HCV Genetic Diversity Can Be Used to Infer Infection Recency and Time since Infection

 , , ,
, , ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Sequencing
2.3. Diversity Score Calculation
- − All codon positions, and only the first/second/third codon positions in turn.
- − Whole open reading frame, individual genes, and 11 overlapping equal regions that the genome was split into (length = 502 or 501 amino acid codons).
2.4. Data Analysis
2.5. Ethics Approval and Consent to Participate
3. Results
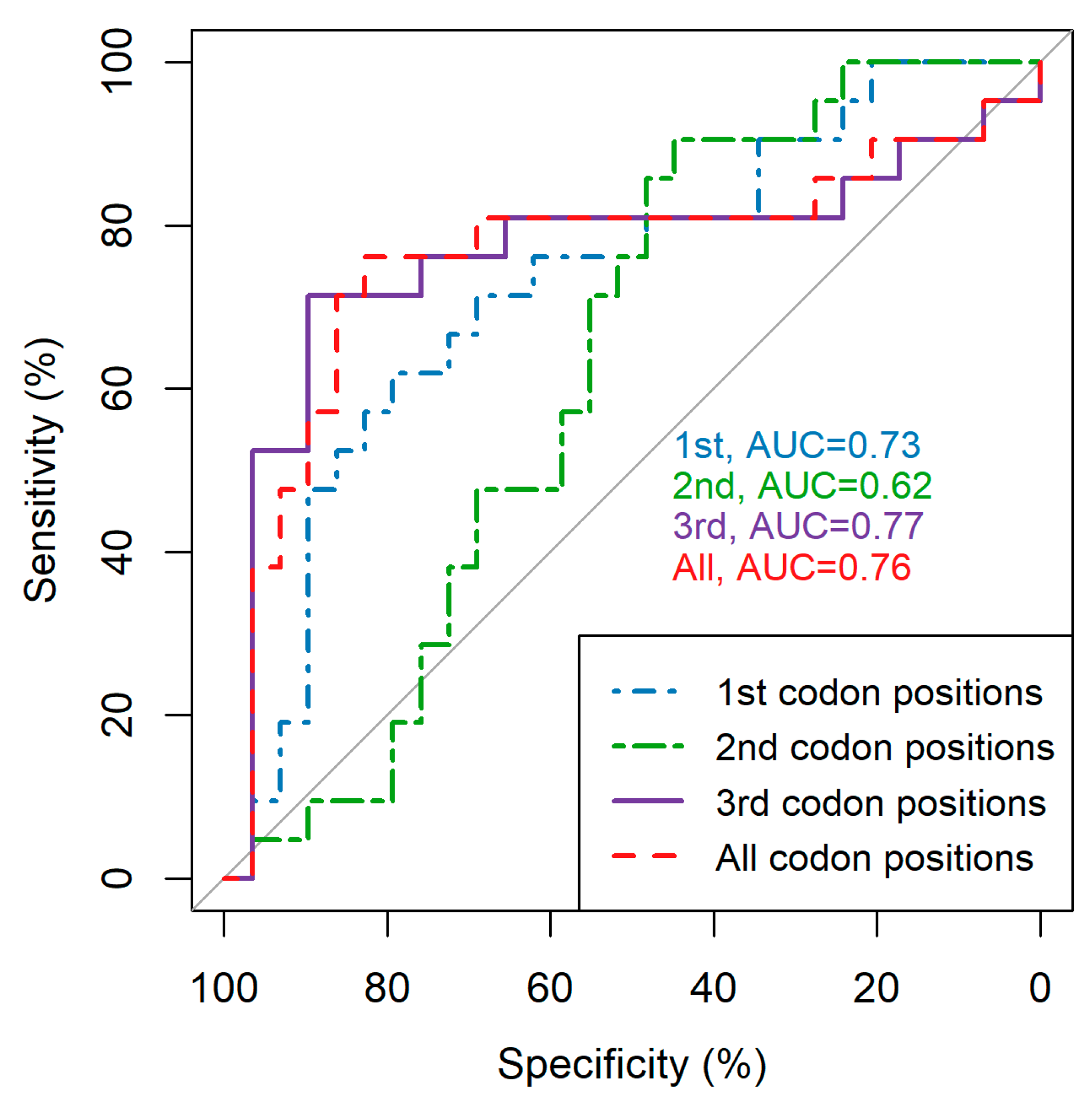
3.1. Codon Position Makes a Notable Difference in Average Pairwise Diversity Scores
3.2. Restricting Average Pairwise Diversity to Certain Regions of the Genome Improves Inference of Time since Infection and Infection Recency
3.3. Sensitivity Analysis Shows Minor Variation between Viral Subtypes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wedemeyer, H.; Duberg, A.S.; Buti, M.; Rosenberg, W.M.; Frankova, S.; Esmat, G.; Ormeci, N.; Van Vlierberghe, H.; Gschwantler, M.; Akarca, U.; et al. Strategies to manage hepatitis C virus (HCV) disease burden. J. Viral Hepat. 2014, 21, 60–89. [Google Scholar] [CrossRef]
- Armstrong, G.L.; Alter, M.J.; McQuillan, G.M.; Margolis, H.S. The past incidence of hepatitis C virus infection: Implications for the future burden of chronic liver disease in the United States. Hepatology 2000, 31, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Cotte, L.; Cua, E.; Reynes, J.; Raffi, F.; Rey, D.; Delobel, P.; Gagneux-Brunon, A.; Jacomet, C.; Palich, R.; Laroche, H.; et al. Hepatitis C virus incidence in HIV-infected and in preexposure prophylaxis (PrEP)-using men having sex with men. Liver Int. 2018, 38, 1736–1740. [Google Scholar] [CrossRef] [PubMed]
- Ghosn, J.; Deveau, C.; Goujard, C.; Garrigue, I.; Saichi, N.; Galimand, J.; Nagy, Z.; Rouzioux, C.; Meyer, L.; Chaix, M.-L.; et al. Increase in hepatitis C virus incidence in HIV-1-infected patients followed up since primary infection. Sex. Transm. Infect. 2006, 82, 458–460. [Google Scholar] [CrossRef][Green Version]
- Giuliani, M.; Caprilli, F.; Gentili, G.; Maini, A.; Lepri, A.C.; Prignano, G.; Palamara, G.; Giglio, A.; Crescimbeni, E.; Rezza, G. Incidence and Determinants of Hepatitis C Virus Infection Among Individuals at Risk of Sexually Transmitted Diseases Attending a Human Immunodeficiency Virus Type 1 Testing Program. Sex. Transm. Dis. 1997, 24, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Prestage, G.P.; Matthews, G.; Zablotska, I.; Rawstorne, P.; Kippax, S.C.; Kaldor, J.M.; Grulich, A.E. Prevalence, incidence and risk factors for hepatitis C in homosexual men: Data from two cohorts of HIV-negative and HIV-positive men in Sydney, Australia. Sex. Transm. Infect. 2009, 86, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.E.; Jarlais, D.C.D.; Arasteh, K.; McKnight, C.; Nash, D.; Perlman, D.C. Incidence and prevalence of hepatitis c virus infection among persons who inject drugs in New York City: 2006–2013. Drug Alcohol Depend. 2015, 152, 194–200. [Google Scholar] [CrossRef]
- Law, M.G.; Dore, G.J.; Bath, N.; Thompson, S.; Crofts, N.; Dolan, K.; Giles, W.; Gow, P.; Kaldor, J.; Loveday, S.; et al. Modelling hepatitis C virus incidence, prevalence and long-term sequelae in Australia, 2001. Int. J. Epidemiol. 2003, 32, 717–724. [Google Scholar] [CrossRef]
- Patrick, D.M.; Tyndall, M.W.; Cornelisse, P.G.; Li, K.; Sherlock, C.H.; Rekart, M.L.; Strathdee, S.A.; Currie, S.L.; Schechter, M.T.; O’Shaughnessy, M.V. Incidence of hepatitis C virus infection among injection drug users during an outbreak of HIV infection. Can. Med. Assoc. J. 2001, 165, 889–895. [Google Scholar]
- Prevost, T.C.; Presanis, A.M.; Taylor, A.; Goldberg, D.J.; Hutchinson, S.J.; De Angelis, D. Estimating the number of people with hepatitis C virus who have ever injected drugs and have yet to be diagnosed: an evidence synthesis approach for Scotland. Addiction 2015, 110, 1287–1300. [Google Scholar] [CrossRef]
- Rauch, A.; Martin, M.; Weber, R.; Hirschel, B.; Tarr, P.E.; Bucher, H.C.; Vernazza, P.; Bernasconi, E.; Zinkernagel, A.S.; Evison, J.; et al. Unsafe Sex and Increased Incidence of Hepatitis C Virus Infection among HIV-Infected Men Who Have Sex with Men: The Swiss HIV Cohort Study. Clin. Infect. Dis. 2005, 41, 395–402. [Google Scholar] [CrossRef]
- Van De Laar, T.J.W.; Van Der Bij, A.K.; Prins, M.; Bruisten, S.M.; Brinkman, K.; Ruys, T.A.; Van Der Meer, J.T.M.; De Vries, H.J.C.; Mulder, J.; Van Agtmael, M.; et al. Increase in HCV Incidence among Men Who Have Sex with Men in Amsterdam Most Likely Caused by Sexual Transmission. J. Infect. Dis. 2007, 196, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Berg, C.V.D.; Smit, C.; Bakker, M.; Geskus, R.B.; Berkhout, B.; Jurriaans, S.; Coutinho, R.A.; Wolthers, K.C.; Prins, M. Major decline of hepatitis C virus incidence rate over two decades in a cohort of drug users. Eur. J. Epidemiol. 2007, 22, 183–193. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kouyos, R.D.; Von Wyl, V.; Yerly, S.; Böni, J.; Rieder, P.; Joos, B.; Taffé, P.; Shah, C.; Bürgisser, P.; Klimkait, T.; et al. Ambiguous Nucleotide Calls From Population-based Sequencing of HIV-1 are a Marker for Viral Diversity and the Age of Infection. Clin. Infect. Dis. 2011, 52, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Ragonnet-Cronin, M.; Aris-Brosou, S.; Joanisse, I.; Merks, H.; Vallée, D.; Caminiti, K.; Rekart, M.; Krajden, M.; Cook, D.; Kim, J.; et al. Genetic Diversity as a Marker for Timing Infection in HIV-Infected Patients: Evaluation of a 6-Month Window and Comparison With BED. J. Infect. Dis. 2012, 206, 756–764. [Google Scholar] [CrossRef]
- Andersson, E.; Shao, W.; Bontell, I.; Cham, F.; Do, C.D.; Wondwossen, A.; Morris, L.; Hunt, G.; Sönnerborg, A.; Bertagnolio, S.; et al. Evaluation of sequence ambiguities of the HIV-1 pol gene as a method to identify recent HIV-1 infection in transmitted drug resistance surveys. Infect. Genet. Evol. 2013, 18, 125–131. [Google Scholar] [CrossRef]
- Puller, V.I.; A Neher, R.; Albert, J. Estimating time of HIV-1 infection from next-generation sequence diversity. PLoS Comput. Biol. 2017, 13, e1005775. [Google Scholar] [CrossRef]
- Carlisle, L.A.; Turk, T.; Kusejko, K.; Leemann, C.; Schenkel, C.D.; Bachmann, N.; Posada, S.; Beerenwinkel, N.; Anagnostopoulos, A.; Bucher, H.C.; et al. Viral Diversity Based on Next-Generation Sequencing of HIV-1 Provides Precise Estimates of Infection Recency and Time Since Infection. J. Infect. Dis. 2019, 220, 254–265. [Google Scholar] [CrossRef]
- Braun, D.L.; Hampel, B.; Kouyos, R.; Nguyen, H.; Shah, C.; Flepp, M.; Stöckle, M.; Conen, A.; Béguelin, C.; Künzler-Heule, P.; et al. High Cure Rates With Grazoprevir-Elbasvir With or Without Ribavirin Guided by Genotypic Resistance Testing Among Human Immunodeficiency Virus/Hepatitis C Virus-coinfected Men Who Have Sex With Men. Clin. Infect. Dis. 2018, 68, 569–576. [Google Scholar] [CrossRef]
- Huber, M.; Metzner, K.J.; Geissberger, F.D.; Shah, C.; Leemann, C.; Klimkait, T.; Böni, J.; Trkola, A.; Zagordi, O. MinVar: A rapid and versatile tool for HIV-1 drug resistance genotyping by deep sequencing. J. Virol. Methods 2017, 240, 7–13. [Google Scholar] [CrossRef]
- Schirmer, M.; Ijaz, U.Z.; D’Amore, R.; Hall, N.; Sloan, W.T.; Quince, C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015, 43, e37. [Google Scholar] [CrossRef]
- A Quail, M.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion torrent, pacific biosciences and illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 1 December 2018).
- Dowle, M.; Srinivasan, A. Data.Table: Extension of ‘Data.Frame’. 2017. Available online: https://CRAN.R-project.org/package=data.table (accessed on 1 December 2018).
- Robin, X.A.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Muller, M.J. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Garbuszus, J.M.; Jeworutzki, S. Readstata13: Import “Stata” Data Files. 2017. Available online: https://CRAN.R-project.org/package=readstata13 (accessed on 1 December 2018).
- Neuwirth, E. RColorBrewer: ColorBrewer Palettes. 2014. Available online: https://CRAN.R-project.org/package=RColorBrewer (accessed on 1 December 2018).
- Gaudieri, S.; Rauch, A.; Pfafferott, K.; Barnes, E.; Cheng, W.; McCaughan, G.; Shackel, N.; Jeffrey, G.P.; Mollison, L.; Baker, R.; et al. Hepatitis C virus drug resistance and immune-driven adaptations: Relevance to new antiviral therapy. Hepatology 2008, 49, 1069–1082. [Google Scholar] [CrossRef]
- Stewart, H.; Bingham, R.; White, S.J.; Dykeman, E.C.; Zothner, C.; Tuplin, A.K.; Stockley, P.G.; Twarock, R.; Harris, M. Identification of novel RNA secondary structures within the hepatitis C virus genome reveals a cooperative involvement in genome packaging. Sci. Rep. 2016, 6, 22952. [Google Scholar] [CrossRef]
- Fricke, M.; Gerst, R.; Ibrahim, B.; Niepmann, M.; Marz, M. Global importance of RNA secondary structures in protein-coding sequences. Bioinformatics 2019, 35, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Mastroianni, C.M.; Lichtner, M.; Mascia, C.; Zuccalà, P.; Vullo, V. Molecular Mechanisms of Liver Fibrosis in HIV/HCV Coinfection. Int. J. Mol. Sci. 2014, 15, 9184–9208. [Google Scholar] [CrossRef]
- Hernandez, M.D.; Sherman, K.E. HIV/HCV coinfection natural history and disease progression, a review of the most recent literature. Curr. Opin. HIV AIDS 2011, 6, 478. [Google Scholar] [CrossRef] [PubMed]
- Benhamou, Y.; Bochet, M.; Di Martino, V.; Charlotte, F.; Azria, F.; Coutellier, A.; Vidaud, M.; Opolon, P.; Katlama, C.; Poynard, T.; et al. Liver fibrosis progression in human immunodeficiency virus and hepatitis C virus coinfected patients. Hepatology 1999, 30, 1054–1058. [Google Scholar] [CrossRef]
- Briat, A.; Dulioust, E.; Galimand, J.; Fontaine, H.; Chaix, M.-L.; Letur-Könirsch, H.; Pol, S.; Jouannet, P.; Rouzioux, C.; Leruez-Ville, M. Hepatitis C virus in the semen of men coinfected with HIV-1: Prevalence and origin. Aids 2005, 19, 1827–1835. [Google Scholar] [CrossRef]
- Hsieh, M.-H.; Tsai, J.-J.; Hsieh, M.-Y.; Huang, C.-F.; Yeh, M.-L.; Yang, J.-F.; Chang, K.; Lin, W.-R.; Lin, C.-Y.; Chen, T.-C.; et al. Hepatitis C Virus Infection among Injection Drug Users with and without Human Immunodeficiency Virus Co-Infection. PLoS ONE 2014, 9, e94791. [Google Scholar] [CrossRef]
- Hagan, H.; Jordan, A.E.; Neurer, J.; Cleland, C.M. Incidence of sexually transmitted hepatitis C virus infection in HIV-positive men who have sex with men: A systematic review and meta-analysis. AIDS 2015, 29, 2335–2345. [Google Scholar] [CrossRef] [PubMed]
- Bottieau, E.; Apers, L.; Van Esbroeck, M.; Vandenbruaene, M.; Florence, E. Hepatitis C virus infection in HIV-infected men who have sex with men: Sustained rising incidence in Antwerp, Belgium, 2001–2009. Eurosurveillance 2010, 15, 19673. [Google Scholar] [PubMed]
- Sanchez, C.; Plaza, Z.; Vispo, E.; De Mendoza, C.; Barreiro, P.; Fernández-Montero, J.V.; Labarga, P.; Poveda, E.; Soriano, V. Scaling up epidemics of acute hepatitis C and syphilis in HIV-infected men who have sex with men in Spain. Liver Int. 2013, 33, 1357–1362. [Google Scholar] [CrossRef]
- Van De Laar, T.J.W.; Matthews, G.V.; Prins, M.; Danta, M. Acute hepatitis C in HIV-infected men who have sex with men: An emerging sexually transmitted infection. Aids 2010, 24, 1799–1812. [Google Scholar] [CrossRef]
- Yaphe, S.; Bozinoff, N.; Kyle, R.; Shivkumar, S.; Pai, N.P.; Klein, M. Incidence of acute hepatitis C virus infection among men who have sex with men with and without HIV infection: A systematic review. Sex. Transm. Infect. 2012, 88, 558–564. [Google Scholar] [CrossRef]
- Wandeler, G.; Gsponer, T.; Bregenzer, A.; Günthard, H.F.; Clerc, O.; Calmy, A.; Stöckle, M.; Bernasconi, E.; Furrer, H.; Rauch, A. Hepatitis C Virus Infections in the Swiss HIV Cohort Study: A Rapidly Evolving Epidemic. Clin. Infect. Dis. 2012, 55, 1408–1416. [Google Scholar] [CrossRef][Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Number | 72 | |
|---|---|---|
| Gender, n (%) | Female | 2 (3) |
| Male | 70 (97) | |
| Age when sample taken (years), median (IQR) | 45 (39, 52) | |
| Ethnicity, n (%) | Asian | 2 (3) |
| Black | 2 (3) | |
| Hispanic | 4 (4) | |
| White | 64 (89) | |
| HIV transmission group, n (%) | HET | 3 (4) |
| MSM | 67 (93) | |
| IDU | 1 (1) | |
| Unclear/unknown | 1 (1) | |
| Recorded history of intravenous drug use ever, n (%) | Yes | 9 (13) |
| No | 63 (88) | |
| HCV Viral subtype, n (%) | 1A | 50 (69) |
| 1B | 5 (7) | |
| 2C | 1 (1) | |
| 3A | 2 (3) | |
| 4D | 14 (19) | |
| Time since HCV infection (years), median (IQR) | 0.82 (0.47, 2.5) | |
| Clearly recent or chronic a, n (%) | True | 50 (69) |
| False | 22 (31) | |
| Full coverage of gene at all codon positions, n (%) | C | 69 (96) |
| E1 | 69 (96) | |
| E2 | 70 (97) | |
| p7 | 70 (97) | |
| NS2 | 70 (97) | |
| NS3 | 71 (99) | |
| NS4A | 71 (99) | |
| NS4B | 70 (97) | |
| NS5A | 69 (96) | |
| NS5B | 65 (90) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carlisle, L.A.; Turk, T.; Metzner, K.J.; Mbunkah, H.A.; Shah, C.; Böni, J.; Huber, M.; Braun, D.L.; Fehr, J.; Salazar-Vizcaya, L.; et al. HCV Genetic Diversity Can Be Used to Infer Infection Recency and Time since Infection. Viruses 2020, 12, 1241. https://doi.org/10.3390/v12111241
Carlisle LA, Turk T, Metzner KJ, Mbunkah HA, Shah C, Böni J, Huber M, Braun DL, Fehr J, Salazar-Vizcaya L, et al. HCV Genetic Diversity Can Be Used to Infer Infection Recency and Time since Infection. Viruses. 2020; 12(11):1241. https://doi.org/10.3390/v12111241
Chicago/Turabian StyleCarlisle, Louisa A., Teja Turk, Karin J. Metzner, Herbert A. Mbunkah, Cyril Shah, Jürg Böni, Michael Huber, Dominique L. Braun, Jan Fehr, Luisa Salazar-Vizcaya, and et al. 2020. "HCV Genetic Diversity Can Be Used to Infer Infection Recency and Time since Infection" Viruses 12, no. 11: 1241. https://doi.org/10.3390/v12111241
APA StyleCarlisle, L. A., Turk, T., Metzner, K. J., Mbunkah, H. A., Shah, C., Böni, J., Huber, M., Braun, D. L., Fehr, J., Salazar-Vizcaya, L., Rauch, A., Yerly, S., Nguyen, A., Cavassini, M., Stoeckle, M., Vernazza, P., Bernasconi, E., Günthard, H. F., & Kouyos, R. D. (2020). HCV Genetic Diversity Can Be Used to Infer Infection Recency and Time since Infection. Viruses, 12(11), 1241. https://doi.org/10.3390/v12111241