Google Scholar] [CrossRef]
- Hattermann, K.; Roedner, C.; Schmitt, C.; Finsterbusch, T.; Steinfeldt, T.; Mankertz, A. Infection studies on human cell lines with porcine circovirus type 1 and porcine circovirus type 2. Xenotransplantation 2004, 11, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K. Transcriptional analysis of porcine circovirus type 2. Virology 2003, 305, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Lekcharoensuk, P.; Morozov, I.; Paul, P.S.; Thangthumniyom, N.; Wajjawalku, W.; Meng, X.J. Epitope Mapping of the Major Capsid Protein of Type 2 Porcine Circovirus (PCV2) by Using Chimeric PCV1 and PCV2. J. Virol. 2004, 78, 8135–8145. [Google Scholar] [CrossRef] [PubMed]
- Nawagitgul, P.; Morozov, I.; Bolin, S.R.; Harms, P.A.; Sorden, S.D.; Paul, P.S. Open reading frame 2 of porcine circovirus type 2 encodes a major capsid protein. J. Gen. Virol. 2000, 81, 2281–2287. [Google Scholar] [CrossRef] [PubMed]
- Segales, J. Porcine circovirus type 2 (PCV2) infections: Clinical signs, pathology and laboratory diagnosis. Virus Res. 2012, 164, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Segales, J.; Allan, G.M.; Domingo, M. Porcine circovirus diseases. Anim. Health Res. Rev. 2005, 6, 119–142. [Google Scholar] [CrossRef] [PubMed]
- Segalés, J.; Domingo, M. Postweaning mulstisystemic wasting syndrome (PMWS) in pigs. A review. Vet. Q. 2002, 24, 109–124. [Google Scholar] [CrossRef]
- Sanchez, R.E., Jr.; Meerts, P.; Nauwynck, H.J.; Ellis, J.A.; Pensaert, M.B. Characteristics of porcine circovirus-2 replication in lymphoid organs of pigs inoculated in late gestation or postnatally and possible relation to clinical and pathological outcome of infection. J. Vet Diagn. Investig. 2004, 16, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.E., Jr.; Meerts, P.; Nauwynck, H.J.; Pensaert, M.B. Change of porcine circovirus 2 target cells in pigs during development from fetal to early postnatal life. Vet. Microbiol. 2003, 95, 15–25. [Google Scholar] [CrossRef]
- Pensaert, M.B.; Sanchez, R.E., Jr.; Ladekjaer-Mikkelsen, A.S.; Allan, G.M.; Nauwynck, H.J. Viremia and effect of fetal infection with porcine viruses with special reference to porcine circovirus 2 infection. Vet. Microbiol. 2004, 98, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Rosell, C.; Segales, J.; Plana-Duran, J.; Balasch, M.; Rodriguez-Arrioja, G.M.; Kennedy, S.; Allan, G.M.; McNeilly, F.; Latimer, K.S.; Domingo, M. Pathological, immunohistochemical, and in-situ hybridization studies of natural cases of postweaning multisystemic wasting syndrome (PMWS) in pigs. J. Comp. Pathol. 1999, 120, 59–78. [Google Scholar] [CrossRef] [PubMed]
- Chianini, F.; Majo, N.; Segales, J.; Dominguez, J.; Domingo, M. Immunohistochemical characterisation of PCV2 associate lesions in lymphoid and non-lymphoid tissues of pigs with natural postweaning multisystemic wasting syndrome (PMWS). Vet. Immunol. Immunopathol. 2003, 94, 63–75. [Google Scholar] [CrossRef]
- Misinzo, G.; Delputte, P.L.; Meerts, P.; Lefebvre, D.J.; Nauwynck, H.J. Porcine circovirus 2 uses heparan sulfate and chondroitin sulfate B glycosaminoglycans as receptors for its attachment to host cells. J. Virol. 2006, 80, 3487–3494. [Google Scholar] [CrossRef] [PubMed]
- Misinzo, G.; Meerts, P.; Bublot, M.; Mast, J.; Weingartl, H.M.; Nauwynck, H.J. Binding and entry characteristics of porcine circovirus 2 in cells of the porcine monocytic line 3D4/31. J. Gen. Virol. 2005, 86, 2057–2068. [Google Scholar] [CrossRef] [PubMed]
- Vincent, I.E.; Carrasco, C.P.; Guzylack-Piriou, L.; Herrmann, B.; McNeilly, F.; Allan, G.M.; Summerfield, A.; McCullough, K.C. Subset-dependent modulation of dendritic cell activity by circovirus type 2. Immunology 2005, 115, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Trus, I.; Yang, B.; Huang, L.; Nauwynck, H. Breed Differences in PCV2 Uptake and Disintegration in Porcine Monocytes. Viruses 2018, 10, 562. [Google Scholar] [CrossRef] [PubMed]
- Darwich, L.; Segales, J.; Mateu, E. Pathogenesis of postweaning multisystemic wasting syndrome caused by Porcine circovirus 2: An immune riddle. Arch. Virol. 2004, 149, 857–874. [Google Scholar] [CrossRef] [PubMed]
- Misinzo, G.; Delputte, P.L.; Lefebvre, D.J.; Nauwynck, H.J. Porcine circovirus 2 infection of epithelial cells is clathrin-, caveolae- and dynamin-independent, actin and Rho-GTPase-mediated, and enhanced by cholesterol depletion. Virus Res. 2009, 139, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, D.J.; Meerts, P.; Costers, S.; Misinzo, G.; Barbe, F.; Van Reeth, K.; Nauwynck, H.J. Increased porcine circovirus type 2 replication in porcine leukocytes in vitro and in vivo by concanavalin A stimulation. Vet. Microbiol. 2008, 132, 74–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauwynck, H.J.; Sanchez, R.; Meerts, P.; Lefebvre, D.J.; Saha, D.; Huang, L.; Misinzo, G. Cell tropism and entry of porcine circovirus 2. Virus Res. 2012, 164, 43–45. [Google Scholar] [CrossRef]
- Van Renne, N.; Wei, R.; Pochet, N.; Nauwynck, H.J. Dissecting clinical outcome of porcine circovirus type 2 with in vivo derived transcriptomic signatures of host tissue responses. BMC Genom. 2018, 19, 831. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.; Hassard, L.; Clark, E.; Harding, J.; Allan, G.; Willson, P.; Strokappe, J.; Martin, K.; McNeilly, F.; Meehan, B.; et al. Isolation of circovirus from lesions of pigs with postweaning multisystemic wasting syndrome. Can. Vet. J. 1998, 39, 44–51. [Google Scholar] [PubMed]
- Meehan, B.M.; McNeilly, F.; McNair, I.; Walker, I.; Ellis, J.A.; Krakowka, S.; Allan, G.M. Isolation and characterization of porcine circovirus 2 from cases of sow abortion and porcine dermatitis and nephropathy syndrome. Arch. Virol. 2001, 146, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Meerts, P.; Nauwynck, H.D.I.; Sanchez, R.E.; Mateusen, B.; Pensaert, M.U. Prevalence of porcine circovirus 2 (PCV2)-related wasting on Belgian farms with or without a history of postweaning multisystemic wasting syndrome. Vlaams Diergeneeskd Tijdschr 2004, 73, 31–38. [Google Scholar]
- McNeilly, F.; McNair, I.; Mackie, D.P.; Meehan, B.M.; Kennedy, S.; Moffett, D.; Ellis, J.; Krakowka, S.; Allan, G.M. Production, characterisation and applications of monoclonal antibodies to porcine circovirus 2. Arch. Virol. 2001, 146, 909–922. [Google Scholar] [CrossRef]
- Lefebvre, D.J.; Costers, S.; Van Doorsselaere, J.; Misinzo, G.; Delputte, P.L.; Nauwynck, H.J. Antigenic differences among porcine circovirus type 2 strains, as demonstrated by the use of monoclonal antibodies. J. Gen. Virol. 2008, 89, 177–187. [Google Scholar] [CrossRef]
- Zanetti, M.; Ratcliffe, A.; Watt, F.M. Two subpopulations of differentiated chondrocytes identified with a monoclonal antibody to keratan sulfate. J. Cell Biol. 1985, 101, 53–59. [Google Scholar] [CrossRef]
- Caterson, B.; Melrose, J. Keratan sulfate, a complex glycosaminoglycan with unique functional capability. Glycobiology 2018, 28, 182–206. [Google Scholar] [CrossRef]
- Nettelbladt, O.; Bergh, J.; Schenholm, M.; Tengblad, A.; Hallgren, R. Accumulation of hyaluronic acid in the alveolar interstitial tissue in bleomycin-induced alveolitis. Am. Rev. Respir. Dis. 1989, 139, 759–762. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Tischer, I.; Peters, D.; Rasch, R.; Pociuli, S. Replication of porcine circovirus: Induction by glucosamine and cell cycle dependence. Arch. Virol. 1987, 96, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Vincent, A.; Opriessnig, T.; Carpenter, S.; Kitikoon, P.; Halbur, P.G.; Thacker, E. Quantification of PCV2 capsid transcript in peripheral blood mononuclear cells (PBMCs) in vitro. Vet. Microbiol. 2007, 123, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W. Biosynthesis of glycosaminoglycans by thymic lymphocytes. Effects of mitogenic activation. Biochemistry 1982, 21, 6088–6096. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Gulping rather than sipping: Macropinocytosis as a way of virus entry. Curr. Opin. Microbiol. 2012, 15, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Kaksonen, M.; Toret, C.P.; Drubin, D.G. Harnessing actin dynamics for clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2006, 7, 404. [Google Scholar] [CrossRef] [PubMed]
- Qualmann, B.; Mellor, H. Regulation of endocytic traffic by Rho GTPases. Biochem. J. 2003, 371, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Marsh, M.; Helenius, A. Virus Entry: Open Sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef] [Green Version]
- Barrett, A.J. Classification of peptidases. Methods Enzymol. 1994, 244, 1–15. [Google Scholar] [PubMed]
- Misinzo, G.; Delputte, P.L.; Nauwynck, H.J. Inhibition of endosome-lysosome system acidification enhances porcine circovirus 2 infection of porcine epithelial cells. J. Virol. 2008, 82, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.; Eley, T.; Browne, C.; Martineau, H.M.; Werling, D. Oral application of freeze-dried yeast particles expressing the PCV2b Cap protein on their surface induce protection to subsequent PCV2b challenge in vivo. Vaccine 2015, 33, 6199–6205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meerts, P.; Misinzo, G.; McNeilly, F.; Nauwynck, H.J. Replication kinetics of different porcine circovirus 2 strains in PK-15 cells, fetal cardiomyocytes and macrophages. Arch. Virol. 2005, 150, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Carino, C.; Duffy, C.; Sanchez-Chardi, A.; McNeilly, F.; Allan, G.M.; Segales, J. Porcine circovirus type 2 morphogenesis in a clone derived from the l35 lymphoblastoid cell line. J. Comp. Pathol. 2011, 144, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, M.; Opriessnig, T.; Halbur, P.G.; Elvinger, F.; Meng, X.J. Two amino acid mutations in the capsid protein of type 2 porcine circovirus (PCV2) enhanced PCV2 replication in vitro and attenuated the virus in vivo. J. Virol. 2004, 78, 13440–13446. [Google Scholar] [CrossRef] [PubMed]
- Clasper, S.; Vekemans, S.; Fiore, M.; Plebanski, M.; Wordsworth, P.; David, G.; Jackson, D.G. Inducible expression of the cell surface heparan sulfate proteoglycan syndecan-2 (fibroglycan) on human activated macrophages can regulate fibroblast growth factor action. J. Biol. Chem. 1999, 274, 24113–24123. [Google Scholar] [CrossRef] [PubMed]
- Khayat, R.; Brunn, N.; Speir, J.A.; Hardham, J.M.; Ankenbauer, R.G.; Schneemann, A.; Johnson, J.E. The 2.3-angstrom structure of porcine circovirus 2. J. Virol. 2011, 85, 7856–7862. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Fu, Y.; Wang, Y.; Lu, Y.; Wei, Y.; Tang, Q.; Fan, P.; Liu, J.; Zhang, L.; Zhang, F.; et al. A porcine circovirus type 2 (PCV2) mutant with 234 amino acids in capsid protein showed more virulence in vivo, compared with classical PCV2a/b strain. PLoS ONE 2012, 7, e41463. [Google Scholar] [CrossRef]
- Zhan, Y.; Wang, N.; Zhu, Z.; Wang, Z.; Wang, A.; Deng, Z.; Yang, Y. In silico analyses of antigenicity and surface structure variation of an emerging porcine circovirus genotype 2b mutant, prevalent in southern China from 2013 to 2015. J. Gen. Virol. 2016, 97, 922–933. [Google Scholar] [CrossRef] [Green Version]
- Schulz, W.L.; Haj, A.K.; Schiff, L.A. Reovirus uses multiple endocytic pathways for cell entry. J. Virol. 2012, 86, 12665–12675. [Google Scholar] [CrossRef] [PubMed]
- Permanyer, M.; Ballana, E.; Este, J.A. Endocytosis of HIV: Anything goes. Trends Microbiol. 2010, 18, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Hasebe, R.; Sasaki, M.; Sawa, H.; Wada, R.; Umemura, T.; Kimura, T. Infectious entry of equine herpesvirus-1 into host cells through different endocytic pathways. Virology 2009, 393, 198–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karasneh, G.A.; Shukla, D. Herpes simplex virus infects most cell types in vitro: Clues to its success. Virol. J. 2011, 8, 481. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-Z.; Xu, Q.-Q.; Wu, D.-G.; Ren, H.; Zhao, P.; Lao, W.-G.; Wang, Y.; Tao, Q.-Y.; Qian, X.-J.; Wei, Y.-H.; et al. Japanese Encephalitis Virus Enters Rat Neuroblastoma Cells via a pH-Dependent, Dynamin and Caveola-Mediated Endocytosis Pathway. J. Virol. 2012, 86, 13407–13422. [Google Scholar] [CrossRef] [Green Version]
- Kalia, M.; Khasa, R.; Sharma, M.; Nain, M.; Vrati, S. Japanese Encephalitis Virus Infects Neuronal Cells through a Clathrin-Independent Endocytic Mechanism. J. Virol. 2013, 87, 148–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, E.; Tscherne, D.M.; Wienholts, M.J.; Cobos-Jimenez, V.; Scholte, F.; Garcia-Sastre, A.; Rottier, P.J.; de Haan, C.A. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 2011, 7, e1001329. [Google Scholar] [CrossRef]
- Aleksandrowicz, P.; Marzi, A.; Biedenkopf, N.; Beimforde, N.; Becker, S.; Hoenen, T.; Feldmann, H.; Schnittler, H.J. Ebola virus enters host cells by macropinocytosis and clathrin-mediated endocytosis. J. Infect. Dis. 2011, 204 (Suppl. 3), S957–S967. [Google Scholar] [CrossRef]
- Timmusk, S.; Fossum, C.; Berg, M. Porcine circovirus type 2 replicase binds the capsid protein and an intermediate filament-like protein. J. Gen. Virol. 2006, 87, 3215–3223. [Google Scholar] [CrossRef]
- Xiao, C.T.; Halbur, P.G.; Opriessnig, T. Global molecular genetic analysis of porcine circovirus type 2 (PCV2) sequences confirms the presence of four main PCV2 genotypes and reveals a rapid increase of PCV2d. J. Gen. Virol. 2015, 96, 1830–1841. [Google Scholar] [CrossRef]
- Wei, R.; Xie, J.; Theuns, S.; Nauwynck, H.J. Changes on the viral capsid surface during the evolution of porcine circovirus type 2 (PCV2) from 2009 till 2018 may lead to a better receptor binding. Virus Evol. 2019, 5, vez206. [Google Scholar] [CrossRef] [PubMed]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, R.; Van Renne, N.; Nauwynck, H.J. Strain-Dependent Porcine Circovirus Type 2 (PCV2) Entry and Replication in T-Lymphoblasts. Viruses 2019, 11, 813. https://doi.org/10.3390/v11090813
Wei R, Van Renne N, Nauwynck HJ. Strain-Dependent Porcine Circovirus Type 2 (PCV2) Entry and Replication in T-Lymphoblasts. Viruses. 2019; 11(9):813. https://doi.org/10.3390/v11090813
Chicago/Turabian StyleWei, Ruifang, Nicolaas Van Renne, and Hans J. Nauwynck. 2019. "Strain-Dependent Porcine Circovirus Type 2 (PCV2) Entry and Replication in T-Lymphoblasts" Viruses 11, no. 9: 813. https://doi.org/10.3390/v11090813
APA StyleWei, R., Van Renne, N., & Nauwynck, H. J. (2019). Strain-Dependent Porcine Circovirus Type 2 (PCV2) Entry and Replication in T-Lymphoblasts. Viruses, 11(9), 813. https://doi.org/10.3390/v11090813