Real-Time Analysis of Individual Ebola Virus Glycoproteins Reveals Pre-Fusion, Entry-Relevant Conformational Dynamics

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Plasmids
2.3. Protein Production and Purification
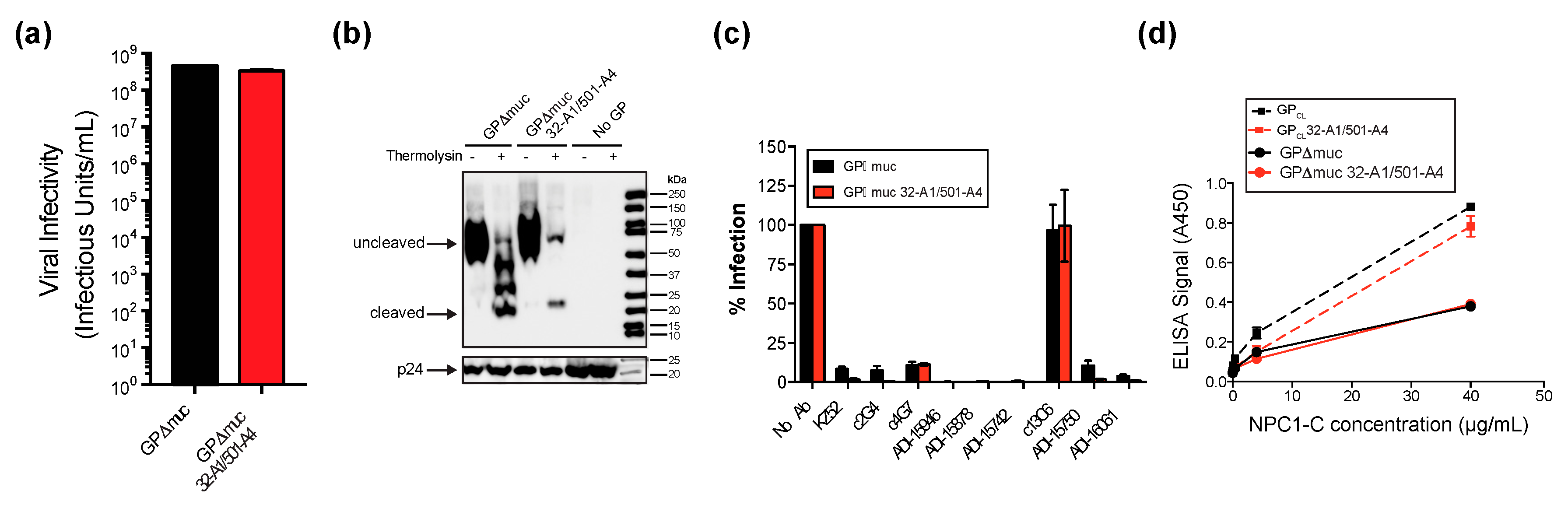
2.4. Infectivity and Neutralization Assays
2.5. sNPC1-C ELISA
2.6. Immunoblots
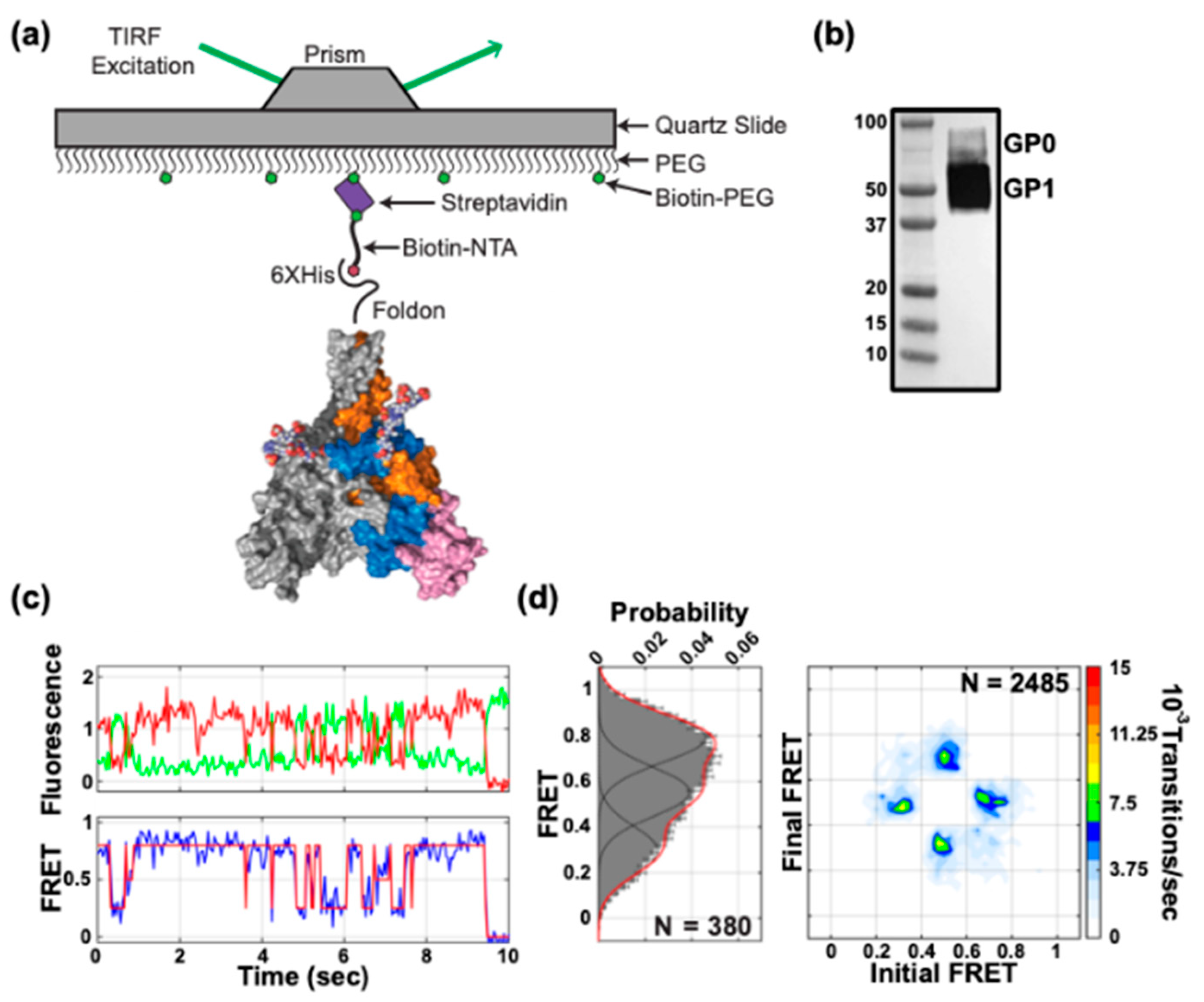
2.7. GPΔTM Production and Labeling for smFRET Imaging
2.8. Pseudovirus Production and Labeling for smFRET Imaging
2.9. smFRET Imaging
2.10. Analysis of smFRET Data
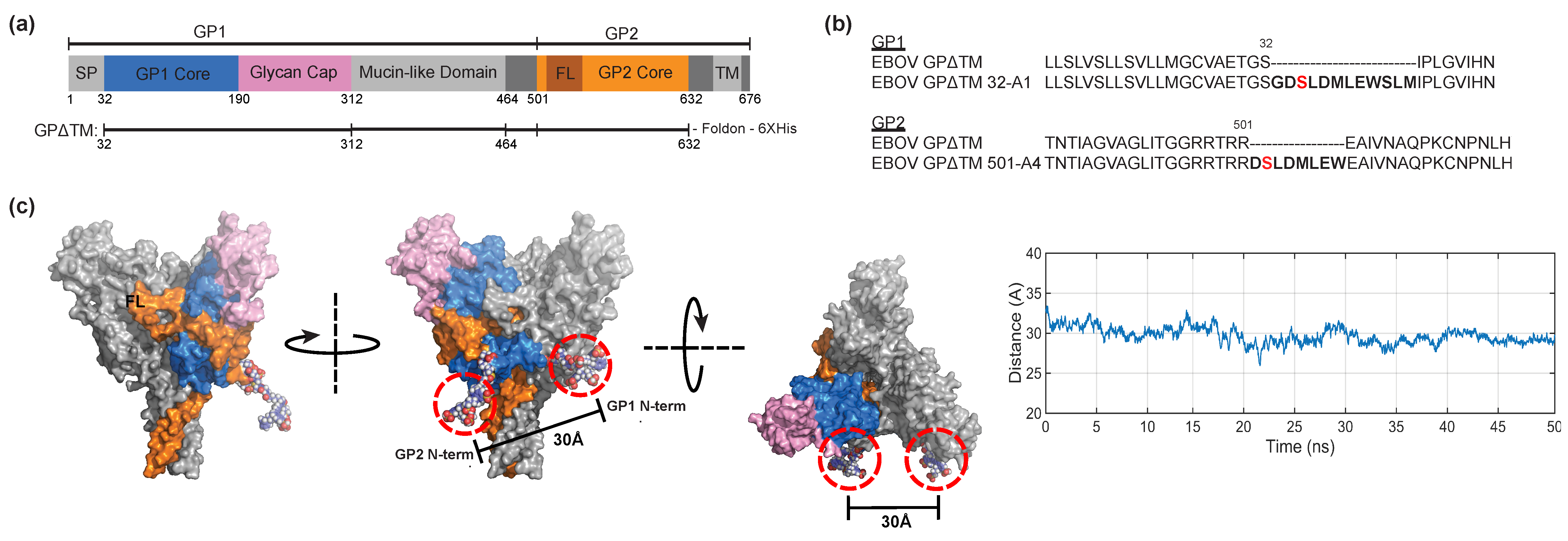
2.11. Molecular Dynamics Simulation of Fluorescently Labeled GPΔTM
3. Results
3.1. Site-Specific Fluorescent Labelling of EBOV GP
3.2. Conformational Dynamics of EBOV GPΔTM
3.3. Conformational Dynamics of Pseudovirion-Associated EBOV GPΔmuc
3.4. Effects of Proteolytic Cleavage and NPC1 Binding on EBOV GPΔmuc Conformation
3.5. Neutralizing Antibodies Mediate GP Conformation
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baseler, L.; Chertow, D.S.; Johnson, K.M.; Feldmann, H.; Morens, D.M. The pathogenesis of Ebola virus disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 387–418. [Google Scholar] [CrossRef]
- Lee, J.E.; Fusco, M.L.; Hessell, A.J.; Oswald, W.B.; Burton, D.R.; Saphire, E. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008, 454, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Chandran, K.; Sullivan, N.; Felbor, U.; Whelan, S.; Cunningham, J. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schornberg, K.; Matsuyama, S.; Kabsch, K.; Delos, S.; Bouton, A.; White, J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006, 80, 4174–4178. [Google Scholar] [CrossRef] [Green Version]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.; Obernosterer, G.; Raaben, M.; Herbert, A.S.; Deffieu, M.S.; Krishnan, A.; Ndungo, E.; Sandesara, R.G.; Carette, J.E.; Kuehne, A.I.; et al. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012, 31, 1947–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Côté, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.S.; Krause, T.B.; Mittler, E.; Jangra, R.K.; Chandran, K. Direct visualization of Ebola virus fusion triggering in the endocytic pathway. mBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Simmons, J.A.; D’Souza, R.S.; Ruas, M.; Galione, A.; Casanova, J.E.; White, J.M. Ebolavirus glycoprotein directs fusion through NPC1(+) endolysosomes. J. Virol. 2016, 90, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Saphire, E.; Aman, J.M. Feverish quest for Ebola immunotherapy: Straight or cocktail? Trends Microbiol. 2016, 24, 684–686. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Ren, J.; Harlos, K.; Jones, D.M.; Zeltina, A.; Bowden, T.A.; Padilla-Parra, S.; Fry, E.E.; Stuart, D.I. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 2016, 535, 169–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misasi, J.; Gilman, M.S.; Kanekiyo, M.; Gui, M.; Cagigi, A.; Mulangu, S.; Corti, D.; Ledgerwood, J.E.; Lanzavecchia, A.; Cunningham, J.; et al. Structural and molecular basis for Ebola virus neutralization by protective human antibodies. Science 2016, 351, 1343–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallesen, J.; Murin, C.D.; de Val, N.; Cottrell, C.A.; Hastie, K.M.; Turner, H.L.; Fusco, M.L.; Flyak, A.I.; Zeitlin, L.; Crowe, J.E.; et al. Structures of Ebola virus GP and sGP in complex with therapeutic antibodies. Nat. Microbiol. 2016, 1, 16128. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, Y.; Song, J.; Qi, J.; Lu, G.; Yan, J.; Gao, G.F. Ebola viral glycoprotein bound to its endosomal receptor Niemann-Pick C1. Cell 2016, 164, 258–268. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Qian, H.; Zhou, X.; Wu, J.; Wan, T.; Cao, P.; Huang, W.; Zhao, X.; Wang, X.; Wang, P.; et al. Structural insights into the Niemann-Pick C1 (NPC1)-mediated cholesterol transfer and Ebola infection. Cell 2016, 165, 1467–1478. [Google Scholar] [CrossRef] [Green Version]
- White, J.M.; Whittaker, G.R. Fusion of enveloped viruses in endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef] [Green Version]
- Weissenhorn, W.; Carfí, A.; Lee, K.-H.; Skehel, J.J.; Wiley, D.C. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell 1998, 2, 605–616. [Google Scholar] [CrossRef]
- Malashkevich, V.N.; Schneider, B.J.; McNally, M.L.; Milhollen, M.A.; Pang, J.X.; Kim, P.S. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9-Å resolution. Proc. Natl. Acad. Sci. USA 1999, 96, 2662–2667. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, T.; Rodriguez, L.; Jahrling, P.; Sanchez, A.; Khan, A.; Nichol, S.; Peters, C.; Parren, P.; Burton, D. Ebola virus can be effectively neutralized by antibody produced in natural human infection. J. Virol. 1999, 73, 6024–6030. [Google Scholar] [CrossRef] [Green Version]
- Bornholdt, Z.A.; Ndungo, E.; Fusco, M.L.; Bale, S.; Flyak, A.I.; Crowe, J.E.; Chandran, K.; Saphire, E. Host-primed Ebola virus GP exposes a hydrophobic NPC1 receptor-binding pocket, revealing a target for broadly neutralizing antibodies. mBio 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- Ou, W.; Delisle, J.; Konduru, K.; Bradfute, S.; Radoshitzky, S.R.; Retterer, C.; Kota, K.; Bavari, S.; Kuhn, J.H.; Jahrling, P.B.; et al. Development and characterization of rabbit and mouse antibodies against ebolavirus envelope glycoproteins. J. Virol. Methods 2011, 174, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munro, J.B.; Gorman, J.; Ma, X.; Zhou, Z.; Arthos, J.; Burton, D.R.; Koff, W.C.; Courter, J.R.; Smith, A.B.; Kwong, P.D.; et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 2014, 346, 759–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, D.; Govindan, R.; Nikić-Spiegel, I.; Krammer, F.; Lemke, E.A.; Munro, J.B. Direct visualization of the conformational dynamics of single influenza hemagglutinin trimers. Cell 2018, 174, 926–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juette, M.F.; Terry, D.S.; Wasserman, M.R.; Tman, R.; Zhou, Z.; Zhao, H.; Blanchard, S.C. Single-molecule imaging of non-equilibrium molecular ensembles on the millisecond timescale. Nat. Methods 2016, 13, 341–344. [Google Scholar] [CrossRef]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Qin, F. Restoration of single-channel currents using the segmental k-means method based on hidden Markov modeling. Biophys. J. 2004, 86, 1488–1501. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Cironi, P.; Lin, A.J.; Xu, Y.; Hrvatin, S.; Golan, D.E.; Silver, P.A.; Walsh, C.T.; Yin, J. Genetically encoded short peptide tags for orthogonal protein labeling by Sfp and AcpS phosphopantetheinyl transferases. ACS Chem. Biol. 2007, 2, 337–346. [Google Scholar] [CrossRef]
- Zhou, Z.; Koglin, A.; Wang, Y.; McMahon, A.P.; Walsh, C.T. An eight residue fragment of an acyl carrier protein suffices for post-translational introduction of fluorescent pantetheinyl arms in protein modification in vitro and in vivo. J. Am. Chem. Soc. 2008, 130, 9925–9930. [Google Scholar] [CrossRef] [Green Version]
- Corti, D.; Misasi, J.; Mulangu, S.; Stanley, D.A.; Kanekiyo, M.; Wollen, S.; Ploquin, A.; Doria-Rose, N.A.; Staupe, R.P.; Bailey, M.; et al. Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science 2016, 351, 1339–1342. [Google Scholar] [CrossRef] [Green Version]
- Shedlock, D.J.; Bailey, M.A.; Popernack, P.M.; Cunningham, J.M.; Burton, D.R.; Sullivan, N.J. Antibody-mediated neutralization of Ebola virus can occur by two distinct mechanisms. Virology 2010, 401, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Murin, C.D.; Fusco, M.L.; Bornholdt, Z.A.; Qiu, X.; Olinger, G.G.; Zeitlin, L.; Kobinger, G.P.; Ward, A.B.; Saphire, E. Structures of protective antibodies reveal sites of vulnerability on Ebola virus. Proc. Natl. Acad. Sci. USA 2014, 111, 17182–17187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Imonti, J.; Melito, L.P.; Fernando, L.; Ströher, U.; Jones, S.M. Characterization of Zaire ebolavirus glycoprotein-specific monoclonal antibodies. Clin. Immunol. 2011, 141, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Wec, A.Z.; Herbert, A.S.; Murin, C.D.; Nyakatura, E.K.; Abelson, D.M.; Fels, M.J.; He, S.; James, R.M.; de Vega, M.-A.; Zhu, W.; et al. Antibodies from a human survivor define sites of vulnerability for broad protection against ebolaviruses. Cell 2017, 169, 878–890. [Google Scholar] [CrossRef]
- Bornholdt, Z.A.; Turner, H.L.; Murin, C.D.; Li, W.; Sok, D.; Souders, C.A.; Piper, A.E.; Goff, A.; Shamblin, J.D.; Wollen, S.E.; et al. Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science 2016, 351, 1078–1083. [Google Scholar] [CrossRef] [Green Version]
- West, B.R.; Wec, A.Z.; Moyer, C.L.; Fusco, M.L.; Ilinykh, P.A.; Huang, K.; Wirchnianski, A.S.; James, R.M.; Herbert, A.S.; Hui, S.; et al. Structural basis of broad ebolavirus neutralization by a human survivor antibody. Nat. Struct. Mol. Biol. 2019, 26, 204–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murin, C.D.; Bruhn, J.F.; Bornholdt, Z.A.; Copps, J.; Stanfield, R.; Ward, A.B. Structural basis of pan-ebolavirus neutralization by an antibody targeting the glycoprotein fusion loop. Cell Rep. 2018, 24, 2723–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, C.M.; Kim, P.S. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 1993, 73, 823–832. [Google Scholar] [CrossRef]
- Das, D.; Bulow, U.; Diehl, W.E.; Durham, N.D.; Senjobe, F.; Chandran, K.; Luban, J.; Munro, J.B. Conformational changes in the Ebola virus membrane fusion machine induced by pH, Ca2+, and receptor binding. 2019; submitted. [Google Scholar]
- Lappala, A.; Nishima, W.; Miner, J.; Fenimore, P.; Fischer, W.; Hraber, P.; Zhang, M.; McMahon, B.; Tung, C.-S. Structural transition and antibody binding of EBOV GP and ZIKV E proteins from pre-fusion to fusion-initiation state. Biomolecules 2018, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Brecher, M.; Schornberg, K.L.; Delos, S.E.; Fusco, M.L.; Saphire, E.; White, J.M. Cathepsin cleavage potentiates the Ebola virus glycoprotein to undergo a subsequent fusion-relevant conformational change. J. Virol. 2012, 86, 364–372. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Occupancies (%) | |||
|---|---|---|---|
| Low FRET | Intermediate FRET | High FRET | |
| GPΔTM | 20.5% | 26.9% | 52.6% |
| GPΔmuc | 31.4% | 23.6% | 45.0% |
| GPCL | 22.2% | 45.8% | 32.0% |
| GPCL + NPC1-C | 19.8% | 53.4% | 26.8% |
| GPΔmuc + KZ52 (15 μg/mL) | 35.7% | 23.3% | 41.0% |
| GPΔmuc + KZ52 (500 μg/mL) | 42.0% | 23.2% | 34.8% |
| GPΔmuc + c2G4 | 46.7% | 24.9% | 28.4% |
| GPΔmuc + c4G7 | 47.2% | 25.6% | 27.2% |
| GPΔmuc + ADI-15946 | 27.1% | 25.0% | 47.9% |
| GPΔmuc + ADI-15878 | 20.8% | 20.2% | 59.0% |
| GPΔmuc + ADI-15742 | 21.8% | 23.3% | 49.3% |
| GPΔmuc + ADI-15750 | 41.4% | 23.6% | 35.0% |
| GPΔmuc + ADI-16061 | 32.0% | 23.1% | 44.9% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durham, N.D.; Howard, A.R.; Govindan, R.; Senjobe, F.; Fels, J.M.; Diehl, W.E.; Luban, J.; Chandran, K.; Munro, J.B. Real-Time Analysis of Individual Ebola Virus Glycoproteins Reveals Pre-Fusion, Entry-Relevant Conformational Dynamics. Viruses 2020, 12, 103. https://doi.org/10.3390/v12010103
Durham ND, Howard AR, Govindan R, Senjobe F, Fels JM, Diehl WE, Luban J, Chandran K, Munro JB. Real-Time Analysis of Individual Ebola Virus Glycoproteins Reveals Pre-Fusion, Entry-Relevant Conformational Dynamics. Viruses. 2020; 12(1):103. https://doi.org/10.3390/v12010103
Chicago/Turabian StyleDurham, Natasha D., Angela R. Howard, Ramesh Govindan, Fernando Senjobe, J. Maximilian Fels, William E. Diehl, Jeremy Luban, Kartik Chandran, and James B. Munro. 2020. "Real-Time Analysis of Individual Ebola Virus Glycoproteins Reveals Pre-Fusion, Entry-Relevant Conformational Dynamics" Viruses 12, no. 1: 103. https://doi.org/10.3390/v12010103
APA StyleDurham, N. D., Howard, A. R., Govindan, R., Senjobe, F., Fels, J. M., Diehl, W. E., Luban, J., Chandran, K., & Munro, J. B. (2020). Real-Time Analysis of Individual Ebola Virus Glycoproteins Reveals Pre-Fusion, Entry-Relevant Conformational Dynamics. Viruses, 12(1), 103. https://doi.org/10.3390/v12010103