
Characterization of Novel Erwinia amylovora Jumbo Bacteriophages from Eneladusvirus Genus



 ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Phage Isolation, Purification, and Propagation
2.2. Propagation, and Purification of Phages
2.3. Transmission Electron Microscopy
2.4. Host Range Analysis
2.5. DNA Preparation and Sequencing
2.6. Genome Analysis
2.7. Proteome Analysis
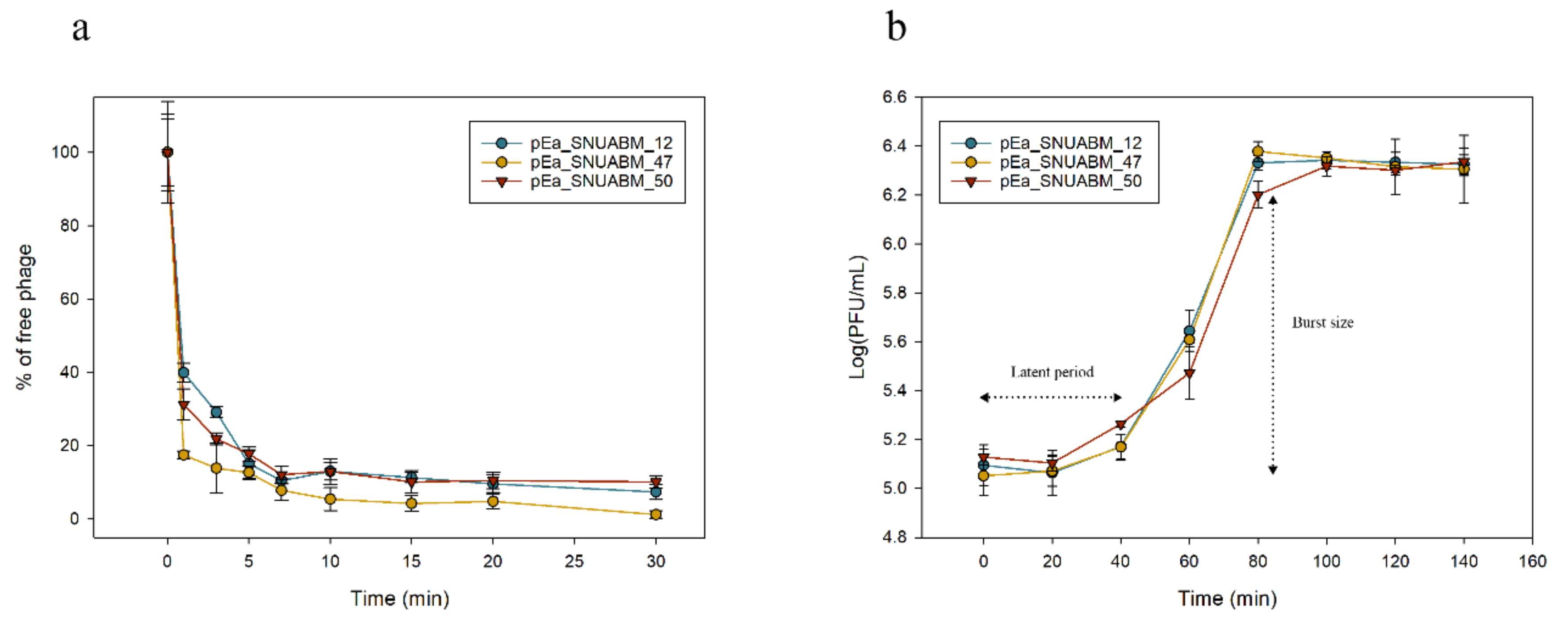
2.8. Adsorption Assay and One-Step Growth Curve
2.9. Data Availability
3. Results and Discussions
3.1. Biological Characteristics of pEa_SNUABM_12, pEa_SNUABM_47, and pEa_SNUABM_50
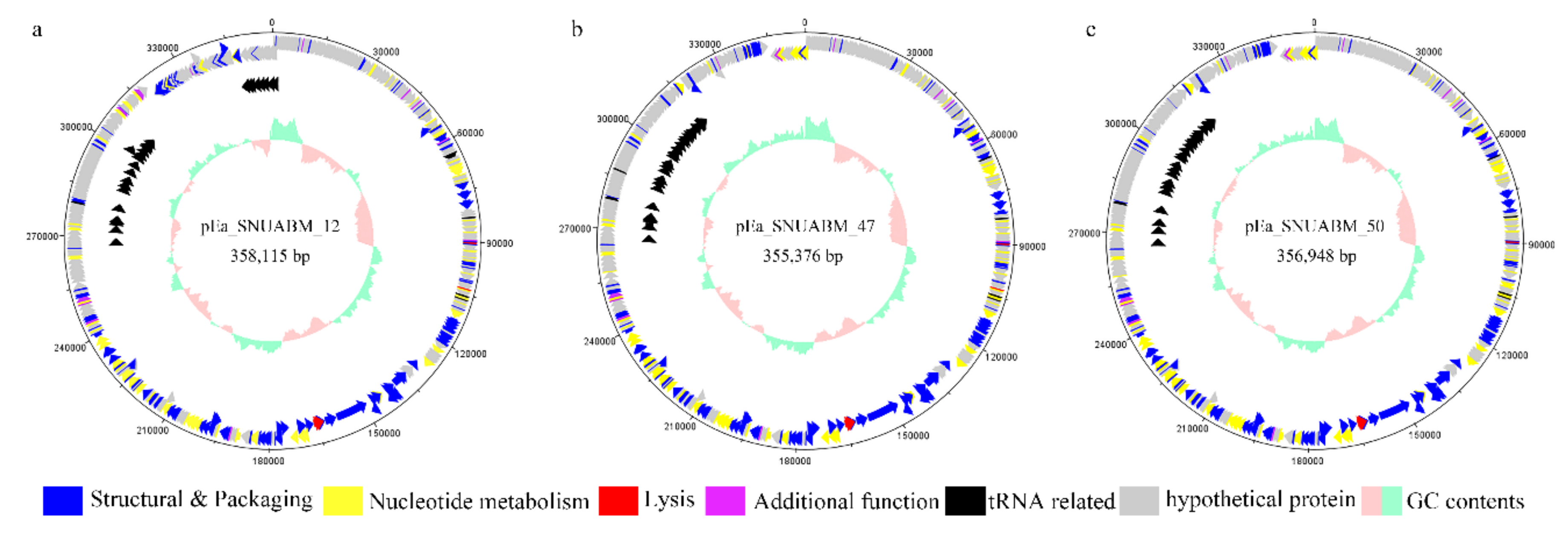
3.2. General Features of the pEa_SNUABM_12, pEa_SNUABM_47, and pEa_SNUABM_50 Genomes
3.3. Comparative Analysis of the pEa_SNUABM_12, pEa_SNUABM_47, and pEa_SNUABM_50 Genomes
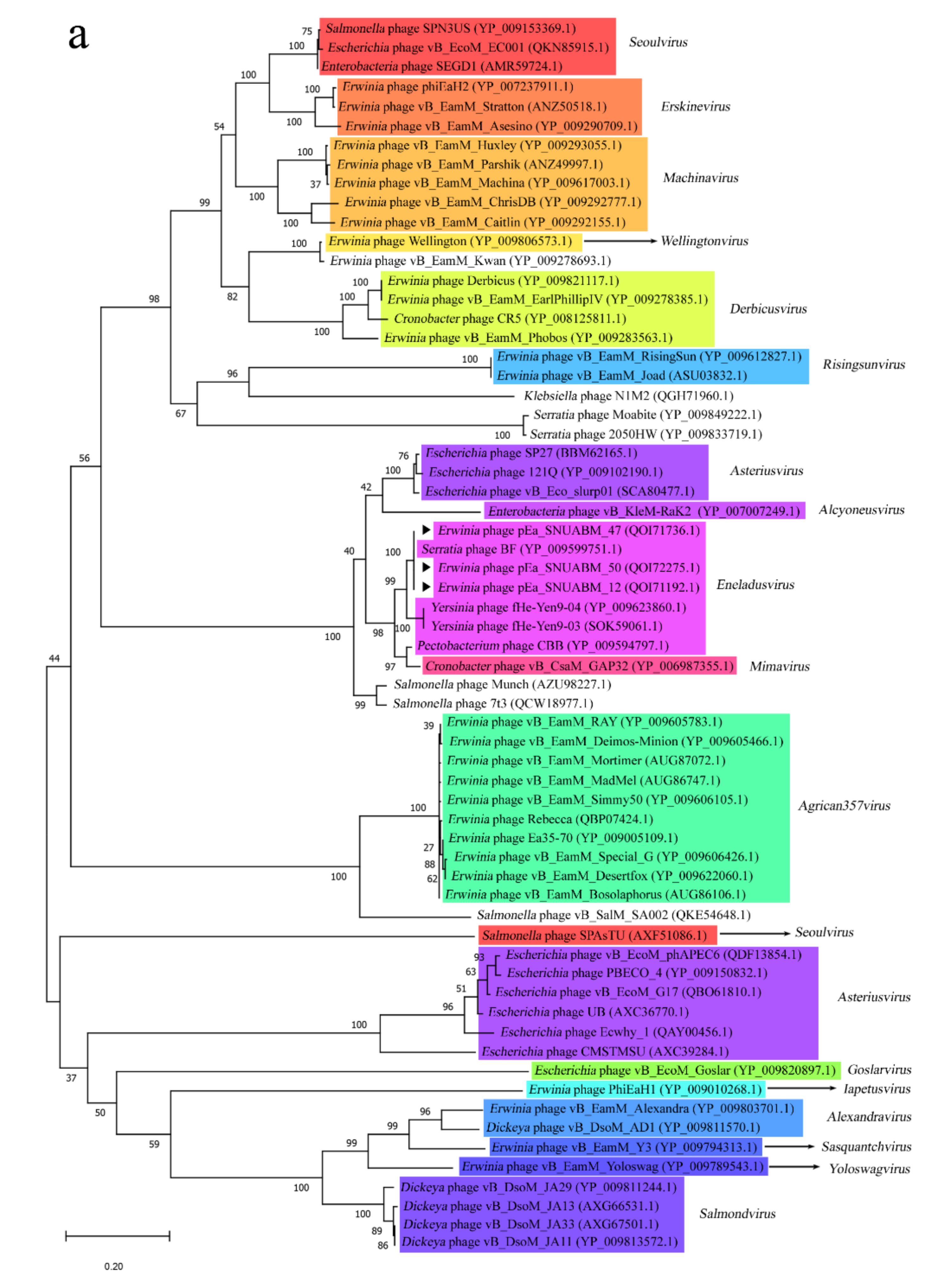
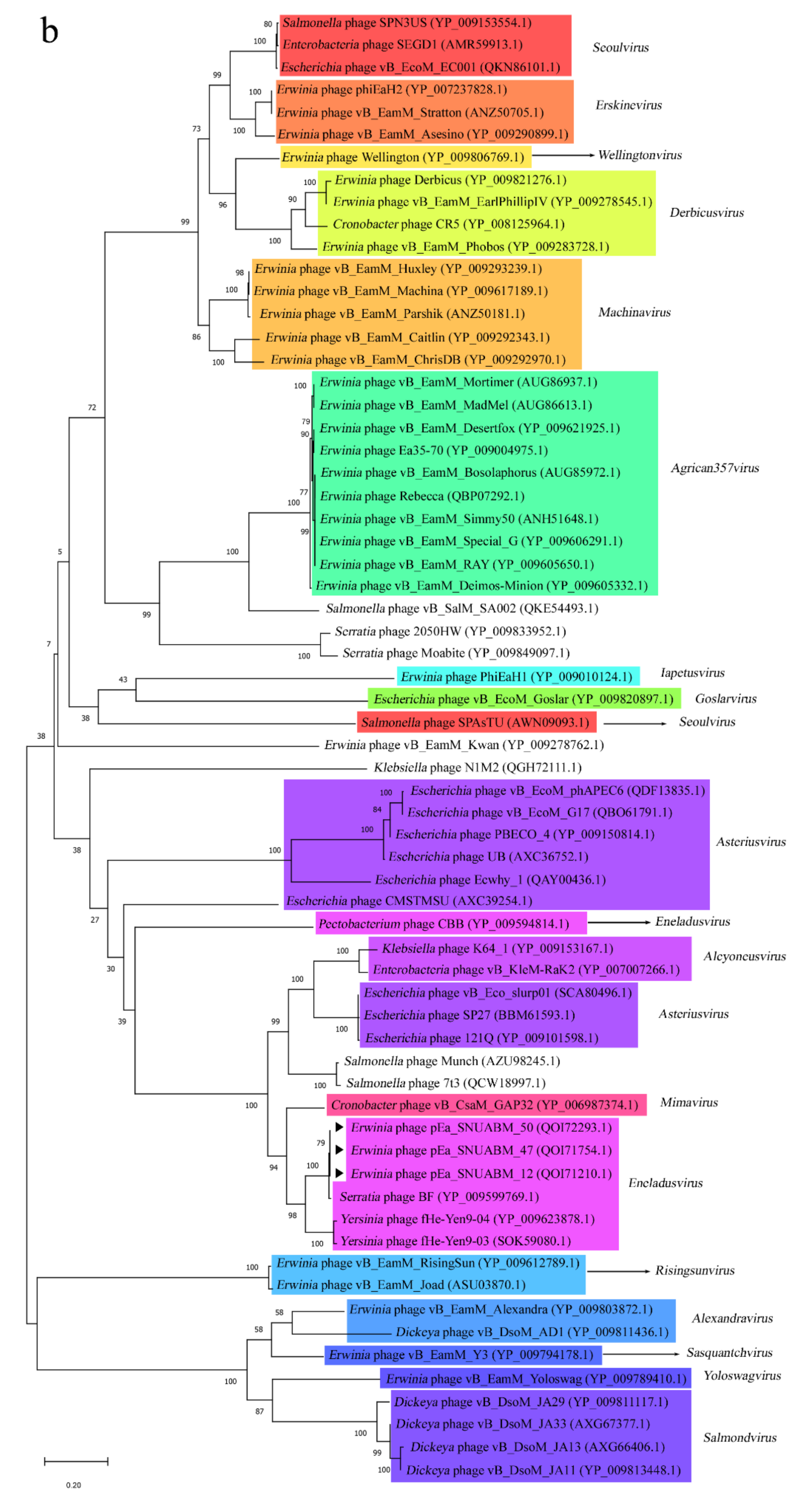
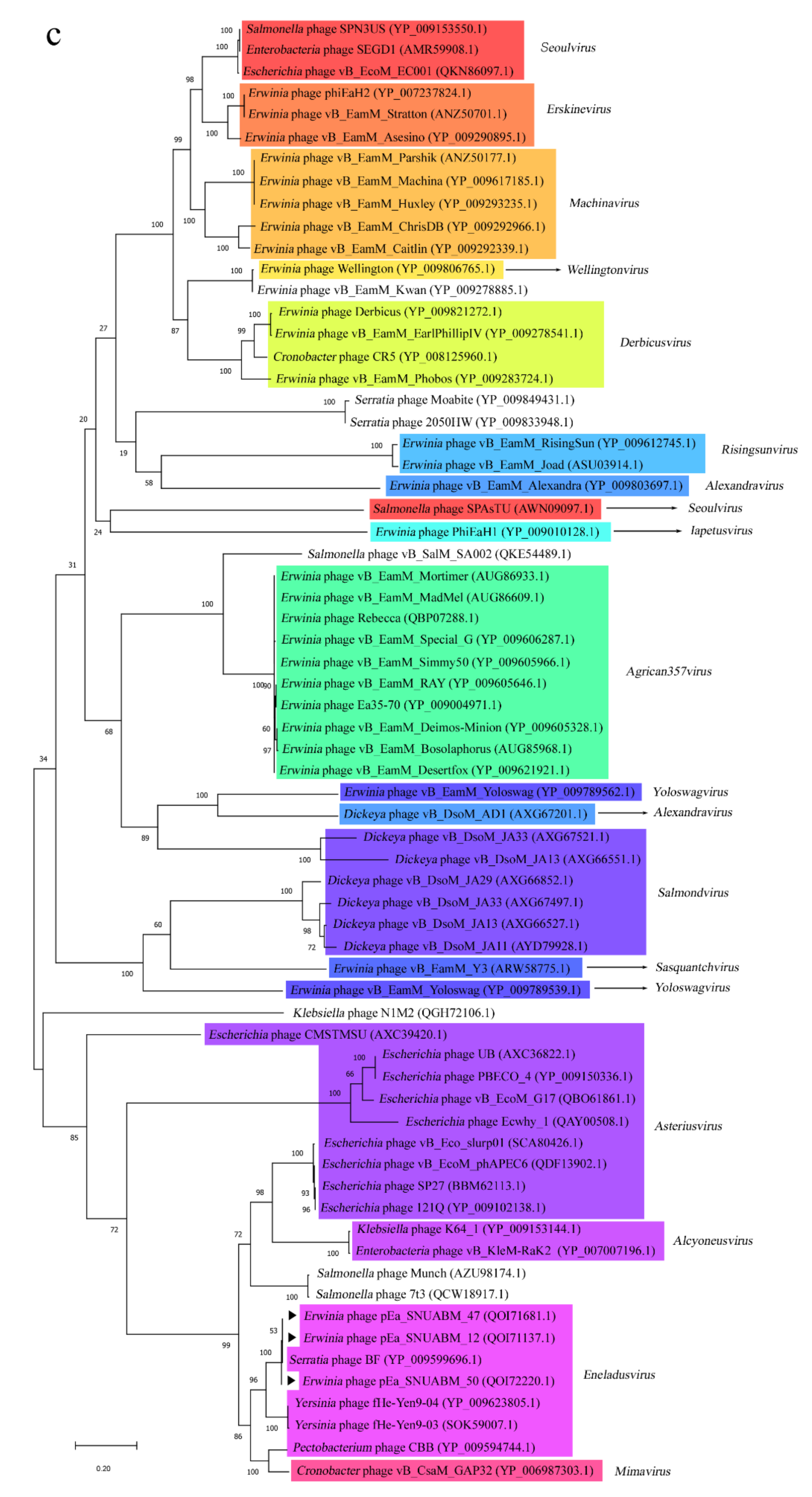
3.4. Phylogeneitc Analysis of the pEa_SNUABM_12, pEa_SNUABM_47, and pEa_SNUABM_50 Genomes
3.5. Functional Analysis of the pEa_SNUABM_12, pEa_SNUABM_47, and pEa_SNUABM_50 Genomes
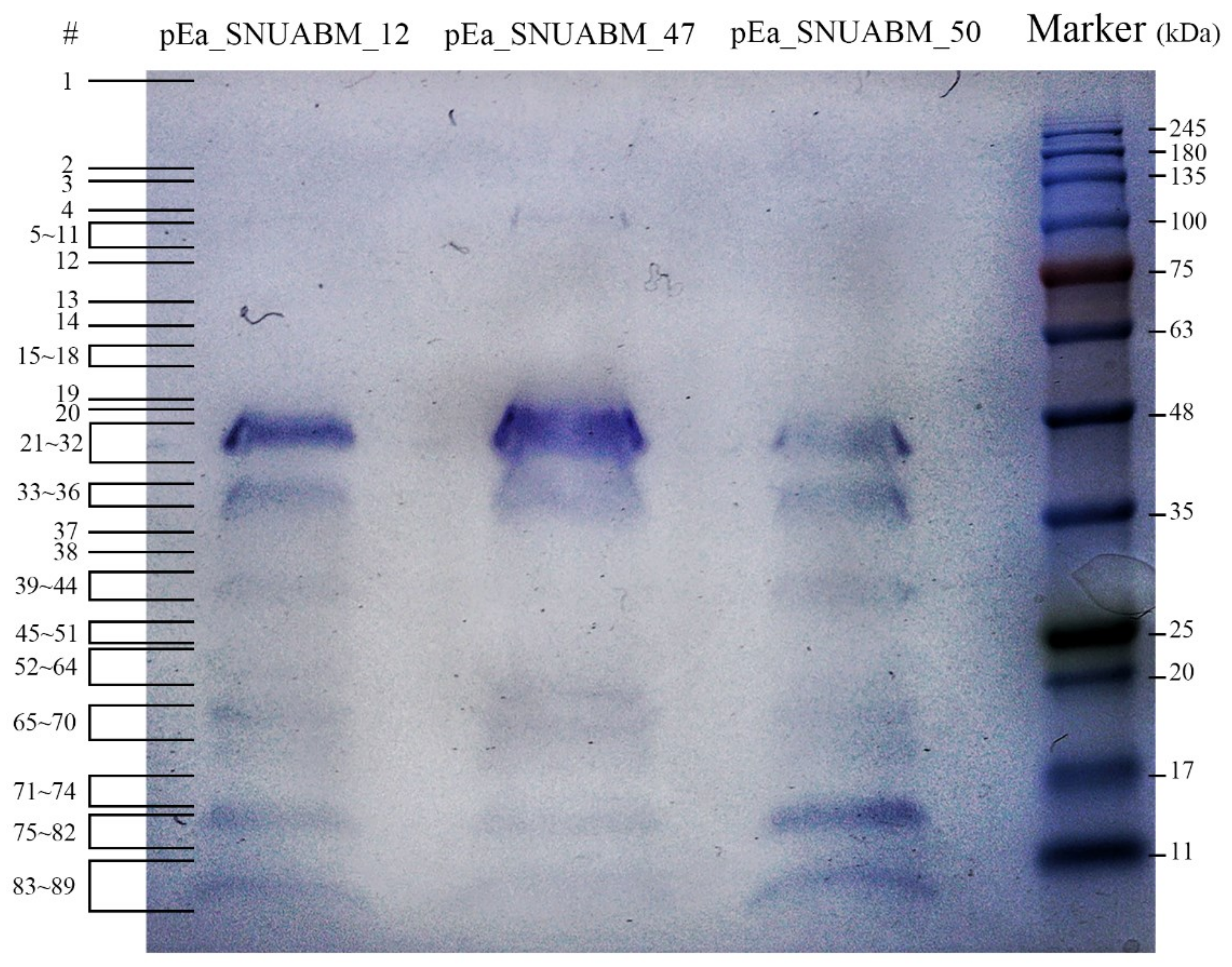
3.5.1. Mass Spectrometry
3.5.2. Structure and Packaging
3.5.3. tRNAs
3.5.4. Nucleotide Metabolism
3.5.5. Lysis
3.5.6. Additional Function
3.6. Adsorption Assay and One-Step Growth Curve
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nagy, J.; Király, L.; Schwarczinger, I. Phage therapy for plant disease control with a focus on fire blight. Open Life Sci. 2012, 7, 1–12. [Google Scholar] [CrossRef]
- Boulé, J.; Sholberg, P.L.; Lehman, S.M.; O’gorman, D.T.; Svircev, A.M. Isolation and characterization of eight bacteriophages infecting Erwinia amylovora and their potential as biological control agents in British Columbia, Canada. Can. J. Plant Pathol. 2011, 33, 308–317. [Google Scholar] [CrossRef]
- Schwarczinger, I.; Nagy, J.K.; Künstler, A.; Szabó, L.; Geider, K.; Király, L.; Pogány, M. Characterization of Myoviridae and Podoviridae family bacteriophages of Erwinia amylovora from Hungary-potential of application in biological control of fire blight. Eur. J. Plant Pathol. 2017, 149, 639–652. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Pielstick, B.A.; Bell, K.A.; Nieman, T.B.; Stubbs, O.A.; Yeates, E.L.; Baltrus, D.A.; Grose, J.H. A novel, highly-related jumbo family of bacteriophages that were isolated against Erwinia. Front. Microbiol. 2019, 10, 1533. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, G.M.; Kim, D.; Park, D.H.; Oh, C.S. Characterization of the lytic bacteriophage phiEaP-8 effective against both Erwinia amylovora and Erwinia pyrifoliae causing severe diseases in apple and pear. Plant Pathol. J. 2018, 34, 445. [Google Scholar]
- Domingo-Calap, P.; Delgado-Martínez, J. Bacteriophages: Protagonists of a post-antibiotic era. J. Antibiot. 2018, 7, 66. [Google Scholar] [CrossRef] [Green Version]
- Evseev, P.; Sykilinda, N.; Gorshkova, A.; Kurochkina, L.; Ziganshin, R.; Drucker, V.; Miroshnikov, K. Pseudomonas Phage PaBG—A Jumbo Member of an Old Parasite Family. Viruses 2020, 12, 721. [Google Scholar] [CrossRef]
- Attai, H.; Boon, M.; Phillips, K.; Noben, J.P.; Lavigne, R.; Brown, P.J. Larger than life: Isolation and genomic characterization of a jumbo phage that infects the bacterial plant pathogen, Agrobacterium tumefaciens. Front. Microbiol. 2018, 9, 1861. [Google Scholar] [CrossRef] [Green Version]
- Lewis, R.; Clooney, A.G.; Stockdale, S.R.; Buttimer, C.; Draper, L.A.; Ross, R.P.; Hill, C. Isolation of a novel jumbo bacteriophage effective against Klebsiella aerogenes. Front. Med. 2020, 7, 67. [Google Scholar] [CrossRef] [Green Version]
- Kawato, Y.; Istiqomah, I.; Gaafar, A.Y.; Hanaoka, M.; Ishimaru, K.; Yasuike, M.; Nishiki, I.; Nakamura, Y.; Fujiwara, A.; Nakai, T. A novel jumbo Tenacibaculum maritimum lytic phage with head-fiber-like appendages. Arch. Virol. 2020, 165, 303–311. [Google Scholar] [CrossRef]
- Arens, D.K.; Brady, T.S.; Carter, J.L.; Pape, J.A.; Robinson, D.M.; Russell, K.A.; Staley, L.A.; Stettler, J.M.; Tateoka, O.B.; Townsend, M.H.; et al. Characterization of two related Erwinia myoviruses that are distant relatives of the PhiKZ-like Jumbo phages. PLoS ONE 2018, 13, e0200202. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Berg, J.A.; Beatty, N.J.; Choi, M.C.; Cowger, A.E.; Cozzens, B.J.; Duncan, S.G.; Fajardo, C.P.; Ferguson, H.P.; Galbraith, T.; et al. Genome sequences of nine Erwinia amylovora bacteriophages. Microbiol. Resour. Announc. 2018, 7, e00944-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esplin, I.N.; Berg, J.A.; Sharma, R.; Allen, R.C.; Arens, D.K.; Ashcroft, C.R.; Bairett, S.R.; Beatty, N.J.; Bickmore, M.; Bloomfield, T.J. Genome sequences of 19 novel Erwinia amylovora bacteriophages. Genome Announc. 2017, 5, e00931-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, D.W.; Casjens, S.R.; Sharma, R.; Grose, J.H. Genomic comparison of 60 completely sequenced bacteriophages that infect Erwinia and/or Pantoea bacteria. Virology 2019, 535, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Buttimer, C.; Hendrix, H.; Oliveira, H.; Casey, A.; Neve, H.; McAuliffe, O.; Ross, R.P.; Hill, C.; Noben, J.; O’Mahony, J.; et al. Things are getting hairy: Enterobacteria bacteriophage vB_PcaM_CBB. Front. Microbiol. 2017, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olszak, T.; Danis-Wlodarczyk, K.; Arabski, M.; Gula, G.; Maciejewska, B.; Wasik, S.; Lood, C.; Higgins, G.; Harvey, B.J.; Lavigne, R.; et al. Pseudomonas aeruginosa PA5oct jumbo phage impacts planktonic and biofilm population and reduces its host virulence. Viruses 2019, 11, 1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza, S.D.; Nieweglowska, E.S.; Govindarajan, S.; Leon, L.M.; Berry, J.D.; Tiwari, A.; Chaikeeratisak, V.; Pogliano, J.; Agard, D.A.; Bondy-Denomy, J. A bacteriophage nucleus-like compartment shields DNA from CRISPR nucleases. Nature 2020, 577, 244–248. [Google Scholar] [CrossRef]
- Hampton, H.G.; Watson, B.N.; Fineran, P.C. The arms race between bacteria and their phage foes. Nature 2020, 577, 327–336. [Google Scholar] [CrossRef]
- Ceyssens, P.J.; Minakhin, L.; Van den Bossche, A.; Yakunina, M.; Klimuk, E.; Blasdel, B.; Smet, J.D.; Noben, J.; Bläsi, U.; Severinov, K.; et al. Development of giant bacteriophage ϕKZ is independent of the host transcription apparatus. J. Virol. 2014, 88, 10501–10510. [Google Scholar] [CrossRef] [Green Version]
- Modell, A.E.; Siriwardena, S.U.; Choudhary, A. A jumbo phage forms a nucleus-like compartment to evade nacterial defense systems. Biochemistry 2020, 59, 1869–1870. [Google Scholar] [CrossRef]
- Malone, L.M.; Warring, S.L.; Jackson, S.A.; Warnecke, C.; Gardner, P.P.; Gumy, L.F.; Fineran, P.C. A jumbo phage that forms a nucleus-like structure evades CRISPR–Cas DNA targeting but is vulnerable to type III RNA-based immunity. Nat. Microbiol. 2020, 5, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Saad, A.M.; Soliman, A.M.; Kawasaki, T.; Fujie, M.; Nariya, H.; Shimamoto, T.; Yamada, T. Systemic method to isolate large bacteriophages for use in biocontrol of a wide-range of pathogenic bacteria. J. Biosci. Bioeng. 2019, 127, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Hesse, S.; Adhya, S. Phage therapy in the twenty-first century: Facing the decline of the antibiotic era; is it finally time for the age of the phage? Annu. Rev. Microbiol. 2019, 73, 155–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaki, S.; Kuronuma, S.; Kawai, Y.; Yamazaki, K. Inhibitory effect of a combination with novel jumbo bacteriophages ΦMV-1 and ΦMV-4 on Morganella morganii subsp. morganii growth and histamine accumulation. Int. J. Food Microbiol. 2020, 317, 108457. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Jun, J.W.; Giri, S.S.; Yun, S.; Kim, H.J.; Kim, S.W.; Kang, J.W.; Han, S.J.; Jeong, D.; Park, S.C. Isolation and characterisation of pVa-21, a giant bacteriophage with anti-biofilm potential against Vibrio alginolyticus. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Giri, S.S.; Yun, S.; Kim, H.J.; Kim, S.W.; Kang, J.W.; Han, S.J.; Kwon, J.; Jun, J.W.; Oh, W.T.; et al. Genomic characterization of bacteriophage pEt-SU, a novel phiKZ-related virus infecting Edwardsiella tarda. Arch. Virol. 2020, 165, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Giri, S.S.; Yun, S.; Kim, H.J.; Kim, S.W.; Kang, J.W.; Han, S.J.; Kwon, J.; Oh, W.T.; Jun, J.W.; et al. Synergistic phage-surfactant combination clears IgE-promoted Staphylococcus aureus aggregation in vitro and enhances the effect in vivo. Int. J. Antimicrob. Agents 2020, 56, 105997. [Google Scholar] [CrossRef]
- Boulanger, P. Purification of bacteriophages and SDS-PAGE analysis of phage structural proteins from ghost particles. Methods Mol. Biol. 2009, 502, 227–238. [Google Scholar]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carver, T.; Thomson, N.; Bleasby, A.; Berriman, M.; Parkhill, J. DNAPlotter: Circular and linear interactive genome visualization. Bioinformatics 2009, 25, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Krumsiek, J.; Arnold, R.; Rattei, T. Gepard: A rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 2007, 23, 1026–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, 636–641. [Google Scholar] [CrossRef] [Green Version]
- Denyes, J.M.; Krell, P.J.; Manderville, R.A.; Ackermann, H.W.; She, Y.M.; Kropinski, A.M. The genome and proteome of Serratia bacteriophage η which forms unstable lysogens. Virol. J. 2014, 11, 6. [Google Scholar] [CrossRef] [Green Version]
- To Create One New Genus, Eneladusvirus, Containing Two Species in the Family Myoviridae 2018.026B. Available online: https://talk.ictvonline.org/files/ictv_official_taxonomy_updates_since_the_8th_report/m/prokaryote-official/8151/ (accessed on 24 February 2019).
- Tolstoy, I.; Kropinski, A.M.; Brister, J.R. Bacteriophage taxonomy: An evolving discipline. In Bacteriophage Therapy; Azeredo, J., Silankorva, S., Eds.; Humana Press: New York, NY, USA, 2018; pp. 57–71. [Google Scholar]
- Sazinas, P.; Smith, C.; Suhaimi, A.; Hobman, J.L.; Dodd, C.E.; Millard, A.D. Draft genome sequence of the bacteriophage vB_Eco_slurp01. Genome Announc. 2016, 4, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasifar, R.; Griffiths, M.W.; Sabour, P.M.; Ackermann, H.W.; Vandersteegen, K.; Lavigne, R.; Noben, J.; Villa, A.A.; Abbasifar, A. Supersize me: Cronobacter sakazakii phage GAP32. Virology 2014, 460, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šimoliūnas, E.; Kaliniene, L.; Truncaitė, L.; Zajančkauskaitė, A.; Staniulis, J.; Kaupinis, A.; Ger, M.; Valius, M.; Meškys, R. Klebsiella phage vB_KleM-RaK2—A giant singleton virus of the family Myoviridae. PLoS ONE 2013, 8, e60717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kala, S.; Cumby, N.; Sadowski, P.D.; Hyder, B.Z.; Kanelis, V.; Davidson, A.R.; Maxwell, K.W. HNH proteins are a widespread component of phage DNA packaging machines. Microbiology 2014, 111, 6022–6027. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Gao, M. Jumbo bacteriophages: An overview. Front. Microbiol. 2017, 8, 403. [Google Scholar] [CrossRef] [Green Version]
- Nelson, S.W.; Zhuang, Z.; Spiering, M.M.; Benkovic, S.J. T4 phage replisome. In Viral Genome Replication; Raney, K.D., Gotte, M., Cameron, C.E., Eds.; Springer: Boston, MA, USA, 2009; pp. 337–364. [Google Scholar]
- Samson, J.E.; Magadán, A.H.; Sabri, M.; Moineau, S. Revenge of the phages: Defeating bacterial defences. Nat. Rev. Microbiol. 2013, 11, 675–687. [Google Scholar] [CrossRef]
- Chinnadurai, L.; Eswaramoorthy, T.; Paramachandran, A.; Paul, S.; Rathy, R.; Arumugaperumal, A.; Sivasubramaniam, S.; Thavasimuthu, C. Draft genome sequence of Escherichia coli phage CMSTMSU, isolated from shrimp farm effluent water. Microbiol. Resour. Announc. 2018, 7, e01034-18. [Google Scholar] [CrossRef] [Green Version]
- Casey, E.; Fitzgerald, B.; Mahony, J.; Lugli, G.A.; Ventura, M.; van Sinderen, D. Genome sequence of Serratia marcescens phage BF. Genome Announc. 2017, 5, e00211-17. [Google Scholar] [CrossRef] [Green Version]
- Kongari, R.; Rajaure, M.; Cahill, J.; Rasche, E.; Mijalis, E.; Berry, J.; Young, R. Phage spanins: Diversity, topological dynamics and gene convergence. BMC Bioinform. 2018, 19, 326. [Google Scholar] [CrossRef] [Green Version]
- Young, R. Phage lysis: Three steps, three choices, one outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef]
- Sewell, E. Characterizing the Intact Prophage of Mycobacterium Chelonae Bergey. Honors Thesis, University of Maine, Orono, ME, USA, May 2017. [Google Scholar]
- Wang, J.; Jiang, Y.; Vincent, M.; Sun, Y.; Yu, H.; Wang, J.; Bao, Q.; Kong, H.; Hu, S. Complete genome sequence of bacteriophage T5. Virology 2005, 332, 45–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.C.; Wu, Y.Q.; Cao, D.; Zhang, W.B.; Wang, H.H. Only acyl carrier protein 1 (AcpP1) functions in Pseudomonas aeruginosa fatty acid synthesis. Front. Microbiol. 2017, 8, 2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, S.; Ma, T.; Zhang, X.; Huang, Y.; Mi, Z.; Sun, Q.; An, X.; Fan, H.; Wu, S.; Wei, L.; et al. First complete genome sequence of a virulent bacteriophage infecting the opportunistic pathogen Serratia rubidaea. Arch. Virol. 2017, 162, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F. Dark matter of the biosphere: The amazing world of bacteriophage diversity. J. Virol. 2015, 89, 8107–8110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pope, W.H.; Hatfull, G.F. Adding pieces to the puzzle: New insights into bacteriophage diversity from integrated research-education programs. Bacteriophage 2015, 5, e1084073. [Google Scholar] [CrossRef] [Green Version]
- Dendooven, T.; Lavigne, R. Dip-a-Dee-Doo-Dah: Bacteriophage-mediated rescoring of a harmoniously orchestrated RNA metabolism. Annu. Rev. Virol. 2019, 6, 199–213. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Strain | Biological Origin | Host Range a | ||
|---|---|---|---|---|
| pEa_12 | pEa_47 | pEa_50 | ||
| Erwinia amylovora | ||||
| SNUABM_01 b | Apple | + | + | + |
| SNUABM_02 | Pear | + | + | + |
| Erwinia pyrifoliae | ||||
| KACC13945 | Apple | + | + | + |
| KACC13946 | Apple | + | + | + |
| KACC13948 | Apple | + | + | + |
| KACC13949 | Apple | + | + | + |
| KACC13952 | Apple | + | + | + |
| Erwinia rhapontici | ||||
| KACC14080 | Pear | - | - | - |
| Erwinia sp. | ||||
| KACC10370 | N/A | - | - | - |
| KACC10403 | N/A | + | + | + |
| Pectobacterium carotovorum | ||||
| KACC17004 | Cabbage | - | LO | LO |
| KACC18645 | Chinese cabbagge | - | LO | LO |
| KACC21701 | Chinese cabbagge | LO | LO | LO |
| Pseudomonas aeruginosa | ||||
| KCCM40395 | N/A | - | - | - |
| Escherichia coli | ||||
| KCTC2571 | Human feces | - | - | - |
| Serratia marcescens | ||||
| KACC10502 | Tropical greenhouse | + | + | + |
| KACC11961 | Pond water | + | + | + |
| Size (bp) | ORFs | tRNAs | GC Content (%) | DNA Circularity | Accession Number | |
|---|---|---|---|---|---|---|
| pEa_SNUABM_12 | 358,115 | 546 | 32 | 34.41 | Circular | MT939486.1 |
| pEa_SNUABM_47 | 355,376 | 540 | 35 | 34.48 | Circular | MT939487.1 |
| pEa_SNUABM_50 | 356,948 | 540 | 34 | 34.45 | Circular | MT939488.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.G.; Lee, S.B.; Giri, S.S.; Kim, H.J.; Kim, S.W.; Kwon, J.; Park, J.; Roh, E.; Park, S.C. Characterization of Novel Erwinia amylovora Jumbo Bacteriophages from Eneladusvirus Genus. Viruses 2020, 12, 1373. https://doi.org/10.3390/v12121373
Kim SG, Lee SB, Giri SS, Kim HJ, Kim SW, Kwon J, Park J, Roh E, Park SC. Characterization of Novel Erwinia amylovora Jumbo Bacteriophages from Eneladusvirus Genus. Viruses. 2020; 12(12):1373. https://doi.org/10.3390/v12121373
Chicago/Turabian StyleKim, Sang Guen, Sung Bin Lee, Sib Sankar Giri, Hyoun Joong Kim, Sang Wha Kim, Jun Kwon, Jungkum Park, Eunjung Roh, and Se Chang Park. 2020. "Characterization of Novel Erwinia amylovora Jumbo Bacteriophages from Eneladusvirus Genus" Viruses 12, no. 12: 1373. https://doi.org/10.3390/v12121373
APA StyleKim, S. G., Lee, S. B., Giri, S. S., Kim, H. J., Kim, S. W., Kwon, J., Park, J., Roh, E., & Park, S. C. (2020). Characterization of Novel Erwinia amylovora Jumbo Bacteriophages from Eneladusvirus Genus. Viruses, 12(12), 1373. https://doi.org/10.3390/v12121373