Phylogenetic Investigation of Norovirus Transmission between Humans and Animals

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phylogenetic Analyses
2.2. BEAST Analyses
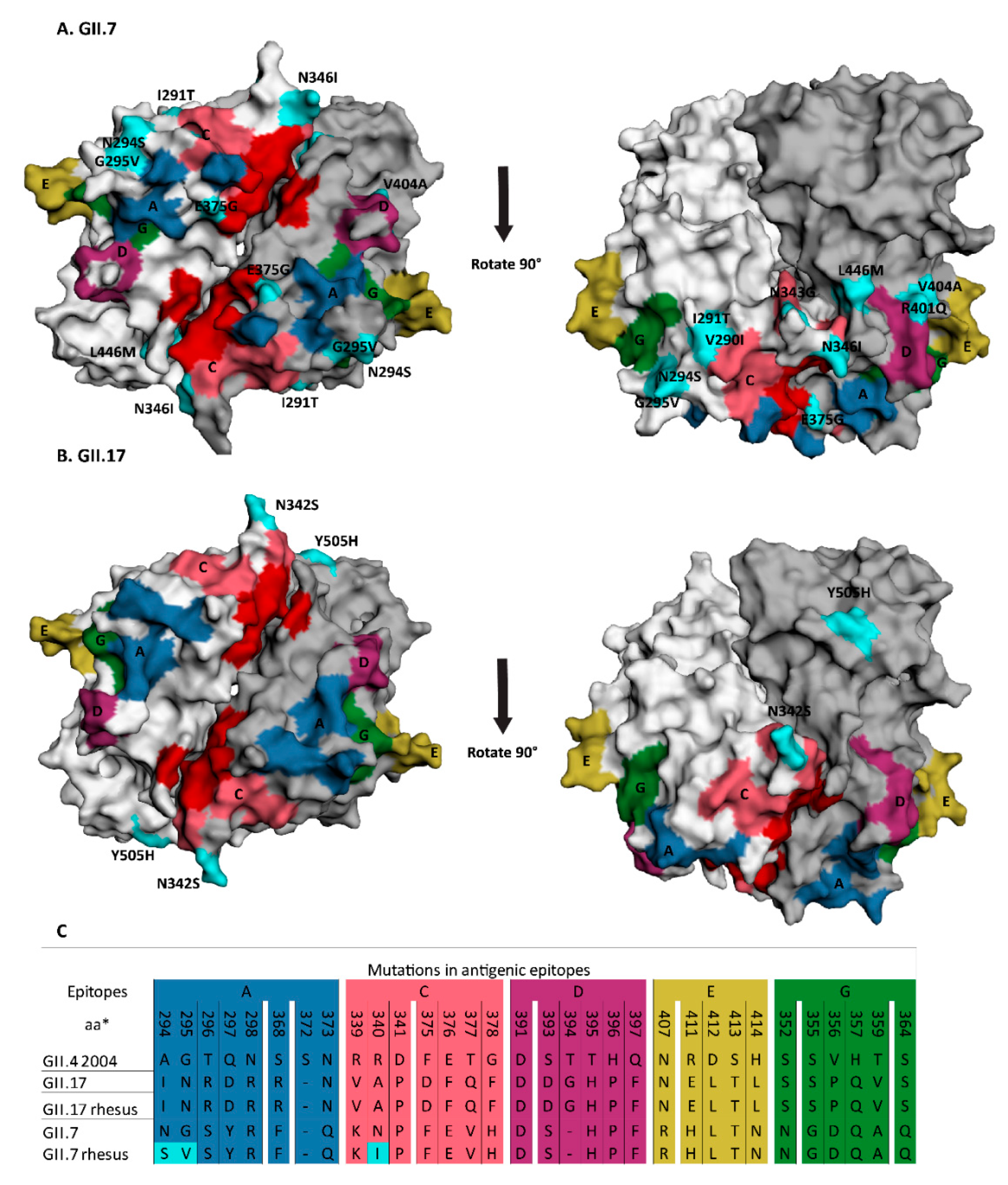
2.3. Mapping of Amino Acid Changes onto 3D Structure
3. Results
3.1. Norovirus Strains, Closely Related to Human Noroviruses, Are Found in Animals
3.2. Molecular Clock Phylogeny of GII.7 and GII.17 Genotypes
3.3. Animal GII.7 and GII.17 Sequences Contain Amino Acid Changes That Are Located either in or Adjacent to Antigenic Epitopes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Vinje, J. Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 2015, 53, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, L.G.; Goodfellow, I.G. Norovirus gene expression and replication. J. Gen. Virol. 2014, 95, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Jiang, X. Norovirus and its histo-blood group antigen receptors: An answer to a historical puzzle. Trends Microbiol. 2005, 13, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Bok, K.; Abente, E.J.; Realpe-Quintero, M.; Mitra, T.; Sosnovtsev, S.V.; Kapikian, A.Z.; Green, K.Y. Evolutionary dynamics of gii.4 noroviruses over a 34-year period. J. Virol. 2009, 83, 11890–11901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.-W.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, jgv001318. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.N.H.; Trainor, E.; Nakagomi, T.; Cunliffe, N.A.; Nakagomi, O. Molecular epidemiology of noroviruses associated with acute sporadic gastroenteritis in children: Global distribution of genogroups, genotypes and gii.4 variants. J. Clin. Virol. 2013, 56, 185–193. [Google Scholar]
- van Beek, J.; de Graaf, M.; Al-Hello, H.; Allen, D.J.; Ambert-Balay, K.; Botteldoorn, N.; Brytting, M.; Buesa, J.; Cabrerizo, M.; Chan, M.; et al. Molecular surveillance of norovirus, 2005–2016: An epidemiological analysis of data collected from the noronet network. Lancet Infect. Dis. 2018, 18, 545–553. [Google Scholar] [CrossRef]
- Cannon, J.L.; Barclay, L.; Collins, N.R.; Wikswo, M.E.; Castro, C.J.; Magaña, L.C.; Gregoricus, N.; Marine, R.L.; Chhabra, P.; Vinjé, J. Genetic and epidemiologic trends of norovirus outbreaks in the united states from 2013 to 2016 demonstrated emergence of novel gii. 4 recombinant viruses. J. Clin. Microbiol. 2017, 55, 2208–2221. [Google Scholar] [CrossRef] [Green Version]
- Villabruna, N.; Koopmans, M.; De Graaf, M. Animals as reservoir for human norovirus. Viruses 2019, 11, 478. [Google Scholar] [CrossRef] [Green Version]
- Todd, K.V.; Tripp, R.A. Human norovirus: Experimental models of infection. Viruses 2019, 11, 151. [Google Scholar] [CrossRef] [Green Version]
- Kroneman, A.; Vega, E.; Vennema, H.; Vinje, J.; White, P.A.; Hansman, G.; Green, K.; Martella, V.; Katayama, K.; Koopmans, M. Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 2013, 158, 2059–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of phyml 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, V.; Longueville, J.E.; Gascuel, O. Sms: Smart model selection in phyml. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using tempest (formerly path-o-gen). Virus. Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using beast 1.10. Virus. Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, C.R.; Islam, S.A.; Sternberg, M.J.E. Ezmol: A web server wizard for the rapid visualization and image production of protein and nucleic acid structures. J. Mol. Biol. 2018, 430, 2244–2248. [Google Scholar] [CrossRef]
- Tan, M.; Jiang, X. Norovirus gastroenteritis, carbohydrate receptors, and animal models. PLoS Pathog. 2010, 6, 3–4. [Google Scholar] [CrossRef]
- Siebenga, J.J.; Vennema, H.; Zheng, D.P.; Vinjé, J.; Lee, B.E.; Pang, X.L.; Ho, E.C.M.; Lim, W.; Choudekar, A.; Broor, S.; et al. Norovirus illness is a global problem: Emergence and spread of norovirus gii. 4 variants, 2001–2007. J. Infect. Dis. 2009, 200, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Tohma, K.; Lepore, C.J.; Gao, Y.; Ford-Siltz, L.A.; Parra, G.I. Population genomics of gii.4 noroviruses reveal complex diversification and new antigenic sites involved in the emergence of pandemic strains. MBio 2019, 10, e02202–e02219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summa, M.; Henttonen, H.; Maunula, L. Human noroviruses in the faeces of wild birds and rodents-new potential transmission routes. Zoonoses Public Health 2018, 65, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Chao, D.Y.; Wei, J.Y.; Chang, W.F.; Wang, J.; Wang, L.C. Detection of multiple genotypes of calicivirus infection in asymptomatic swine in taiwan. Zoonoses Public Health 2012, 59, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Saga, Y.; Iwai, M.; Obara, M.; Horimoto, E.; Hasegawa, S.; Kurata, T.; Okumura, H.; Nagoshi, M.; Takizawa, T. Frequent detection of noroviruses and sapoviruses in swine and high genetic diversity of porcine sapovirus in japan during fiscal year 2008. J. Clin. Microbiol. 2010, 48, 1215–1222. [Google Scholar] [CrossRef] [Green Version]
- Summa, M.; von Bonsdorff, C.H.; Maunula, L. Pet dogs-a transmission route for human noroviruses? J. Clin. Virol. 2012, 53, 244–247. [Google Scholar] [CrossRef]
- Charoenkul, K.; Nasamran, C.; Janetanakit, T.; Tangwangvivat, R.; Bunpapong, N.; Boonyapisitsopa, S.; Suwannakarn, K.; Theamboonler, A.; Chuchaona, W.; Poovorawan, Y. Human norovirus infection in dogs, thailand. Emerg. Infect. Dis. 2020, 26, 350–353. [Google Scholar] [CrossRef]
- Mattison, K.; Shukla, A.; Cook, A.; Pollari, F.; Friendship, R.; Kelton, D.; Bidawid, S.; Farber, J.M. Human noroviruses in swine and cattle. Emerg. Infect. Dis. 2007, 13, 1184–1188. [Google Scholar] [CrossRef]
- Duarte, M.A.; Silva, F.; João, M.; Brito, C.R.; Teixeira, D.S.; Melo, F.L.; Ribeiro, B.M.; Nagata, T.; Campos, F.S. Faecal virome analysis of wild animals from brazil. Viruses 2019, 11, 803. [Google Scholar] [CrossRef] [Green Version]
- Farkas, T. Natural norovirus infections in rhesus macaques. Emerg. Infect. Dis. 2016, 22, 1272–1274. [Google Scholar] [CrossRef]
- Wolf, S.; Reetz, J.; Johne, R.; Heiberg, A.C.; Petri, S.; Kanig, H.; Ulrich, R.G. The simultaneous occurrence of human norovirus and hepatitis e virus in a norway rat (rattus norvegicus). Arch. Virol. 2013, 158, 1575–1578. [Google Scholar] [CrossRef]
- Sisay, Z.; Djikeng, A.; Berhe, N.; Belay, G.; Abegaz, W.E.; Wang, Q.H.; Saif, L.J. First detection and molecular characterization of sapoviruses and noroviruses with zoonotic potential in swine in ethiopia. Arch. Virol. 2016, 161, 2739–2747. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Tao, Y.; Li, C.; Li, X.; Liu, J.; He, Z.; Xia, M.; Jiang, X.; Tan, M.; Liu, H. Complete genome sequence of a gii.17 norovirus isolated from a rhesus monkey in china. Genome Announc. 2016, 4, e00904–e00916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkas, T.; Cross, R.W.; Hargitt, I.E.; Lerche, N.W.; Morrow, A.L.; Sestak, K. Genetic diversity and histo-blood group antigen interactions of rhesus enteric caliciviruses. J. Virol. 2010, 84, 8617–8625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Graaf, M.; van Beek, J.; Vennema, H.; Podkolzin, A.T.; Hewitt, J.; Bucardo, F.; Templeton, K.; Nordgren, J.; Reuter, G.; Lynch, M.; et al. Emergence of a novel gii.17 norovirus—End of the gii.4 era? Euro. Surveill. 2015, 20, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zhou, N.; Lin, X.; Wang, S.; Tao, Z.; Xiong, P.; Wang, H.; Liu, Y.; Song, Y.; Xu, A. Molecular epidemiology of gi and gii noroviruses in sewage: 1-year surveillance in eastern china. J. Appl. Microbiol. 2016, 121, 1172–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mans, J.; Netshikweta, R.; Magwalivha, M.; Van Zyl, W.B.; Taylor, M.B. Diverse norovirus genotypes identified in sewage-polluted river water in south africa. Epidemiol. Infect. 2013, 141, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindesmith, L.C.; Kocher, J.F.; Donaldson, E.F.; Debbink, K.; Mallory, M.L.; Swann, E.W.; Brewer-Jensen, P.D.; Baric, R.S. Emergence of novel human norovirus gii.17 strains correlates with changes in blockade antibody epitopes. J. Infect. Dis. 2017, 216, 1227–1234. [Google Scholar] [CrossRef]
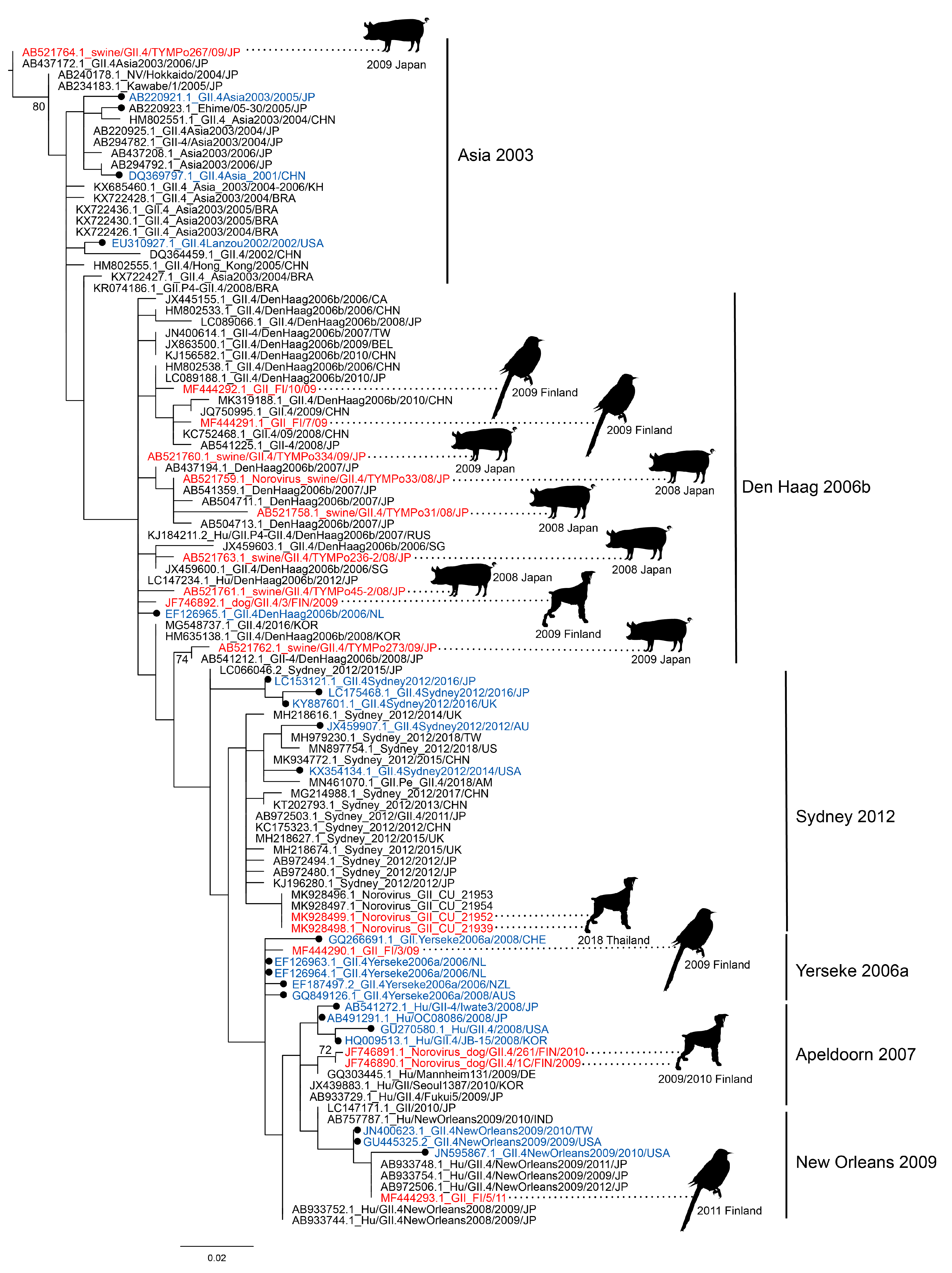
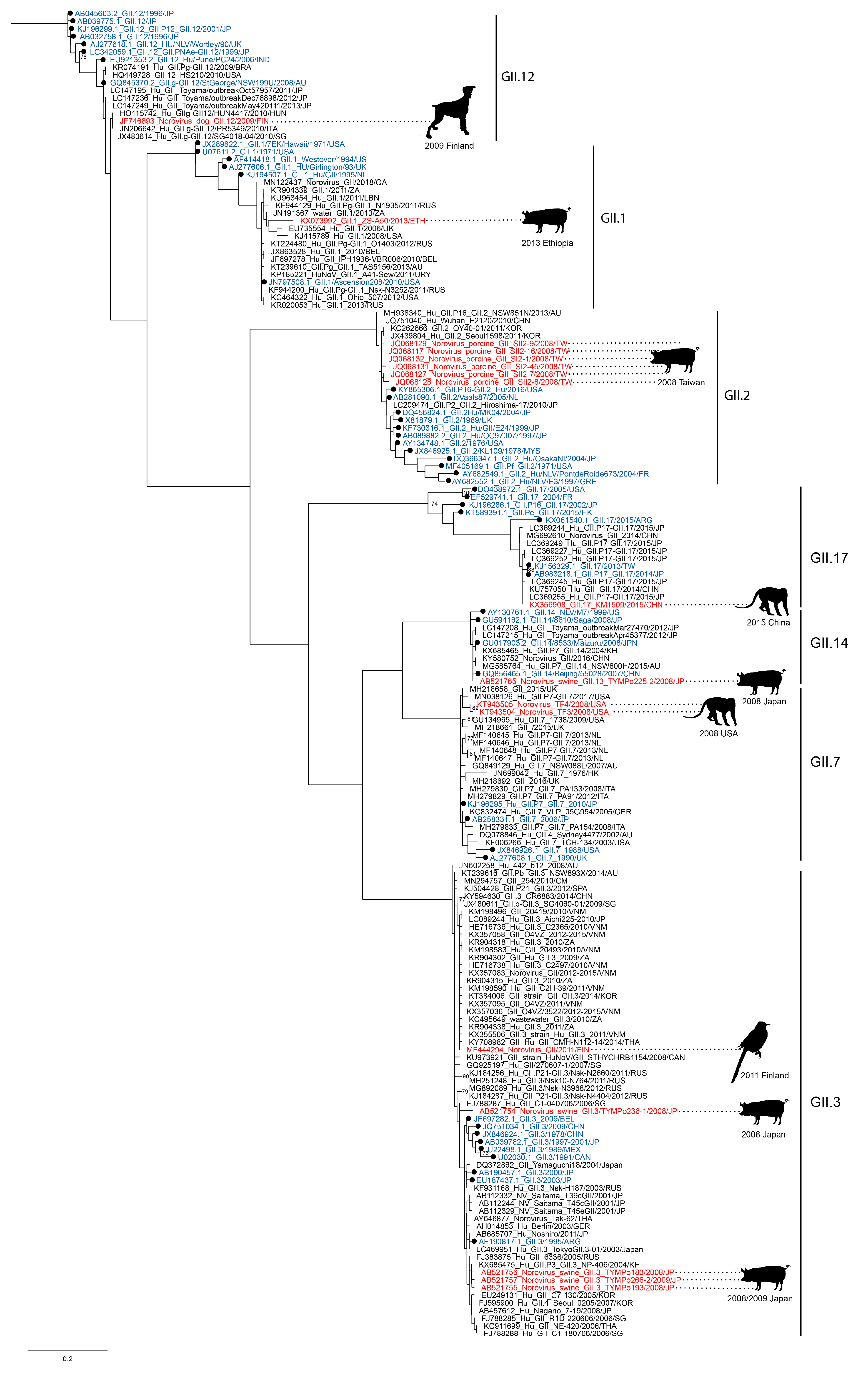
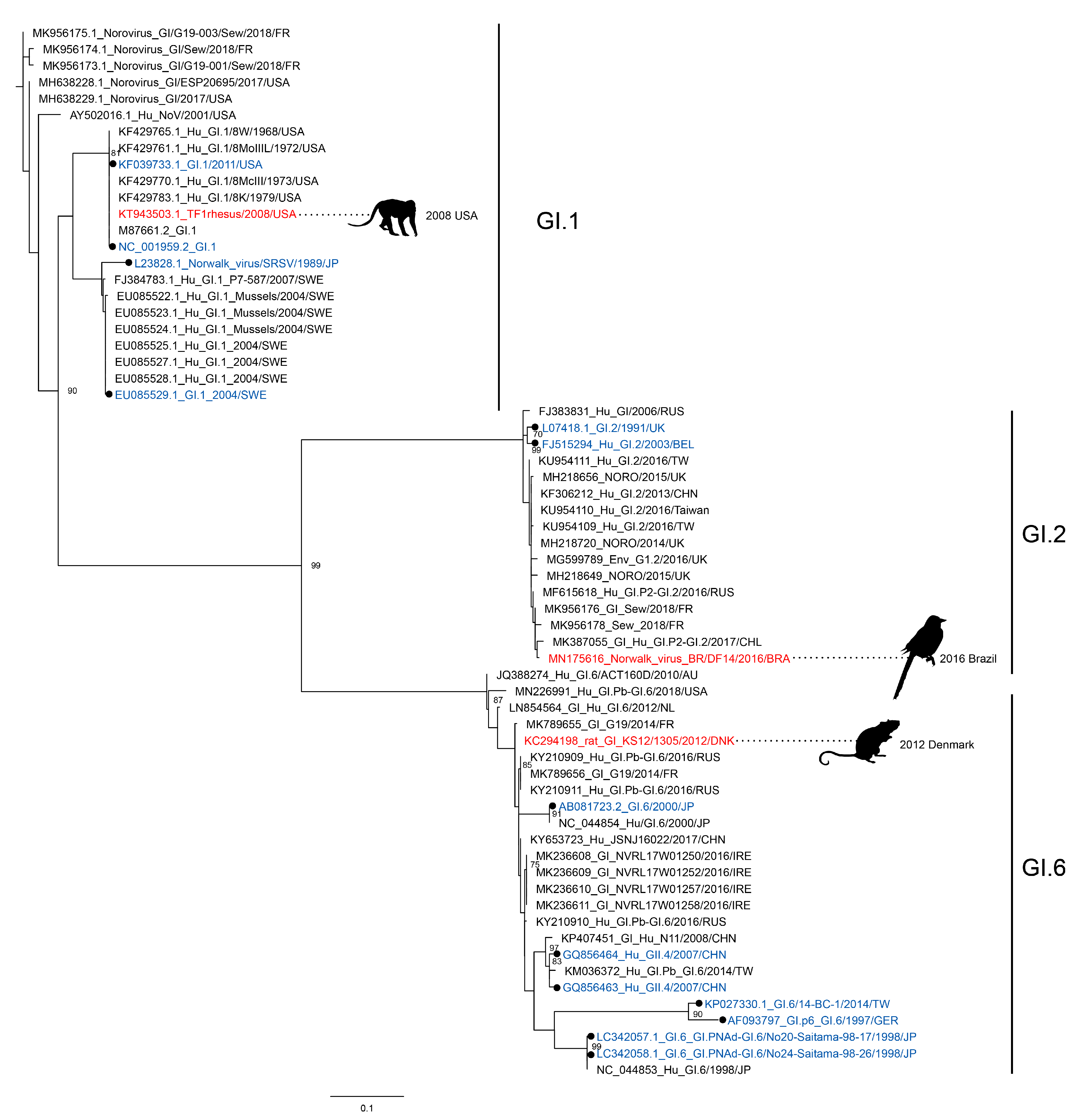
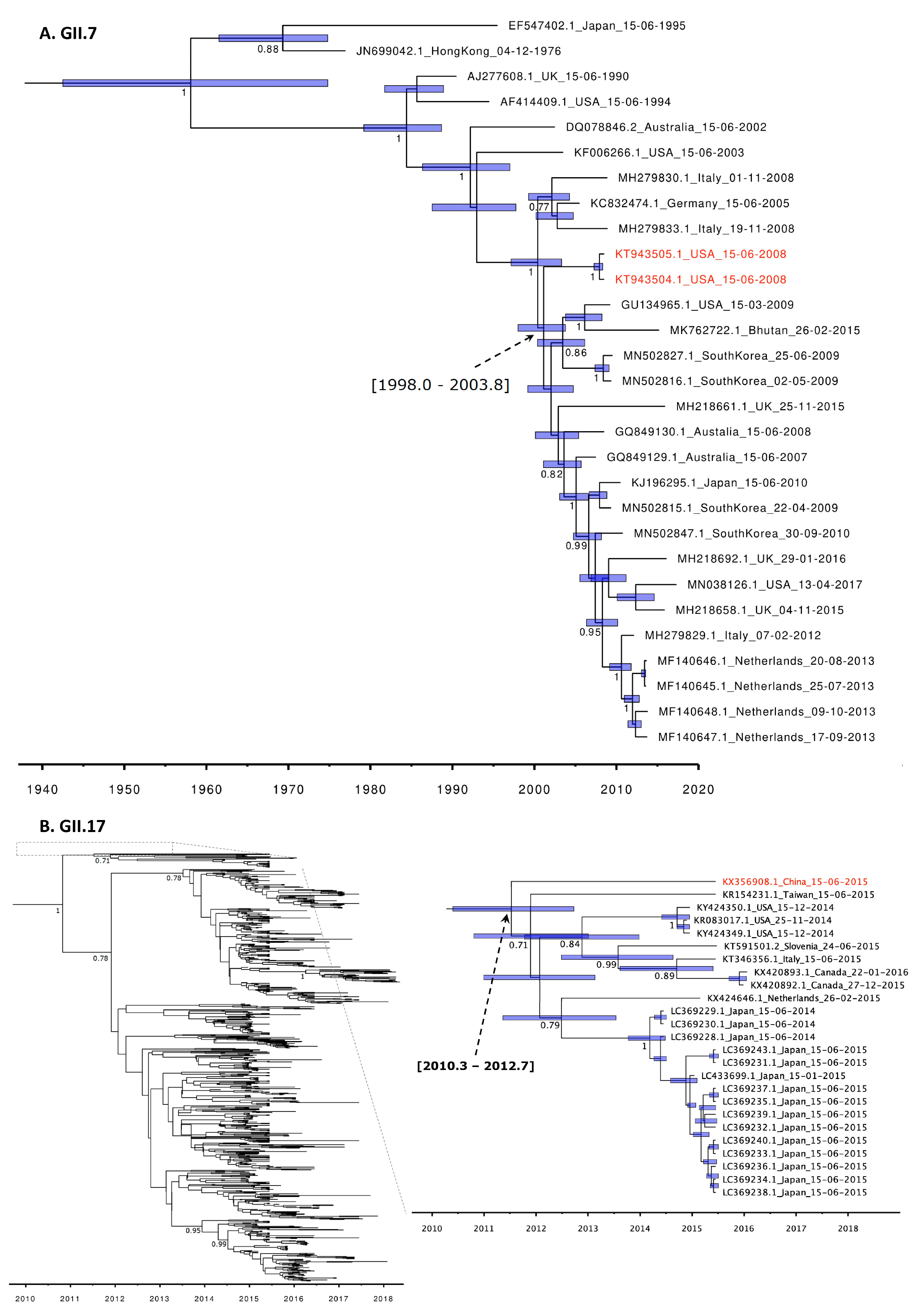

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VP1 = Virus Capsid Protein 1 (ORF2), VP2 = Virus Capsid Protein 2 (ORF3), RdRp = RNA-Dependent RNA Polymerase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Accession Number | Host | Location | Year | Circulation Based on References [20,21] | Norovirus Typing | Sequence Length (bp) | Genome Region Covered | Similarity to Best Blast Hit | Ref |
| MF444290 | Bird | Finland | 2009 | NA | GII.4 could not assign | 223 | VP1 | 222/223 (99.5%) | [22] |
| MF444291 | Bird | Finland | 2009 | NA | GII.4 could not assign | 223 | VP1 | 222/223 (99.5%) | [22] |
| MF444292 | Bird | Finland | 2009 | 2006–8 | GII.4 Den Haag 2006b | 223 | VP1 | 222/223 (99.5%) | [22] |
| MF444293 | Bird | Finland | 2011 | 2009–12 | GII.4 New Orleans 2009 | 223 | VP1 | 223/223 (100%) | [22] |
| JQ068133 * | Pig | Taiwan | 2008 | 2006–8 | GII.4 Den Haag 2006b | 239 | RdRp/VP1 | 239/239 (100%) | [23] |
| AB521758-63 | Pig | Japan | 2008/9 | 2006–8 | GII.4 Den Haag 2006b | 302 | VP1 | 298/302 (98.9%)–302/302 (100%) | [24] |
| AB521764 | Pig | Japan | 2009 | 2002–6 | GII.4 Asia 2003 | 302 | VP1 | 302/302 (100%) | [24] |
| JF7468901 | Dog | Finland | 2009 | NA | GII.4 could not assign | 283 | VP1 | 281/283 (99.3%) | [25] |
| JF746891 | Dog | Finland | 2010 | NA | GII.4 could not assign | 283 | VP1 | 281/283 (99.3%) | [25] |
| JF746892 | Dog | Finland | 2009 | 2006–8 | GII.4 Den Haag 2006b | 283 | VP1 | 281/283 (99.3%) | [25] |
| MK928498-99 1 | Dog | Thailand | 2018 | 2012–20 | GII.4 Sydney 2012[P31] | 7513 | Genome | 7457/7513 (99.3%) | [26] |
| EF175441 ** | Pig | Canada | 2005 | 2002–4 | GII.P4 Farmington Hills 2002 | 172 | RdRp | 172/172 (100%) | [27] |
| CE-M-05–0114 **,6 | Pig | Canada | 2005 | NA | GII could not assign | 172 | RdRp | 172/172 (100%) | [27] |
| CE-M-06–0013 **,6 | Pig | Canada | 2005 | 2002–4 | GII.P4 Farmington Hills 2002 | 172 | RdRp | 169/172 (98.3%) | [27] |
| CE-M-06–0509 **,6 | Cattle | Canada | 2005 | 2006–8 | GII.P4 Den Haag 2006b | 172 | RdRp | 168/172 (98.3%) | [27] |
| GU556160-66 ** | Pig | Taiwan | 2008 | 2006–08 | GII.P4 Den Haag 2006b | 260 | RdRp | 241/256 (94%) | [23] |
| MN175616 | Bird | Brazil | 2016 | NA | GI.2 | 492 | VP1 | 475/478 (99.4%) | [28] |
| KT943503 | Macaque 2 | USA | 2008 | NA | GI.1 | 1670 | VP1-VP2 | 1670/1670 (100%) | [29] |
| KC294198 | Rat 3 | Denmark | 2012 | NA | GI.6[GI.Pb] | 4012 | RdRp-VP1/2 | 3959/3993 (99.1%) | [30] |
| MF444294 | Crow 4 | Finland | 2011 | NA | GII.3 | 223 | VP1 | 223/223 (100%) | [22] |
| KX073992 | Pig | Ethiopia | 2013 | NA | GII.1 | 291 | VP1 | 279/291 (95.9%) | [31] |
| KT943504 | Macaque | USA | 2008 | NA | GII.7 | 1623 | VP1 | 1570/1632 (96%) | [29] |
| KT943505 | Macaque 2 | USA | 2008 | NA | GII.7 | 1623 | VP1 | 1570/1632 (96%) | [29] |
| JF746893 | Dog | Finland | 2009 | NA | GII.12 | 283 | VP1 | 282/283 (99.7%) | [25] |
| KX356908 | Macaque | China | 2015 | 2014–15 | GII.17[P17] | 7556 | Genome | 7496/7556 (99.2%) | [32] |
| AB521765 | Pig | Japan | 2008 | NA | GII.14 | 302 | VP1 | 291/302 (96.4%) | [24] |
| AB521754-57 | Pig | Japan | 2008 | NA | GII.3 | 302 | VP1 | 297/302 (98.3%)–301/302 (99.7%) | [24] |
| JQ068117-132 | Pig | Taiwan | 2008 | NA | GII.2 | 239 | VP1 | 238/239 (99.6%) | [23] |
| HM035148 ** | Macaque 2 | USA | 2008 | NA | GII.7 could not assign 5 | 274 | RdRp | 259/274 (94.5%) to GII.P7 | [29,33] |
| MN175617 ** | Bird | Brazil | 2016 | NA | GII.P31 could not assign 5 | 438 | p48 | 416/419 (99.3%) (gap) | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villabruna, N.; Izquierdo Lara, R.W.; Szarvas, J.; Koopmans, M.P.G.; Graaf, M.d. Phylogenetic Investigation of Norovirus Transmission between Humans and Animals. Viruses 2020, 12, 1287. https://doi.org/10.3390/v12111287
Villabruna N, Izquierdo Lara RW, Szarvas J, Koopmans MPG, Graaf Md. Phylogenetic Investigation of Norovirus Transmission between Humans and Animals. Viruses. 2020; 12(11):1287. https://doi.org/10.3390/v12111287
Chicago/Turabian StyleVillabruna, Nele, Ray W. Izquierdo Lara, Judit Szarvas, Marion P. G. Koopmans, and Miranda de Graaf. 2020. "Phylogenetic Investigation of Norovirus Transmission between Humans and Animals" Viruses 12, no. 11: 1287. https://doi.org/10.3390/v12111287