Block-And-Lock Strategies to Cure HIV Infection

{kind=link}
Abstract
:1. Introduction
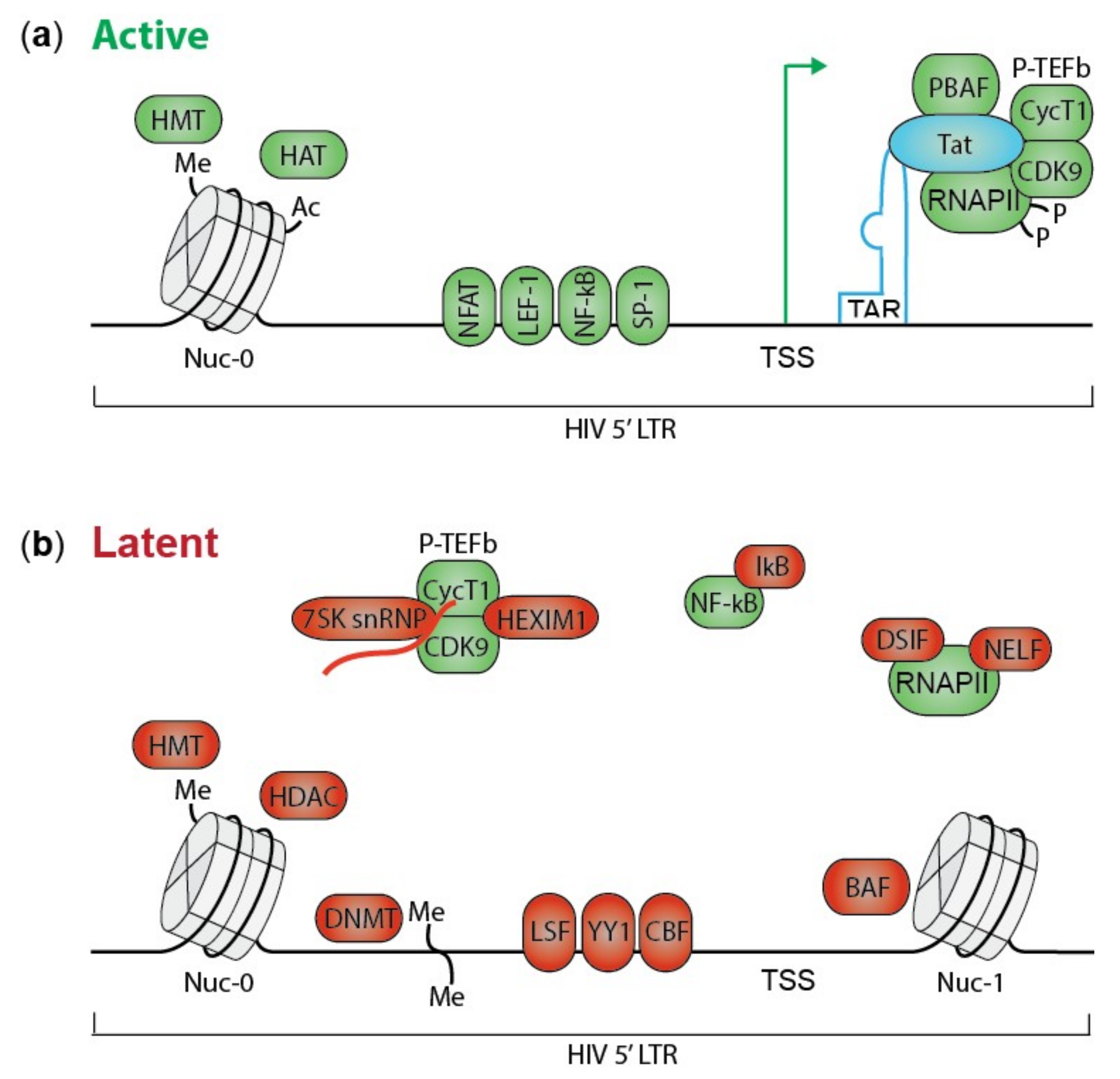
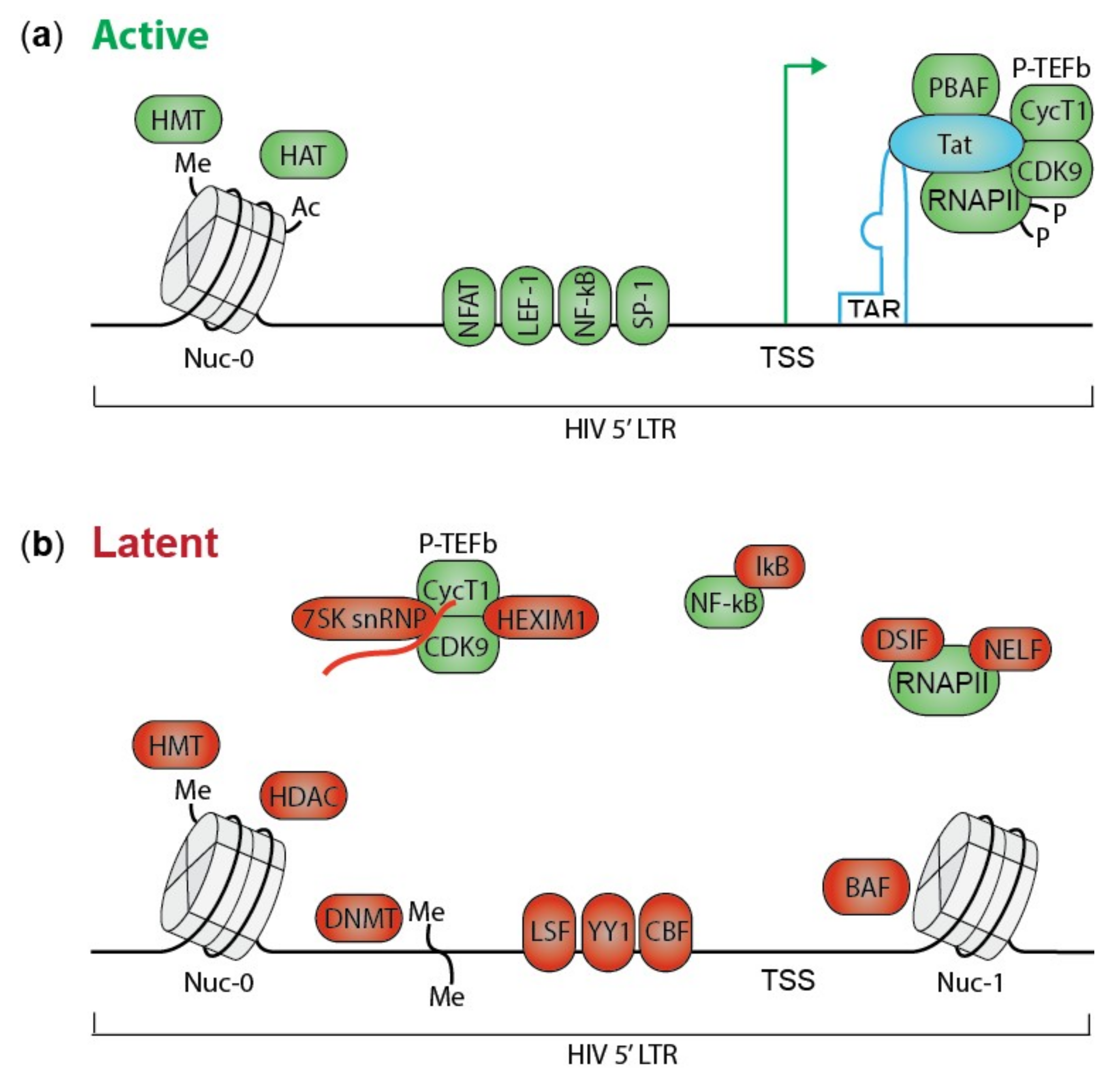
2. HIV Transcription and Silencing
3. Block-And-Lock Strategies
3.1. Tat Inhibition by Didehydro-Cortistatin A
3.2. LEDGINs
3.3. FACT Inhibition by Curaxin CBL0100
3.4. RNA-Induced Epigenetic Silencing
3.5. HSP90 Inhibitors
3.6. Jak-STAT Inhibitors
3.7. BRD4 Modulators
3.8. mTOR Inhibitors
3.9. Kinase Inhibitors
3.10. Triptolide
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a Reservoir for HIV-1 in Patients on Highly Active Antiretroviral Therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Barton, K.; Winckelmann, A.; Palmer, S. HIV-1 Reservoirs During Suppressive Therapy. Trends Microbiol. 2016, 24, 345–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewica, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512. [Google Scholar] [CrossRef] [PubMed]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müßig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-Term Control of HIV by CCR5 Delta32/Delta32 Stem-Cell Transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez Piicado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Kordelas, L.; Verheyen, J.; Esser, S. Shift of HIV Tropism in Stem-Cell Transplantation with CCR5 Delta32 Mutation. N. Engl. J. Med. 2014, 371, 880–882. [Google Scholar] [CrossRef]
- Verheyen, J.; Thielen, A.; Lübke, N.; Dirks, M.; Widera, M.; Dittmer, U.; Kordelas, L.; Daumer, M.; De Jong, D.C.M.; Wensing, A.M.J.; et al. Rapid Rebound of a Preexisting CXCR4-tropic Human Immunodeficiency Virus Variant After Allogeneic Transplantation With CCR5 Δ32 Homozygous Stem Cells. Clin. Infect. Dis. 2019, 68, 684–687. [Google Scholar] [CrossRef]
- Henrich, T.J.; Hanhauser, E.; Marty, F.M.; Sirignano, M.N.; Keating, S.; Lee, T.H.; Robles, Y.P.; Davis, B.T.; Li, J.Z.; Heisey, A.; et al. Antiretroviral-Free HIV-1 Remission and Viral Rebound After Allogeneic Stem Cell Transplantation. Ann. Intern. Med. 2014, 161, 319. [Google Scholar] [CrossRef]
- Liao, H.K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Hishida, T.; Chang, C.J.; Esteban, C.R.; et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015, 6, 6413. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas9 Can Inhibit HIV-1 Replication but NHEJ Repair Facilitates Virus Escape. Mol. Ther. 2016, 24, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Peterson, C.W.; Kiem, H.-P. Cell and Gene Therapy for HIV Cure BT—HIV-1 Latency; Silvestri, G., Lichterfeld, M., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 211–248. [Google Scholar]
- Herrera-Carrillo, E.; Gao, Z.; Berkhout, B. CRISPR therapy towards an HIV cure. Brief. Funct. Genomics 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Van Driessche, B.; Van Lint, C. Preclinical shock strategies to reactivate latent HIV-1: An update. Curr. Opin. HIV AIDS 2016, 11, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Søgaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schmeimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef] [Green Version]
- Matalon, S.; Rasmussen, T.A.; Dinarello, C.A. Histone deacetylase inhibitors for purging HIV-1 from the latent reservoir. Mol. Med. 2011, 17, 466–472. [Google Scholar] [CrossRef]
- Battivelli, E.; Dahabieh, M.S.; Abdel-Mohsen, M.; Svensson, J.P.; Da Silva, I.T.; Cohn, L.B.; Gramatica, A.; Deeks, S.; Greene, W.; Pillai, S.K.; et al. Chromatin Functional States Correlate with HIV Latency Reversal in Infected Primary CD4+ T Cells. Elife 2018.
- Chen, H.C.; Martinez, J.P.; Zorita, E.; Meyerhans, A.; Filion, G.J. Position effects influence HIV latency reversal. Nat. Struct. Mol. Biol. 2017, 24, 47. [Google Scholar] [CrossRef]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef] [Green Version]
- Zaikos, T.D.; Painter, M.M.; Kettinger, N.T.S.; Terry, V.H.; Collins, K.L. Class 1-Selective Histone Deacetylase (HDAC) Inhibitors Enhance HIV Latency Reversal while Preserving the Activity of HDAC Isoforms Necessary for Maximal HIV Gene Expression. J. Virol. 2018, 92, e02110–e02117. [Google Scholar] [CrossRef] [Green Version]
- Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; Van Driessche, B.; Corazza, F.; Catot, J.S.; Melard, A.; Vanhulle, C.; Kabeya, K.; et al. Sequential treatment with 5-aza-2’-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol. Med. 2016, 8, 117–138. [Google Scholar] [CrossRef]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-specific cytolytic T-lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the ‘kill’ into ‘shock and kill’: Strategies to eliminate latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Deeks, S.G. Immunologic Strategies for HIV-1 Remission and Eradication. Science 2014, 345, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macedo, A.B.; Novis, C.L.; Bosque, A. Targeting Cellular and Tissue HIV Reservoirs With Toll-Like Receptor Agonists. Front. Immunol. 2019, 10, 2450. [Google Scholar] [CrossRef] [Green Version]
- Wykes, M.N.; Lewin, S.R. Immune checkpoint blockade in infectious diseases. Nat. Rev. Immunol. 2018, 18, 91–104. [Google Scholar] [CrossRef]
- Stephenson, K.E. Therapeutic vaccination for HIV: Hopes and challenges. Curr. Opin. HIV AIDS 2018, 13, 408–415. [Google Scholar] [CrossRef]
- Kumar, R.; Qureshi, H.; Deshpande, S.; Bhattacharya, J. Broadly neutralizing antibodies in HIV-1 treatment and prevention. Ther. Adv. Vaccines Immunother. 2018, 6, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, K.E.; Barouch, D.H. Broadly Neutralizing Antibodies for HIV Eradication. Curr. HIV AIDS Rep. 2016, 13, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Hattori, S.I.; Matsuda, K.; Tsuchiya, K.; Gatanaga, H.; Oka, S.; Yoshimura, K.; Mitsuya, H.; Maeda, K. Combination of a Latency-Reversing Agent With a Smac Mimetic Minimizes Secondary HIV-1 Infection in vitro. Front. Microbiol. 2018, 9, 2022. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.L.; Trout, R.N.; Spector, S.A. SMAC Mimetics Induce Autophagy-Dependent Apoptosis of HIV-1-Infected Resting Memory CD4+ T Cells. Cell Host Microbe 2018, 24, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Spector, S.A. DIABLO/SMAC mimetics selectively kill HIV-1-infected resting memory CD4+ T cells: A potential role in a cure strategy for HIV-1 infection. Autophagy 2019, 15, 744–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, N.W.; Sainski-Nguyen, A.M.; Natesampillai, S.; Aboulnasr, F.; Kaufmann, S.; Badley, A.D. Maintenance of the HIV Reservoir Is Antagonized by Selective BCL2 Inhibition. J. Virol. 2017, 91, e00012–e00017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, N.W.; Sainski, A.M.; Dai, H.; Natesampillai, S.; Pang, Y.P.; Bren, G.D.; de Araujo Correia, M.C.M.; Sampath, R.; Rizza, S.A.; O’Brien, D.; et al. Prime, Shock, and Kill: Priming CD4 T Cells from HIV Patients with a BCL-2 Antagonist before HIV Reactivation Reduces HIV Reservoir Size. J. Virol. 2016, 90, 4032–4048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, A.; Kim, Y.; Rivera-Pabon, O.; Chae, S.; Kim, D.H.; Kim, B. Targeting the PI3K/Akt Cell Survival Pathway to Induce Cell Death of HIV-1 Infected Macrophages with Alkylphospholipid Compounds. PLoS ONE 2010, 5, e13121. [Google Scholar] [CrossRef]
- Darcis, G.; Van Driessche, B.; Van Lint, C. HIV Latency: Should We Shock or Lock? Trends Immunol. 2017, 38, 217–228. [Google Scholar] [CrossRef]
- Pierson, T.C.; Zhou, Y.; Kieffer, T.L.; Ruff, C.T.; Buck, C.; Siliciano, R.F. Molecular Characterization of Preintegration Latency in Human Immunodeficiency Virus Type 1 Infection. J. Virol. 2002, 76, 8518–8531. [Google Scholar] [CrossRef] [Green Version]
- Pierson, T.C.; Kieffer, T.L.; Ruff, C.T.; Buck, C.; Gange, S.J.; Siliciano, R.F. Intrinsic stability of episomal circles formed during human immunodeficiency virus type 1 replication. J. Virol. 2002, 76, 4138–4144. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.; Beddall, M.H.; Yu, D.; Iyer, S.R.; Marsh, J.W.; Wu, Y. Human macrophages support persistent transcription from unintegrated HIV-1 DNA. Virology 2008, 372, 300–312. [Google Scholar] [CrossRef] [Green Version]
- Pang, S.; Koyanagi, Y.; Miles, S.; Wiley, C.; Vinters, H.V.; Chen, I.S. High levels of unintegrated HIV-1 DNA in brain tissue of AIDS dementia patients. Nature 1990, 343, 85–89. [Google Scholar] [CrossRef]
- Roy, S.; Delling, U.; Chen, C.H.; Rosen, C.A.; Sonenberg, N. A bulge structure in HIV-1 TAR RNA is required for Tat binding and Tat-mediated trans-activation. Genes Dev. 1990, 4, 1365–1373. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Pe’ery, T.; Peng, J.; Ramanathan, Y.; Marshall, N.; Marshall, T.; Amendit, B.; Mathews, M.B.; Price, D.H. Transcription elongation factor P-TEFb is required for HIV-1 Tat transactivation in vitro. Genes Dev. 1997, 11, 2622–2632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, Y.H.; Rana, T.M. DSIF and NELF Interact with RNA Polymerase II Elongation Complex and HIV-1 Tat Stimulates P-TEFb-mediated Phosphorylation of RNA Polymerase II and DSIF during Transcription Elongation. J. Biol. Chem. 2001, 276, 12951–12958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easley, R.; Carpio, L.; Dannenberg, L.; Choi, S.; Alani, D.; Van Duyne, R.; Guendel, I.; Klase, Z.; Agbottah, E.; Kehn-Hall, K.; et al. Transcription through the HIV-1 nucleosomes: Effects of the PBAF complex in Tat activated transcription. Virology 2010, 405, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammosova, T.; Berro, R.; Jerebtsova, M.; Jackson, A.; Charles, S.; Klase, Z.; Southerland, W.; Gordeuk, V.R.; Kashanchi, F.; Nekhai, S. Phosphorylation of HIV-1 Tat by CDK2 in HIV-1 transcription. Retrovirology 2006, 3, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Zhu, Q.; Luo, K.; Zhou, Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 2001, 414, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Yik, J.H.; Chen, R.; Nishimura, R.; Jennings, J.L.; Link, A.J.; Zhou, Q. Inhibition of P-TEFb (CDK9/Cyclin T) Kinase and RNA Polymerase II Transcription by the Coordinated Actions of HEXIM1 and 7SK snRNA. Mol. Cell 2003, 12, 971–982. [Google Scholar] [CrossRef]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef] [Green Version]
- West, M.J.; Lowe, A.D.; Karn, J. Activation of human immunodeficiency virus transcription in T cells revisited: NF-kappaB p65 stimulates transcriptional elongation. J. Virol. 2001, 75, 8524–8537. [Google Scholar] [CrossRef] [Green Version]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, S.; Su, L.; Amano, M.; Timmerman, L.A.; Kaneshima, H.; Nolan, G.P. The T Cell Activation Factor NF-ATc Positively Regulates HIV-1 Replication and Gene Expression in T Cells. Immunity 1997, 6, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Taube, R.; Peterlin, M. Lost in transcription: Molecular mechanisms that control HIV latency. Viruses 2013, 5, 902–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delannoy, A.; Poirier, M.; Bell, B. Cat and Mouse: HIV Transcription in Latency, Immune Evasion and Cure/Remission Strategies. Viruses 2019, 11, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbonye, U.; Karn, J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu. Rev. Virol. 2017, 4, 261–285. [Google Scholar] [CrossRef]
- Khoury, G.; Darcis, G.; Lee, M.Y.; Bouchat, S.; Van Driessche, B.; Purcell, D.F.J.; Van Lint, C. The Molecular Biology of HIV Latency BT—HIV Vaccines and Cure: The Path Towards Finding an Effective Cure and Vaccine; Zhang, L., Lewin, S.R., Eds.; Springer: Singapore, 2018; pp. 187–212. [Google Scholar]
- Coull, J.J.; Romerio, F.; Sun, J.M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The Human Factors YY1 and LSF Repress the Human Immunodeficiency Virus Type 1 Long Terminal Repeat via Recruitment of Histone Deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, M.; Karn, J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007, 26, 4985–4995. [Google Scholar] [CrossRef]
- He, G.; Margolis, D.M. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol. Cell. Biol. 2002, 22, 2965–2973. [Google Scholar] [CrossRef] [Green Version]
- Pearson, R.; Kim, Y.K.; Hokello, J.; Lassen, K.; Friedman, J.; Tyagi, M.; Karn, J. Epigenetic Silencing of Human Immunodeficiency Virus (HIV) Transcription by Formation of Restrictive Chromatin Structures at the Viral Long Terminal Repeat Drives the Progressive Entry of HIV into Latency. J. Virol. 2008, 82, 12291–12303. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, M.; Pearson, R.J.; Karn, J. Establishment of HIV Latency in Primary CD4+ Cells Is due to Epigenetic Transcriptional Silencing and P-TEFb Restriction. J. Virol. 2010, 84, 6425–6437. [Google Scholar] [CrossRef] [Green Version]
- Krajewski, W.A.; Becker, P.B. Reconstitution of hyperacetylated, DNase I-sensitive chromatin characterized by high conformational flexibility of nucleosomal DNA. Proc. Natl. Acad. Sci. USA 1998, 95, 1540–1545. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Schneider, R.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Spatial Distribution of Di- and Tri-methyl Lysine 36 of Histone H3 at Active Genes. J. Biol. Chem. 2005, 280, 17732–17736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du Chéné, I.; Basyuk, E.; Lin, Y.L.; Triboulet, R.; Knezevich, A.; Chable-Bessia, C.; Mettling, C.; Baillat, V.; Reynes, J.; Corbeau, P.; et al. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007, 26, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Trejbalová, K.; Kovářová, D.; Blažková, J.; Machala, L.; Jilich, D.; Weber, J.; Kučerová, D.; Vencalek, O.; Hirsch, I.; Hejnar, J. Development of 5’ LTR DNA methylation of latent HIV-1 provirus in cell line models and in long-term-infected individuals. Clin. Epigenetics 2016, 8, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazkova, J.; Trejbalova, K.; Gondois-Rey, F.; Halfon, P.; Philibert, P.; Guiguen, A.; Verdin, E.; Olive, D.; Van Lint, C.; Hejnar, J.; et al. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 2009, 5, e1000554. [Google Scholar] [CrossRef] [Green Version]
- Verdin, E.R.I.C. DNase I-hypersensitive sites are associated with both long terminal repeats and with the intragenic enhancer of integrated human immunodeficiency virus type 1. J. Virol. 1991, 65, 6790–6799. [Google Scholar] [CrossRef] [Green Version]
- Yukl, S.A.; Kaiser, P.; Kim, P.; Telwatte, S.; Joshi, S.K.; Vu, M.; Lampiris, H.; Wong, J.K. HIV latency in isolated patient CD4+ T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci. Transl. Med. 2018, 10, eaap9927. [Google Scholar] [CrossRef] [Green Version]
- Telwatte, S.; Lee, S.; Somsouk, M.; Hatano, H.; Baker, C.; Kaiser, P.; Kim, P.; Chen, T.H.; Milush, J.; Hunt, P.W.; et al. Gut and blood differ in constitutive blocks to HIV transcription, suggesting tissue-specific differences in the mechanisms that govern HIV latency. PLoS Pathog. 2018, 14, e1007357. [Google Scholar] [CrossRef] [Green Version]
- Misteli, T. Beyond the Sequence: Cellular Organization of Genome Function. Cell 2007, 128, 787–800. [Google Scholar] [CrossRef] [Green Version]
- Hübner, M.R.; Spector, D.L. Chromatin dynamics. Annu. Rev. Biophys. 2010, 39, 471–489. [Google Scholar] [CrossRef]
- Lusic, M.; Siliciano, R.F. Nuclear landscape of HIV-1 infection and integration. Nat. Rev. Microbiol. 2016, 15, 69. [Google Scholar] [CrossRef]
- Debyser, Z.; Christ, F.; De Rijck, J.; Gijsbers, R. Host factors for retroviral integration site selection. Trends Biochem. Sci. 2015, 40, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavillo, F.; et al. Nuclear architecture dictates HIV-1 integration site selection. Nature 2015, 521, 227. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Echer, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287. [Google Scholar] [CrossRef] [PubMed]
- Vranckx, L.S.; Demeulemeester, J.; Saleh, S.; Boll, A.; Vansant, G.; Schrijvers, R.; Weydert, C.; Batticelli, E.; Verdin, E.; Ceresedo, A.; et al. LEDGIN-mediated Inhibition of Integrase–LEDGF/p75 Interaction Reduces Reactivation of Residual Latent HIV. EBioMedicine 2016, 8, 248–264. [Google Scholar] [CrossRef] [Green Version]
- Schrijvers, R.; Vets, S.; De Rijck, J.; Malani, N.; Bushman, F.D.; Debyser, Z.; Gijsbers, R. HRP-2 determines HIV-1 integration site selection in LEDGF/p75 depleted cells. Retrovirology 2012, 9, 84. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.R.; Perreira, J.M.; Savidis, G.; Portmann, J.M.; Aker, A.M.; Feeley, E.M.; Smith, M.C.; Brass, A.L. Direct Visualization of HIV-1 Replication Intermediates Shows that Capsid and CPSF6 Modulate HIV-1 Intra-nuclear Invasion and Integration. Cell Rep. 2015, 13, 1717–1731. [Google Scholar] [CrossRef] [Green Version]
- Rasheedi, S.; Shun, M.C.; Serrao, E.; Sowd, G.A.; Qian, J.; Hao, C.; Dasgupta, T.; Engelman, A.N.; Skoronski, J. The Cleavage and Polyadenylation Specificity Factor 6 (CPSF6) Subunit of the Capsid-recruited Pre-messenger RNA Cleavage Factor I (CFIm) Complex Mediates HIV-1 Integration into Genes. J. Biol. Chem. 2016, 291, 11809–11819. [Google Scholar] [CrossRef] [Green Version]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S.; et al. Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration. Cell Host Microbe 2018, 24, 392–404. [Google Scholar] [CrossRef] [Green Version]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef] [Green Version]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Cohn, L.B.; Silva, I.T.; Oliveira, T.Y.; Rosales, R.A.; Parrish, E.H.; Learn, G.H.; Hahn, B.H.; Czartoski, J.L.; McElarch, M.J.; Lehmann, C.; et al. HIV-1 Integration Landscape during Latent and Active Infection. Cell 2015, 160, 420–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesana, D.; de Sio, F.R.S.; Rudilosso, L.; Gallina, P.; Calabria, A.; Beretta, S.; Merelli, I.; Bruzzesi, E.; Passerini, L.; Nozza, S.; et al. HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nat. Commun. 2017, 8, 498. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Taki, T.; Chinen, Y.; Tsutsumi, Y.; Ohshiro, M.; Kobayashi, T.; Matsumoto, Y.; Kuroda, J.; Horiike, S.; Nishida, K.; et al. Identification of IGHCδ–BACH2 fusion transcripts resulting from cryptic chromosomal rearrangements of 14q32 with 6q15 in aggressive B-cell lymphoma/leukemia. Genes Chromosom. Cancer 2011, 50, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Flucke, U.; Tops, B.B.; de Saint Aubain Somerhausen, N.; Bras, J.; Creytens, D.H.; Küsters, B.; Groenen, P.J.T.A.; Verdijik, M.A.J.; Suurmeijier, A.J.H.; Mentzel, T. Presence of C11orf95–MKL2 fusion is a consistent finding in chondroid lipomas: A study of eight cases. Histopathology 2013, 62, 925–930. [Google Scholar] [CrossRef]
- Rice, A.P. The HIV-1 Tat Protein: Mechanism of Action and Target for HIV-1 Cure Strategies. Curr. Pharm. Des. 2017, 23, 4098–4102. [Google Scholar] [CrossRef] [Green Version]
- Mousseau, G.; Clementz, M.A.; Bakeman, W.N.; Nagarsheth, N.; Cameron, M.; Shi, J.; Baran, P.; Fromentin, R.; Chomont, N.; Valente, S.T. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell Host Microbe 2012, 12, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. MBio 2015, 6, e00465–e00515. [Google Scholar] [CrossRef] [Green Version]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L.; et al. In vivo suppression of HIV rebound by didehydro-Cortistatin A, a ‘block-and-lock’ strategy for HIV-1 cure. Cell Rep. 2017, 21, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Mousseau, G.; Valente, S.T. Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat. Epigenetics Chromatin 2019, 12, 23. [Google Scholar] [CrossRef]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van der Veken, N.J.; Van Remoortel, B.; De Maeyer, M.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef]
- Al-Mawsawi, L.Q.; Neamati, N. Allosteric Inhibitor Development Targeting HIV-1 Integrase. ChemMedChem 2011, 6, 228–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessl, J.J.; Jena, N.; Koh, Y.; Taskent-Sezgin, H.; Slaughter, A.; Feng, L.; de Silva, S.; Wu, L.; Le Grice, S.F.J.; Engelman, A.; et al. Multimode, Cooperative Mechanism of Action of Allosteric HIV-1 Integrase Inhibitors. J. Biol. Chem. 2012, 287, 16801–16811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsiang, M.; Jones, G.S.; Niedziela-Majka, A.; Kan, E.; Lansdon, E.B.; Huang, W.; Hung, M.; Samuel, D.; Novikov, N.; Xu, Y.; et al. New Class of HIV-1 Integrase (IN) Inhibitors with a Dual Mode of Action. J. Biol. Chem. 2012, 287, 21189–21203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenwick, C.; Bailey, M.D.; Bethell, R.; Bös, M.; Bonneau, P.; Cordingley, M.; Coulombe, R.; Duan, J.; Edwards, P.; Fader, L.D.; et al. Preclinical profile of BI 224436, a novel HIV-1 non-catalytic-site integrase inhibitor. Antimicrob. Agents Chemother. 2014, 58, 3233–3244. [Google Scholar] [CrossRef] [Green Version]
- Christ, F.; Shaw, S.; Demeulemeester, J.; Desimmie, B.A.; Marchand, A.; Butler, S.; Smets, W.; Chaltin, P.; Westby, M.; Debyser, Z.; et al. Small-Molecule Inhibitors of the LEDGF/p75 Binding Site of Integrase Block HIV Replication and Modulate Integrase Multimerization. Antimicrob. Agents Chemother. 2012, 56, 4365–4374. [Google Scholar] [CrossRef] [Green Version]
- Desimmie, B.A.; Schrijvers, R.; Demeulemeester, J.; Borrenberghs, D.; Weydert, C.; Thys, W.; Vets, S.; Van Remoortel, B.; Hofkens, J.; Bannert, N.; et al. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 2013, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Jurado, K.A.; Wang, H.; Slaughter, A.; Feng, L.; Kessl, J.J.; Koh, Y.; Wang, W.; Ballandras-Colas, A.; Patel, P.A.; Fuchs, J.R.; et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. USA 2013, 110, 8690–8695. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, M.; Yant, S.R.; Tsai, L.; O’Sullivan, C.; Bam, R.A.; Tsai, A.; Niedziela-Majka, A.; Stray, K.M.; Sakowicz, R.; Cihlar, T. Non-Catalytic Site HIV-1 Integrase Inhibitors Disrupt Core Maturation and Induce a Reverse Transcription Block in Target Cells. PLoS ONE 2013, 8, e74163. [Google Scholar] [CrossRef]
- Le Rouzic, E.; Bonnard, D.; Chasset, S.; Bruneau, J.M.; Chevreuil, F.; Le Strat, F.; Nguyen, J.; Beauvoir, R.; Amadori, C.; Brias, J.; et al. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology 2013, 10, 144. [Google Scholar] [CrossRef] [Green Version]
- Debyser, Z.; Vansant, G.; Bruggemans, A.; Janssens, J.; Christ, F. Insight in HIV Integration Site Selection Provides a Block-and-Lock Strategy for a Functional Cure of HIV Infection. Viruses 2018, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Vansant, G.; Vranckx, L.S.; Zurnic, I.; Van Looveren, D.; Van de Velde, P.; Nobles, C.; Gijsbers, R.; Christ, F.; Debyser, Z. Impact of LEDGIN treatment during virus production on residual HIV-1 transcription. Retrovirology 2019, 16, 8. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS (Joint United Nations Programme on HIV/AIDS). Global HIV & AIDS Statistics: 2019 Fact Sheet; UNAIDS: Geneva, Switzerland, 2019; pp. 1–6. [Google Scholar]
- Orphanides, G.; Wu, W.H.; Lane, W.S.; Hampsey, M.; Reinberg, D. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 1999, 400, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Orphanides, G.; LeRoy, G.; Chang, C.H.; Luse, D.S.; Reinberg, D. Reinberg, FACT, a Factor that Facilitates Transcript Elongation through Nucleosomes. Cell 1998, 92, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Gasparian, A.V.; Burkhart, C.A.; Purmal, A.A.; Brodsky, L.; Pal, M.; Saranadasa, M.; Bosykh, D.A.; Commane, M.; Guryanova, O.A.; Pal, S.; et al. Curaxins: Anticancer compounds that simultaneously suppress NF-κB and activate p53 by targeting FACT. Sci. Transl. Med. 2011, 3, 95ra74. [Google Scholar] [CrossRef]
- Jean, M.J.; Hayashi, T.; Huang, H.; Brennan, J.; Simpson, S.; Purmal, A.; Gurova, K.; Keefer, M.C.; Kobie, J.J.; Santoso, N.G.; et al. Curaxin CBL0100 Blocks HIV-1 Replication and Reactivation through Inhibition of Viral Transcriptional Elongation. Front. Microbiol. 2017, 8, 2007. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Juelich, T.; Lim, H.; Ishida, T.; Watanebe, T.; Cooper, D.A.; Rao, S.; Kelleher, A.D. Closed chromatin architecture is induced by an RNA duplex targeting the HIV-1 promoter region. J. Biol. Chem. 2008, 283, 23353–23363. [Google Scholar] [CrossRef] [Green Version]
- Ahlenstiel, C.; Mendez, C.; Lim, S.T.; Marks, K.; Turville, S.; Cooper, D.A.; Kelleher, A.D.; Suzuki, K. Novel RNA Duplex Locks HIV-1 in a Latent State via Chromatin-mediated Transcriptional Silencing. Mol. Ther. Nucleic Acids 2015, 4, e261. [Google Scholar] [CrossRef]
- Méndez, C.; Ledger, S.; Petoumenos, K.; Ahlenstiel, C.; Kelleher, A.D. RNA-induced epigenetic silencing inhibits HIV-1 reactivation from latency. Retrovirology 2018, 15, 67. [Google Scholar] [CrossRef] [Green Version]
- Ringrose, J.H.; Jeeninga, R.E.; Berkhout, B.; Speijer, D. Proteomic studies reveal coordinated changes in T-cell expression patterns upon infection with human immunodeficiency virus type 1. J. Virol. 2008, 82, 4320–4330. [Google Scholar] [CrossRef] [Green Version]
- Boukli, N.M.; Shetty, V.; Cubano, L.; Ricaurte, M.; Coelho-dos-Reis, J.; Nickens, Z.; Shah, P.; Talal, A.H.; Philip, R.; Jain, P. Unique and differential protein signatures within the mononuclear cells of HIV-1 and HCV mono-infected and co-infected patients. Clin. Proteomics 2012, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Vozzolo, L.; Loh, B.; Gane, P.J.; Tribak, M.; Zhou, L.; Anderson, I.; Nyakatura, E.; Jenner, R.G.; Selwood, D.; Fassati, A. Gyrase B inhibitor impairs HIV-1 replication by targeting Hsp90 and the capsid protein. J. Biol. Chem. 2010, 285, 39314–39328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, P.; Stoddart, C.A. Impaired infectivity of ritonavir-resistant HIV is rescued by heat shock protein 90AB1. J. Biol. Chem. 2011, 286, 24581–24592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, I.; Low, J.S.; Weston, S.; Weinberger, M.; Zhyvoloup, A.; Labokha, A.A.; Corazza, G.; Kitson, R.A.; Moody, J.C.; Marcello, A.; et al. Heat shock protein 90 controls HIV-1 reactivation from latency. Proc. Natl. Acad. Sci. USA 2014, 111, E1528–E1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Choi, M.S.; Inn, K.S.; Kim, B.J. Inhibition of HIV-1 reactivation by a telomerase-derived peptide in a HSP90-dependent manner. Sci. Rep. 2016, 6, 28896. [Google Scholar] [CrossRef] [Green Version]
- Roesch, F.; Meziane, O.; Kula, A.; Nisole, S.; Porrot, F.; Anderson, I.; Mammano, F.; Fassati, A.; Marcello, A.; Benkirane, M.; et al. Hyperthermia Stimulates HIV-1 Replication. PLOS Pathog. 2012, 8, e1002792. [Google Scholar] [CrossRef] [Green Version]
- Joshi, P.; Maidji, E.; Stoddart, C.A. Inhibition of Heat Shock Protein 90 Prevents HIV Rebound. J. Biol. Chem. 2016, 291, 10332–10346. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.J.; Yeh, K.H.; Chiu, C.F.; Chen, J.S.; Yen, C.C.; Lee, K.D.L.L.; Bai, L.Y.; Chen, M.H.; Lin, J.S.; Yang, Y.; et al. Results of Phase II trial of AUY922, a novel heat shock protein inhibitor in patients with metastatic gastrointestinal stromal tumor (GIST) and imatinib and sunitinib therapy. J. Clin. Oncol. 2016, 34, 134. [Google Scholar] [CrossRef]
- Oki, Y.; Younes, A.; Knickerbocker, J.; Samaniego, F.; Nastoupil, L.; Hagemeister, F.; Romaguera, J.; Fowler, N.; Kwak, L.; Westin, J. Experience with HSP90 inhibitor AUY922 in patients with relapsed or refractory non-Hodgkin lymphoma. Haematologica 2015, 100, e272–e274. [Google Scholar] [CrossRef] [Green Version]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 742–755. [Google Scholar] [CrossRef] [Green Version]
- Talaei, S.; Mellatyar, H.; Asadi, A.; Akbarzadeh, A.; Sheervalilou, R.; Zarghami, N. Spotlight on 17-AAG as an Hsp90 inhibitor for molecular targeted cancer treatment. Chem. Biol. Drug Des. 2019, 93, 760–786. [Google Scholar] [CrossRef]
- Gavegnano, C.; Detorio, M.; Montero, C.; Bosque, A.; Planelles, V.; Schinazi, R.F. Ruxolitinib and Tofacitinib Are Potent and Selective Inhibitors of HIV-1 Replication and Virus Reactivation in vitro. Antimicrob. Agents Chemother. 2014, 58, 1977–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavegnano, C.; Brehm, J.H.; Dupuy, F.P.; Talla, A.; Ribeiro, S.P.; Kulpa, D.A.; Cameron, C.; Santos, S.; Hurwitz, S.J.; Marconi, V.C.; et al. Novel mechanisms to inhibit HIV reservoir seeding using Jak inhibitors. PLoS Pathog. 2017, 13, e1006740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Niu, Q.; Liu, Z.; Alamer, E.; Fan, X.; Chen, H.; Endsley, J.; Gelman, B.B.; Tian, B.; Kim, J.H.; Michael, N.L.; et al. Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J. Clin. Investg. 2019, 129, 3361–3373. [Google Scholar] [CrossRef] [PubMed]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR complex controls HIV Latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacca, M. HIV Latency TORn Down. Cell Host Microbe 2016, 20, 700–702. [Google Scholar] [CrossRef] [Green Version]
- Kampmann, M.; Bassik, M.C.; Weissman, J.S. Integrated platform for genome-wide screening and construction of high-density genetic interaction maps in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, E2317–E2326. [Google Scholar] [CrossRef] [Green Version]
- Vargas, B.; Giacobbi, N.S.; Sanyal, A.; Venkatachari, N.J.; Han, F.; Gupta, P.; Sluis-Cremer, N. Inhibitors of Signaling Pathways That Block Reversal of HIV-1 Latency. Antimicrob. Agents Chemother. 2019, 63, e01744–e01818. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q. Triptolide and its expanding multiple pharmacological functions. Int. Immunopharmacol. 2011, 11, 377–383. [Google Scholar] [CrossRef]
- Wan, Z.; Chen, X. Triptolide inhibits human immunodeficiency virus type 1 replication by promoting proteasomal degradation of Tat protein. Retrovirology 2014, 11, 88. [Google Scholar] [CrossRef]
- Baxter, A.E.; O’Doherty, U.; Kaufmann, D.E. Beyond the replication-competent HIV reservoir: Transcription and translation-competent reservoirs. Retrovirology 2018, 15, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiardini, M.; Müller-Trutwin, M. HIV-associated chronic immune activation. Immunol. Rev. 2013, 254, 78–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamichi, H.; Dewar, R.L.; Adelsberger, J.W.; Rehm, C.A.; O’Doherty, U.; Paxinos, E.E.; Fauci, A.S.; Lane, H.C. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 8783–8789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Tellez, T.; Huot, N.; Ploquin, M.J.; Rascle, P.; Jacquelin, B.; Müller-Trutwin, M. Non-human primates in HIV research: Achievements, limits and alternatives. Infect. Genet. Evol. 2016, 46, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Jern, P.; Coffin, J.M. Effects of Retroviruses on Host Genome Function. Annu. Rev. Genet. 2008, 42, 709–732. [Google Scholar] [CrossRef] [Green Version]
- Mediouni, S.; Jablonski, J.; Paris, J.J.; Clementz, M.A.; Thenin-Houssier, S.; McLaughlin, J.P.; Valente, S.T. Didehydro-Cortistatin A Inhibits HIV-1 Tat Mediated Neuroinflammation and Prevents Potentiation of Cocaine Reward in Tat Transgenic Mice. Curr. HIV Res. 2015, 13, 64–79. [Google Scholar] [CrossRef] [Green Version]
- Mousseau, G.; Aneja, R.; Clementz, M.A.; Mediouni, S.; Lima, N.S.; Haregot, A.; Kessing, C.F.; Jablonski, J.A.; Thenin-Houssier, S.; Nagarsheth, N.; et al. Resistance to the tat inhibitor didehydro-cortistatin a is mediated by heightened basal HIV-1 transcription. MBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, L. Ruxolitinib in Myelofibrosis and Polycythemia Vera. J. Adv. Pract. Oncol. 2016, 7, 436–444. [Google Scholar]
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 234–243. [Google Scholar] [CrossRef]
- Hasinoff, B.B.; Patel, D. The lack of target specificity of small molecule anticancer kinase inhibitors is correlated with their ability to damage myocytes in vitro. Toxicol. Appl. Pharmacol. 2010, 249, 132–139. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, Z.; Liu, L.; Zhang, Y.; Zhang, S.; Xiao, J.; Ma, M.; Zhang, L. Triptolide induces adverse effect on reproductive parameters of female Sprague-Dawley rats. Drug Chem. Toxicol. 2011, 34, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.A.; Biggs III, W.H.; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule–kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vansant, G.; Bruggemans, A.; Janssens, J.; Debyser, Z. Block-And-Lock Strategies to Cure HIV Infection. Viruses 2020, 12, 84. https://doi.org/10.3390/v12010084
Vansant G, Bruggemans A, Janssens J, Debyser Z. Block-And-Lock Strategies to Cure HIV Infection. Viruses. 2020; 12(1):84. https://doi.org/10.3390/v12010084
Chicago/Turabian StyleVansant, Gerlinde, Anne Bruggemans, Julie Janssens, and Zeger Debyser. 2020. "Block-And-Lock Strategies to Cure HIV Infection" Viruses 12, no. 1: 84. https://doi.org/10.3390/v12010084
APA StyleVansant, G., Bruggemans, A., Janssens, J., & Debyser, Z. (2020). Block-And-Lock Strategies to Cure HIV Infection. Viruses, 12(1), 84. https://doi.org/10.3390/v12010084