Mouse Cytomegalovirus Differentially Exploits Cell Surface Glycosaminoglycans in a Cell Type-Dependent and MCK-2-Independent Manner


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cells
2.3. Viruses
2.4. Luciferase-Based Infectivity Assays
2.5. Heparin-Binding Assays
2.6. B18 Cell Surface Binding Assays
2.7. qPCR Analysis of Acinar Cell Markers
2.8. MCMV Cell Binding Assays
3. Results
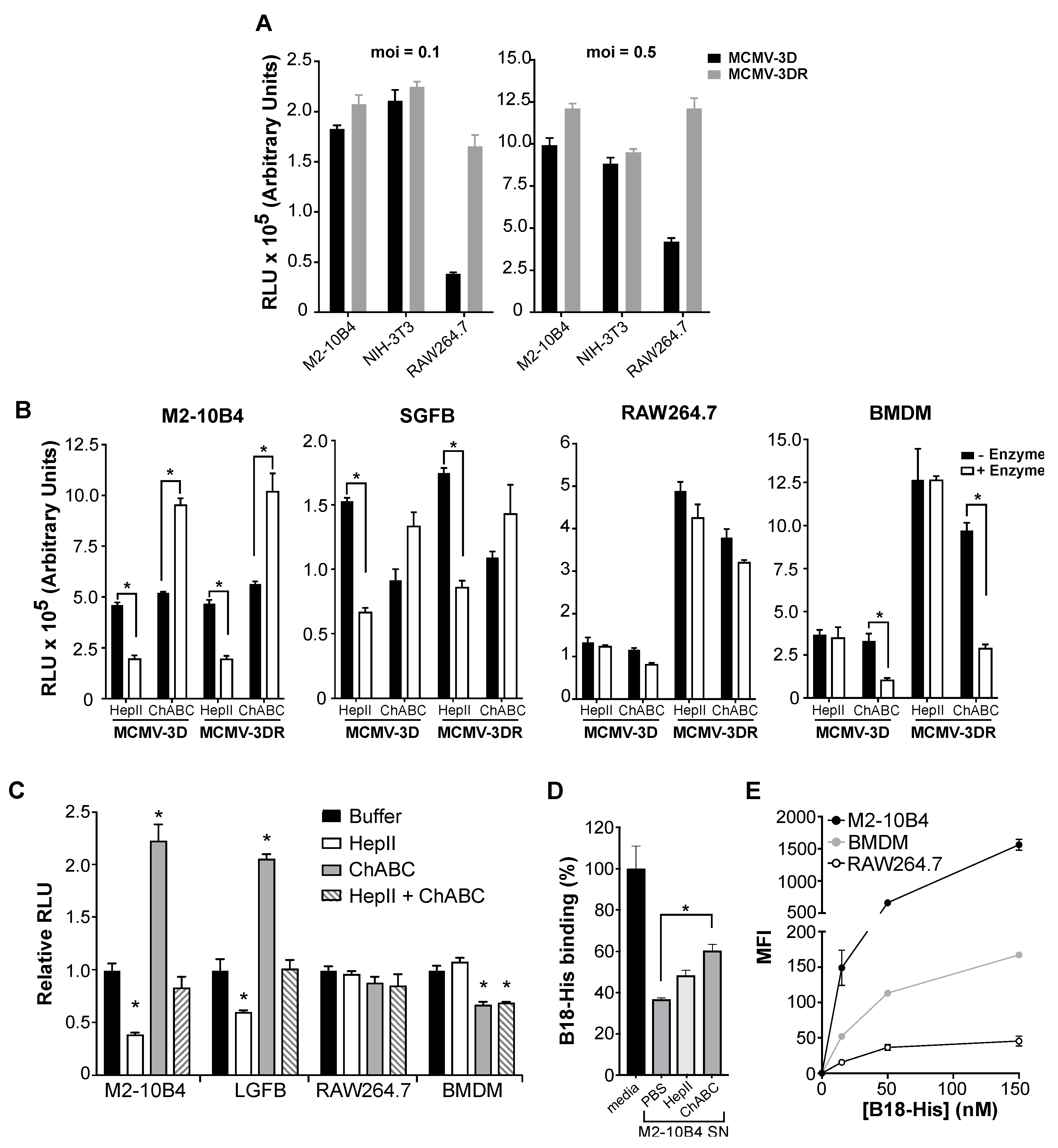
3.1. MCMV Infectivity Is Differentially Influenced by Target Cell Type-Specific GAGs in an MCK-2 Independent Manner
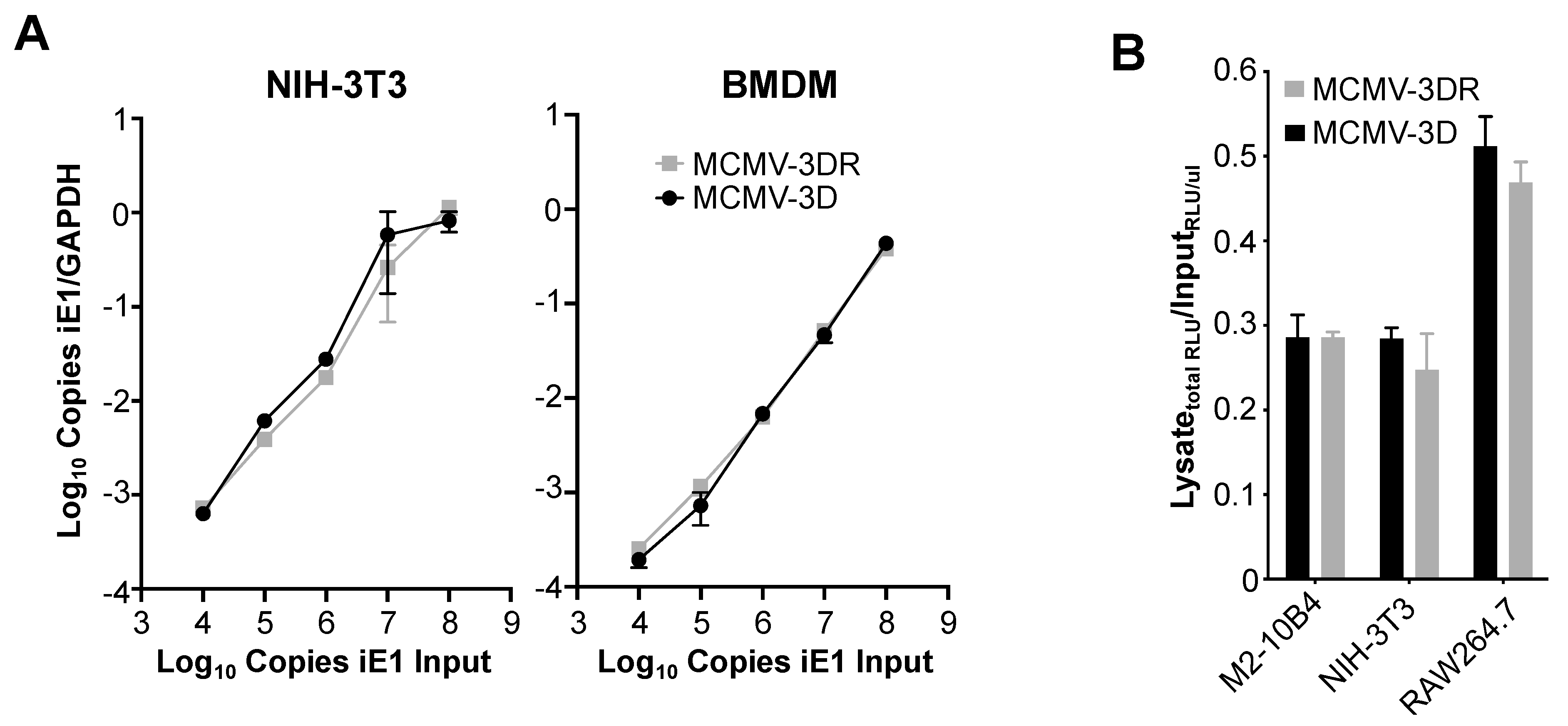
3.2. MCK-2 Is Not Required for the MCMV Infection of Mouse Salivary Gland Acinar Cells (SGACs)
3.3. MCK-2 is Dispensable for MCMV Attachment to the Surface of Fibroblasts and Macrophages
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Britt, W. Manifestations of human cytomegalovirus infection: Proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 2008, 325, 417–470. [Google Scholar] [PubMed]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvin, A.M.; Fast, P.; Myers, M.; Plotkin, S.; Rabinovich, R. National Vaccine Advisory Committee Vaccine development to prevent cytomegalovirus disease: Report from the National Vaccine Advisory Committee. Clin. Infect. Dis. 2004, 39, 233–239. [Google Scholar]
- Nguyen, C.C.; Kamil, J.P. Pathogen at the gates: Human cytomegalovirus entry and cell tropism. Viruses 2018, 10, 704. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Lanchy, J.-M.; Ryckman, B.J. Human Cytomegalovirus gH/gL/gO Promotes the Fusion Step of Entry into All Cell Types, whereas gH/gL/UL128-131 Broadens Virus Tropism through a Distinct Mechanism. J. Virol. 2015, 89, 8999–9009. [Google Scholar] [CrossRef] [Green Version]
- Huber, M.T.; Compton, T. The human cytomegalovirus UL74 gene encodes the third component of the glycoprotein H-glycoprotein L-containing envelope complex. J. Virol. 1998, 72, 8191–8197. [Google Scholar]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef] [Green Version]
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; et al. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 2004, 78, 10023–10033. [Google Scholar] [CrossRef] [Green Version]
- Gerna, G.; Percivalle, E.; Lilleri, D.; Lozza, L.; Fornara, C.; Hahn, G.; Baldanti, F.; Revello, M.G. Dendritic-cell infection by human cytomegalovirus is restricted to strains carrying functional UL131-128 genes and mediates efficient viral antigen presentation to CD8+ T cells. J. Gen. Virol. 2005, 86, 275–284. [Google Scholar] [CrossRef]
- Akter, P.; Cunningham, C.; McSharry, B.P.; Dolan, A.; Addison, C.; Dargan, D.J.; Hassan-Walker, A.F.; Emery, V.C.; Griffiths, P.D.; Wilkinson, G.W.G.; et al. Two novel spliced genes in human cytomegalovirus. J. Gen. Virol. 2003, 84, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Malkowska, M.; Kokoszynska, K.; Dymecka, M.; Rychlewski, L.; Wyrwicz, L.S. Alphaherpesvirinae and Gammaherpesvirinae glycoprotein L and CMV UL130 originate from chemokines. Virol. J. 2013, 10, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straschewski, S.; Patrone, M.; Walther, P.; Gallina, A.; Mertens, T.; Frascaroli, G. Protein pUL128 of human cytomegalovirus is necessary for monocyte infection and blocking of migration. J. Virol. 2011, 85, 5150–5158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Tao, R.; Zheng, Q.; Xu, J.; Shang, S. Recombinant HCMV UL128 expression and functional identification of PBMC-attracting activity in vitro. Arch. Virol. 2013, 158, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Rapp, M.; Messerle, M.; Bühler, B.; Tannheimer, M.; Keil, G.M.; Koszinowski, U.H. Identification of the murine cytomegalovirus glycoprotein B gene and its expression by recombinant vaccinia virus. J. Virol. 1992, 66, 4399–4406. [Google Scholar]
- Scrivano, L.; Esterlechner, J.; Mühlbach, H.; Ettischer, N.; Hagen, C.; Grünewald, K.; Mohr, C.A.; Ruzsics, Z.; Koszinowski, U.; Adler, B. The m74 gene product of murine cytomegalovirus (MCMV) is a functional homolog of human CMV gO and determines the entry pathway of MCMV. J. Virol. 2010, 84, 4469–4480. [Google Scholar] [CrossRef] [Green Version]
- Wagner, F.M.; Brizic, I.; Prager, A.; Trsan, T.; Arapovic, M.; Lemmermann, N.A.W.; Podlech, J.; Reddehase, M.J.; Lemnitzer, F.; Bosse, J.B.; et al. The viral chemokine MCK-2 of murine cytomegalovirus promotes infection as part of a gH/gL/MCK-2 complex. PLoS Pathog. 2013, 9, e1003493. [Google Scholar] [CrossRef] [Green Version]
- Pontejo, S.M.; Murphy, P.M. Chemokines encoded by herpesviruses. J. Leukoc. Biol. 2017, 102, 1199–1217. [Google Scholar] [CrossRef]
- Jordan, S.; Krause, J.; Prager, A.; Mitrovic, M.; Jonjic, S.; Koszinowski, U.H.; Adler, B. Virus progeny of murine cytomegalovirus bacterial artificial chromosome pSM3fr show reduced growth in salivary Glands due to a fixed mutation of MCK-2. J. Virol. 2011, 85, 10346–10353. [Google Scholar] [CrossRef] [Green Version]
- Fleming, P.; Davis-Poynter, N.; Degli-Esposti, M.; Densley, E.; Papadimitriou, J.; Shellam, G.; Farrell, H. The murine cytomegalovirus chemokine homolog, m131/129, is a determinant of viral pathogenicity. J. Virol. 1999, 73, 6800–6809. [Google Scholar]
- Noda, S.; Aguirre, S.A.; Bitmansour, A.; Brown, J.M.; Sparer, T.E.; Huang, J.; Mocarski, E.S. Cytomegalovirus MCK-2 controls mobilization and recruitment of myeloid progenitor cells to facilitate dissemination. Blood 2006, 107, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saederup, N.; Lin, Y.C.; Dairaghi, D.J.; Schall, T.J.; Mocarski, E.S. Cytomegalovirus-encoded beta chemokine promotes monocyte-associated viremia in the host. Proc. Natl. Acad. Sci. USA 1999, 96, 10881–10886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderholm, K.M.; Bierle, C.J.; Schleiss, M.R. Cytomegalovirus vaccines: Current status and future prospects. Drugs 2016, 76, 1625–1645. [Google Scholar] [CrossRef] [PubMed]
- Plachter, B. Prospects of a vaccine for the prevention of congenital cytomegalovirus disease. Med. Microbiol. Immunol. 2016, 205, 537–547. [Google Scholar] [CrossRef]
- Kabanova, A.; Marcandalli, J.; Zhou, T.; Bianchi, S.; Baxa, U.; Tsybovsky, Y.; Lilleri, D.; Silacci-Fregni, C.; Foglierini, M.; Fernandez-Rodriguez, B.M.; et al. Platelet-derived growth factor-α receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat. Microbiol. 2016, 1, 16082. [Google Scholar] [CrossRef]
- Wu, Y.; Prager, A.; Boos, S.; Resch, M.; Brizic, I.; Mach, M.; Wildner, S.; Scrivano, L.; Adler, B. Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-α as a key for entry. PLoS Pathog. 2017, 13, e1006281. [Google Scholar] [CrossRef]
- Martinez-Martin, N.; Marcandalli, J.; Huang, C.S.; Arthur, C.P.; Perotti, M.; Foglierini, M.; Ho, H.; Dosey, A.M.; Shriver, S.; Payandeh, J.; et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell 2018, 174, 1158–1171.e19. [Google Scholar] [CrossRef] [Green Version]
- Aquino, R.S.; Park, P.W. Glycosaminoglycans and infection. Front. Biosci. 2016, 21, 1260–1277. [Google Scholar]
- Pontejo, S.M.; Murphy, P.M. Two glycosaminoglycan-binding domains of the mouse cytomegalovirus-encoded chemokine MCK-2 are critical for oligomerization of the full-length protein. J. Biol. Chem. 2017, 292, 9613–9626. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, A.E.I.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.C.; Kosco-Vilbois, M.H. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.S.; Hsiao, J.C.; Chang, Y.S.; Chang, W. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 1998, 72, 1577–1585. [Google Scholar] [PubMed]
- WuDunn, D.; Spear, P.G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989, 63, 52–58. [Google Scholar] [PubMed]
- Patel, M.; Yanagishita, M.; Roderiquez, G.; Bou-Habib, D.C.; Oravecz, T.; Hascall, V.C.; Norcross, M.A. Cell-surface heparan sulfate proteoglycan mediates HIV-1 infection of T-cell lines. AIDS Res. Hum. Retrovir. 1993, 9, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Compton, T.; Nowlin, D.M.; Cooper, N.R. Initiation of human cytomegalovirus infection requires initial interaction with cell surface heparan sulfate. Virology 1993, 193, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Dhondt, K.P.; Châlons, M.; Mély, S.; Raoul, H.; Negre, D.; Cosset, F.-L.; Gerlier, D.; Vivès, R.R.; Horvat, B. Heparan sulfate-dependent enhancement of henipavirus infection. MBio 2015, 6, e02427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Maguire, T.; Hileman, R.E.; Fromm, J.R.; Esko, J.D.; Linhardt, R.J.; Marks, R.M. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 1997, 3, 866–871. [Google Scholar] [CrossRef]
- Kalia, M.; Chandra, V.; Rahman, S.A.; Sehgal, D.; Jameel, S. Heparan sulfate proteoglycans are required for cellular binding of the hepatitis E virus ORF2 capsid protein and for viral infection. J. Virol. 2009, 83, 12714–12724. [Google Scholar] [CrossRef] [Green Version]
- Dogra, P.; Martin, E.B.; Williams, A.; Richardson, R.L.; Foster, J.S.; Hackenback, N.; Kennel, S.J.; Sparer, T.E.; Wall, J.S. Novel heparan sulfate-binding peptides for blocking herpesvirus entry. PLoS ONE 2015, 10, e0126239. [Google Scholar] [CrossRef] [Green Version]
- Pitt, E.A.; Dogra, P.; Patel, R.S.; Williams, A.; Wall, J.S.; Sparer, T.E. The D-form of a novel heparan binding peptide decreases cytomegalovirus infection in vivo and in vitro. Antivir. Res. 2016, 135, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.W.; Hancock, T.J.; Dogra, P.; Patel, R.; Arav-Boger, R.; Williams, A.D.; Kennel, S.J.; Wall, J.S.; Sparer, T.E. Anticytomegalovirus peptides point to new insights for CMV entry mechanisms and the limitations of in vitro screenings. MSphere 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, J.; Maus, E.; Zanotti, B.; Volin, M.V.; Tandon, R.; Shukla, D.; Tiwari, V. A role for 3-O-sulfated heparan sulfate in promoting human cytomegalovirus infection in human iris cells. J. Virol. 2015, 89, 5185–5192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marim, F.M.; Silveira, T.N.; Lima, D.S.; Zamboni, D.S. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS ONE 2010, 5, e15263. [Google Scholar] [CrossRef] [PubMed]
- Bittencourt, F.M.; Wu, S.-E.; Bridges, J.P.; Miller, W.E. The M33 G protein-coupled receptor encoded by murine cytomegalovirus is dispensable for hematogenous dissemination but is required for growth within the salivary gland. J. Virol. 2014, 88, 11811–11824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquardt, A.; Halle, S.; Seckert, C.K.; Lemmermann, N.A.W.; Veres, T.Z.; Braun, A.; Maus, U.A.; Förster, R.; Reddehase, M.J.; Messerle, M.; et al. Single cell detection of latent cytomegalovirus reactivation in host tissue. J. Gen. Virol. 2011, 92, 1279–1291. [Google Scholar] [CrossRef]
- Stahl, F.R.; Keyser, K.A.; Heller, K.; Bischoff, Y.; Halle, S.; Wagner, K.; Messerle, M.; Förster, R. Mck2-dependent infection of alveolar macrophages promotes replication of MCMV in nodular inflammatory foci of the neonatal lung. Mucosal Immunol. 2015, 8, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Zurbach, K.A.; Moghbeli, T.; Snyder, C.M. Resolving the titer of murine cytomegalovirus by plaque assay using the M2-10B4 cell line and a low viscosity overlay. Virol. J. 2014, 11, 71. [Google Scholar] [CrossRef] [Green Version]
- Montanuy, I.; Alejo, A.; Alcami, A. Glycosaminoglycans mediate retention of the poxvirus type I interferon binding protein at the cell surface to locally block interferon antiviral responses. FASEB J. 2011, 25, 1960–1971. [Google Scholar] [CrossRef] [Green Version]
- Linhardt, R.J.; Turnbull, J.E.; Wang, H.M.; Loganathan, D.; Gallagher, J.T. Examination of the substrate specificity of heparin and heparan sulfate lyases. Biochemistry 1990, 29, 2611–2617. [Google Scholar] [CrossRef]
- Prabhakar, V.; Capila, I.; Bosques, C.J.; Pojasek, K.; Sasisekharan, R. Chondroitinase ABC I from Proteus vulgaris: Cloning, recombinant expression and active site identification. Biochem. J. 2005, 386, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; McCarthy, J.B.; Verfaillie, C.M. Stromal fibroblast heparan sulfate is required for cytokine-mediated ex vivo maintenance of human long-term culture-initiating cells. Blood 1996, 87, 3229–3236. [Google Scholar] [CrossRef] [Green Version]
- Kari, B.; Gehrz, R. A human cytomegalovirus glycoprotein complex designated gC-II is a major heparin-binding component of the envelope. J. Virol. 1992, 66, 1761–1764. [Google Scholar] [PubMed]
- Lemmermann, N.A.W.; Krmpotic, A.; Podlech, J.; Brizic, I.; Prager, A.; Adler, H.; Karbach, A.; Wu, Y.; Jonjic, S.; Reddehase, M.J.; et al. Non-redundant and Redundant Roles of Cytomegalovirus gH/gL Complexes in Host Organ Entry and Intra-tissue Spread. PLoS Pathog. 2015, 11, e1004640. [Google Scholar] [CrossRef] [PubMed]
- Michelacci, Y.M.; Petricevich, V.L. Glycosaminoglycan profile of peritoneal and bone marrow-derived macrophages. Changes associated with macrophage activation. Comp. Biochem. Physiol. B 1991, 100, 617–625. [Google Scholar] [CrossRef]
- Kaplan, M.; Aviram, M. Macrophage plasma membrane chondroitin sulfate proteoglycan binds oxidized low-density lipoprotein. Atherosclerosis 2000, 149, 5–17. [Google Scholar] [CrossRef]
- Edwards, I.J.; Xu, H.; Obunike, J.C.; Goldberg, I.J.; Wagner, W.D. Differentiated macrophages synthesize a heparan sulfate proteoglycan and an oversulfated chondroitin sulfate proteoglycan that bind lipoprotein lipase. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 400–409. [Google Scholar] [CrossRef]
- Borges, F.T.; Michelacci, Y.M.; Aguiar, J.A.K.; Dalboni, M.A.; Garófalo, A.S.; Schor, N. Characterization of glycosaminoglycans in tubular epithelial cells: Calcium oxalate and oxalate ions effects. Kidney Int. 2005, 68, 1630–1642. [Google Scholar] [CrossRef] [Green Version]
- Rapraeger, A.; Jalkanen, M.; Endo, E.; Koda, J.; Bernfield, M. The cell surface proteoglycan from mouse mammary epithelial cells bears chondroitin sulfate and heparan sulfate glycosaminoglycans. J. Biol. Chem. 1985, 260, 11046–11052. [Google Scholar]
- Vogel, K.G.; Kendall, V.F. Cell-surface glycosaminoglycans: Turnover in cultured human embryo fibroblasts (IMR-90). J. Cell Physiol. 1980, 103, 475–487. [Google Scholar] [CrossRef]
- Shimada, K.; Ozawa, T. Evidence that cell surface heparan sulfate is involved in the high affinity thrombin binding to cultured porcine aortic endothelial cells. J. Clin. Investig. 1985, 75, 1308–1316. [Google Scholar] [CrossRef]
- Stramm, L.E. Synthesis and secretion of glycosaminoglycans in cultured retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 1987, 28, 618–627. [Google Scholar]
- Vannucchi, S.; Chiarugi, V.P. Surface exposure of glycosaminoglycans in resting, growing and virus transformed 3T3 cells. J. Cell Physiol. 1977, 90, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Luganini, A.; Giuliani, A.; Pirri, G.; Pizzuto, L.; Landolfo, S.; Gribaudo, G. Peptide-derivatized dendrimers inhibit human cytomegalovirus infection by blocking virus binding to cell surface heparan sulfate. Antivir. Res. 2010, 85, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Farrell, H.E.; Bruce, K.; Lawler, C.; Stevenson, P.G. Murine cytomegalovirus spread depends on the infected myeloid cell type. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley-Bauer, L.P.; Roback, L.J.; Wynn, G.M.; Mocarski, E.S. Cytomegalovirus hijacks CX3CR1(hi) patrolling monocytes as immune-privileged vehicles for dissemination in mice. Cell Host Microbe 2014, 15, 351–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, J.W.; Hancock, T.J.; LaPrade, E.; Dogra, P.; Gann, E.R.; Masi, T.J.; Panchanathan, R.; Miller, W.E.; Wilhelm, S.W.; Sparer, T.E. The Human Cytomegalovirus Chemokine vCXCL-1 Modulates Normal Dissemination Kinetics of Murine Cytomegalovirus In Vivo. MBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, H.E.; Bruce, K.; Lawler, C.; Oliveira, M.; Cardin, R.; Davis-Poynter, N.; Stevenson, P.G. Murine cytomegalovirus spreads by dendritic cell recirculation. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegrowski, Y.; Milard, A.L.; Kotlarz, G.; Toulmonde, E.; Maquart, F.X.; Bernard, J. Cell surface proteoglycan expression during maturation of human monocytes-derived dendritic cells and macrophages. Clin. Exp. Immunol. 2006, 144, 485–493. [Google Scholar] [CrossRef]
- Petersen, F.; Brandt, E.; Lindahl, U.; Spillmann, D. Characterization of a neutrophil cell surface glycosaminoglycan that mediates binding of platelet factor 4. J. Biol. Chem. 1999, 274, 12376–12382. [Google Scholar] [CrossRef] [Green Version]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pontejo, S.M.; Murphy, P.M. Mouse Cytomegalovirus Differentially Exploits Cell Surface Glycosaminoglycans in a Cell Type-Dependent and MCK-2-Independent Manner. Viruses 2020, 12, 31. https://doi.org/10.3390/v12010031
Pontejo SM, Murphy PM. Mouse Cytomegalovirus Differentially Exploits Cell Surface Glycosaminoglycans in a Cell Type-Dependent and MCK-2-Independent Manner. Viruses. 2020; 12(1):31. https://doi.org/10.3390/v12010031
Chicago/Turabian StylePontejo, Sergio M, and Philip M Murphy. 2020. "Mouse Cytomegalovirus Differentially Exploits Cell Surface Glycosaminoglycans in a Cell Type-Dependent and MCK-2-Independent Manner" Viruses 12, no. 1: 31. https://doi.org/10.3390/v12010031