Abstract
In recent years, next generation sequencing (NGS) technology has been widely used for the discovery of novel human papillomavirus (HPV) genotypes, variant characterization and genotyping. Here, we compared the analytical performance of NGS with a commercial PCR-based assay (Anyplex II HPV28) in cervical samples of 744 women. Overall, HPV positivity was 50.2% by the Anyplex and 45.5% by the NGS. With the NGS, we detected 25 genotypes covered by Anyplex and 41 additional genotypes. Agreement between the two methods for HPV positivity was 80.8% (kappa = 0.616) and 84.8% (kappa = 0.652) for 28 HPV genotypes and 14 high-risk genotypes, respectively. We recovered and characterized 243 complete HPV genomes from 153 samples spanning 40 different genotypes. According to phylogenetic analysis and pairwise distance, we identified novel lineages and sublineages of four high-risk and 16 low-risk genotypes. In total, 17 novel lineages and 14 novel sublineages were proposed, including novel lineages of HPV45, HPV52, HPV66 and a novel sublineage of HPV59. Our study provides important genomic insights on HPV types and lineages, where few complete genomes were publicly available.
1. Introduction
Human papillomavirus (HPV) is one of the most common sexually transmitted infections and the principal cause of cervical cancer [1]. HPVs belong to the Papillomaviridae family and are highly diverse. According to the International Committee on the Taxonomy of Viruses Study Group HPVs are classified in genera, species and types based on the L1 open reading frame (ORF), being the most conserved region of the HPV genome [2]. Different HPV types share less than 90% of the L1 gene nucleotide sequence [2]. HPV types with a difference in complete viral nucleotide sequence of 1% to 10% and of 0.5% to 1% have been further classified into lineages and sublineages denoted by letters and numbers, respectively [2,3,4]. Additionally, HPVs are classified according to their oncogenic potential into high-risk (hr) and low-risk (lr) genotypes [5,6].
Recent studies indicate that lineages of HPV16, 31 and 58 genotypes have different carcinogenic properties [3,7,8,9,10,11]. For example, sublineage D2 of HPV16 and lineages A/B of HPV31 are associated with an increased risk of cervical precancer compared to lineages A and C, respectively [8,9,12]. While the scientific community is mainly focusing on the characterization of hr-HPV, only a limited number of lr-HPV have been classified and characterized into lineages [13,14,15].
Modern technological advances have resulted in the development of more than 250 HPV detection and genotyping methods [16]. The Anyplex II HPV28 (Seegene, Seoul, Korea) is a widely used commercial PCR-based assay for the simultaneous detection and genotyping of 14 hr and 14 lr HPV types. This assay with high analytical performance has been clinically validated for cervical cancer screening and used in HPV vaccine surveillance studies [17,18,19,20].
While PCR-based HPV assays detect by design only a limited number of targeted genotypes, next generation sequencing (NGS) may be an alternative approach for universal HPV detection and characterization [16,21,22,23,24,25]. This technology facilitated the detection of novel HPV genotypes and variants [14,22,26] resulting in the identification of 222 HPV types and more than 100 lineages/sublineages [27,28]. Rolling-circle amplification (RCA) has been developed for the random amplification of circular DNA genomes, including viruses from the Papillomaviridae family [29]. It was first used for HPV amplification and sequencing by Rector et al. before the availability of NGS [30], and since then few studies have used this approach for investigating HPV [14,31,32]. The RCA technology allows unbiased random amplification of HPV genome without prior knowledge of the sequence and therefore has a great potential for improving characterization of both known and novel HPV genotypes [26,32,33].
In this study, we aimed to investigate and characterize HPV diversity circulating in healthy young women attending vaccine surveillance study in Luxembourg using NGS-RCA technology. In addition, we compared the analytical performance of the Anyplex II HPV28 assay and NGS using RCA for HPV detection and genotyping. To the best of our knowledge, this is the first large scale study to present HPV diversity using RCA-NGS in healthy women and compare the analytical performance of a PCR based genotyping assay with NGS-RCA.
2. Materials and Methods
A total of 744 cervical samples of women (median age: 22, range 18–43) participating in an HPV vaccination surveillance project were included in the study. More details on the study population and recruitment process have been published elsewhere [20]. Briefly, cervical samples were collected with a cervical broom by gynecologists or other physicians and placed in a vial containing PreservCyt medium of ThinPrep (Hologic, Inc., Bedford, MA, USA) [20,34].
2.1. DNA Extraction
DNA extraction from ThinPrep samples for sequencing and genotyping by the Anyplex II HPV28 was performed using the QIAamp DNA mini kit according to manufacturer’s instructions (Qiagen, Hilden, Germany) [35]. Prior to genotyping/sequencing DNA extracts were stored at −20 °C.
2.2. Anyplex II HPV28
Initially, HPV detection was performed by the Anyplex II HPV28, targeting the L1 gene. The Anyplex assay can simultaneously identify and distinguish of 14 carcinogenic or hr-HPV types (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68), and 14 lr-HPV types (6, 11, 26, 40, 42, 43, 44, 53, 54, 61, 69, 70, 73, 82) in two sets. The viral load is defined as high (+++) if a positive signal occurs before 31 polymerase chain reaction (PCR) cycles, as medium (++) if a positive signal occurs within 31 to 40 PCR cycles, and low if a positive signal occurs after 41 PCR cycles [36].
Real time PCR was performed in two multiplex reactions on the CFX96 real-time thermocycler (Bio-Rad, Hercules, CA, USA). The human beta-globin housekeeping gene was amplified along with L1 viral gene, as an internal positive control while water was used as a negative control. All reactions were performed according to the manufacturer’s instructions and the Seegene viewer software was used for data recording and interpretation [19].
2.3. Next Generation Sequencing
2.3.1. Library Preparation
Total DNA extracts were enriched using rolling-circle amplification (RCA) technology with the TempliPhi 100 kit according to manufacturer’s instructions (GE Healthcare Life Sciences, New Jersey, NJ, USA) [30]. Libraries were prepared using Nextera XT DNA Library Prep Kits (Illumina Inc., San Diego, CA, USA) as recommended by manufacturer’s instructions followed by sequencing on Illumina MiniSeq Platform (Illumina Inc., San Diego, CA, USA). Up to 96 samples were multiplexed using standard Nextera DNA CD Illumina indices in one run (2 × 150 bp paired-end).
2.3.2. Bioinformatic Analysis
Full Genome Assembly and Annotation
In order to assemble full HPV genome sequences, we followed two complementary approaches. For both approaches we performed an initial QC step, including visual inspection of FastQC report for each sample [37]. The strictness of applied quality criteria depended on the subsequent approach followed (Supplementary Figure S1A,B).
The first approach was to “blindly” assemble all reads from 744 samples (irrelevant of being HPV positive or negative) remaining after filtering against human, bacterial, plasmid and fungal reference sequences. The second approach was based on assemblies done on only dehumanized reads, which have been pre-mapped to all available HPV reference genome sequences. In both approaches mapping was performed with Bowtie2 [38] and de novo assembly with SPAdes (v. 3.13.0) [38,39]. Annotation of genomes was completed by means of an adapted version of VAPiD [40]. Detailed information on methods and annotation is provided in Supplementary Materials.
We obtained 75 identical genomes by both methods, 86 not fully identical, 42 only with blind assembly and 43 only with bowtie assembly. We selected 75 identical genomes, while not identical genomes were compared and further processed to allow assembly selection. All assemblies were checked by remapping fastq reads to assembled genomes and by aligning to the respective reference genomes. Of the total 246 genomes assessed, three (HPV42, HPV53, HPV56) were discarded from further analysis due to chimeric artefacts.
HPV Detection
For HPV detection and genotyping, quality controlled and dehumanized fastq-files from (trimmed and filtered against human genome as described above) were mapped with Bowtie2 to a reference set of 318 HPV sequences downloaded from PaVE (https://pave.niaid.nih.gov/, accessed in November 2018) and one novel genotype detected in our laboratory. Accession numbers of reference genomes are provided in Supplementary Table S1 [27].
Samples were considered positive if at least one concordantly mapping read pair was detected covering a minimum of 150 bp of the reference genome. To avoid possible mapping artefacts reads covering < 200 bp with the mutation rate > 0.03 (~4.5 variants on 150 bp read) were blasted against NCBI-based database (built on July 2019) and manually/visually checked. Artefacts were removed if the blast provided different results.
2.4. Phylogenetic Analysis and Lineage Classification
Complete HPV genomes obtained in this study (n = 243) were assessed to investigate HPV variant distribution in healthy women in Luxembourg. Complete genomes were linearized according to the respective reference sequence available in PaVE (https://pave.niaid.nih.gov/) [27] and aligned using MAFFT [41]. Each HPV isolate genome was classified into lineage or sublineage using the p-distance method and phylogenetic analysis with respective reference [3,4,13]. For the phylogenetic analysis the evolutionary history was inferred using RAxML method employing 1000 bootstrap values [42]. P-distance calculation was done in MEGA7 [43]. For HPV genomes with well-established lineages/sublineages we used the respective reference lineages and sublineages in PaVE and Chen et al. [13], whereas for HPVs with no established lineages/sublineages all complete genomes available in NCBI were downloaded for analysis (Supplementary Table S2). Phylogenetic trees were visualized in iTOL5.3 and Dendroscope 3 [44,45]. Plots were constructed in R (scripts available on request).
2.5. Statistical Analysis
Statistical analysis was performed using STATA 14 (College Station, TX, USA). We estimated HPV genotyping concordance by the percentage agreement and Cohen’s kappa (k). Cohen’s kappa values less than zero were classified as no agreement, 0.00–0.19 as poor, 0.20–0.39 as fair, 0.40–0.59 as moderate, 0.60–0.79 as good and 0.80–1.00 as excellent, as proposed in VALGENT [46,47]. Agreement, concordance and discordance were assessed restricting analysis to 28 genotypes included in the Anyplex II HPV28 assay and 729/744 samples with results available by both methods. Fully concordant samples were defined when all genotypes were detected by both Anyplex and NGS. Partially concordant samples were defined if at least one genotypes was shared by the two methods and discordant samples were defined as those where no genotypes were shared.
2.6. Data Access
Complete genomes generated during the study have been submitted to the European Nucleotide Archive (ENA) repository and are available under accession numbers LR861810, LR861811, LR861836–LR861981, LR861983, LR861984, LR861986, LR861987, LR861989, LR861990, LR861992–LR862007, LR862010, LR862011, LR862014, LR862015, LR862017, LR862018, LR862020–LR862086 (Supplementary Table S3). Sequence alignments are included in supplementary files. Accession numbers for genomes downloaded from GenBank are provided in Supplementary Tables S1 and S2.
2.7. Ethical Approval
The study was approved by the Comité National d’Ethique de Recherche (CNER # 201501/02 and 201909/01) and authorized by the Commission Nationale pour la Protection des Données (CNPD 288/2016).
3. Results
3.1. NGS and Anyplex II HPV 28
Overall, HPV positivity was 366/729 (50.2%) by Anyplex and 332/729 (45.5%) by NGS. From 28 genotypes covered by Anyplex, NGS detected 25 (all except HPV11, HPV26, HPV69) plus 41 additional genotypes in 171 (23.5%) samples, of which 43/171 (25.1%) were negative by Anyplex. Anyplex detected 146 genotypes in 97 samples negative by NGS. Overall, Anyplex detected 778 and NGS detected 888 type-specific infections, corresponding to an average of 1.1 and 1.2 genotypes per sample, respectively.
When considering only positive samples, the average number of genotypes detected per sample was 2.1 for Anyplex and 2.7 for NGS. Multiple genotypes were detected in 200/366 (54.6%, range 0–7) of the samples with Anyplex and 204/332 (61.4%, range 0–14) with NGS. Anyplex viral load was significantly associated with the count of reads mapping to HPV by NGS (p < 0.001) (Table 1). The top three most-frequently detected hr genotypes by Anyplex were HPV51 (5.8%), HPV58 (5.7%) and HPV68 (5.3%), while the top three genotypes by NGS were HPV51 (6.1%), HPV56 (4.3%) and HPV66 (3.7%) (Figure 1A). The most-frequently detected lr-HPV by both methods were HPV42 and HPV53 (Figure 1B). HPV67 (3.6%), HPV90 (3.0%), HPV62 (2.6%), HPV89 (2.5%) and HPV87 (2.3%) were the most-frequently observed genotypes detected by NGS (not targeted by Anyplex) (Figure 1C).

Table 1.
Proportion of genotypes detected by next generation sequencing (NGS) stratified by viral load as determined by Anyplex II HPV28.
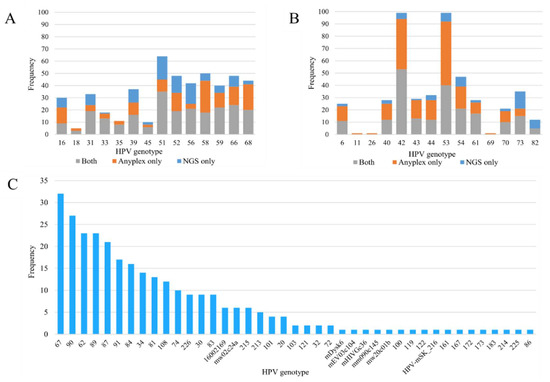
Figure 1.
(A,B) Frequency distribution of human papillomavirus (HPV) genotypes detected by Anyplex II HPV28 and next-generation sequencing: (A) high-risk genotypes, (B) low-risk genotypes. Grey bars indicate genotypes detected by both methods, orange bars indicate genotypes detected by Anyplex II HPV28 only, and blue bars indicate genotypes detected by next-generation sequencing only. (C) Frequency distribution of HPV genotypes detected by next-generation sequencing only.
General agreement for overall HPV positivity of 28 genotypes between the two methods was 80.8% (kappa = 0.616) (good concordance). When restricting the analysis to medium and high viral load results (++/+++), agreement was higher (86.3%) with a kappa value of 0.712 (good concordance). Agreement for 14 hr HPV positivity was 84.8% with kappa value of 0.65 (good concordance). Similarly, when restricting analysis to medium and high viral load (++/+++) agreement for 14 hr-HPV was slightly higher (87.1%, kappa = 0.69) (good concordance) (Table 2). Type-specific agreement is reported in Supplementary Table S4. Among 398/729 (54.6%) HPV positive samples by either method, we observed 115/398 (28.9%) fully concordant samples down to genotype level, also considering infections with multiple genotypes.
Table 2.
Overall agreement for HPV detection between Anyplex II HPV28 and next-generation sequencing.
3.2. HPV Variant Distribution and Lineage Classification
We obtained 232 complete alpha HPV and 11 gamma HPV genomes from 153 samples (Figure 2) spanning 40 different HPV types. The most-frequently obtained complete genomes were HPV51 (9.1%), HPV42 (8.6%), HPV53 (8.6%) and HPV59 (7.4%) (Table 3 and Table 4 and Supplementary Table S3). Phylogenetic trees were created for each isolate and topologies were evaluated to classify our genomes to the existing or novel lineages/sublineages as described before [4]. Reference HPV genomes were considered as the prototype sequence, which were always assigned as lineage A or sublineage A1 (Supplementary Table S2).
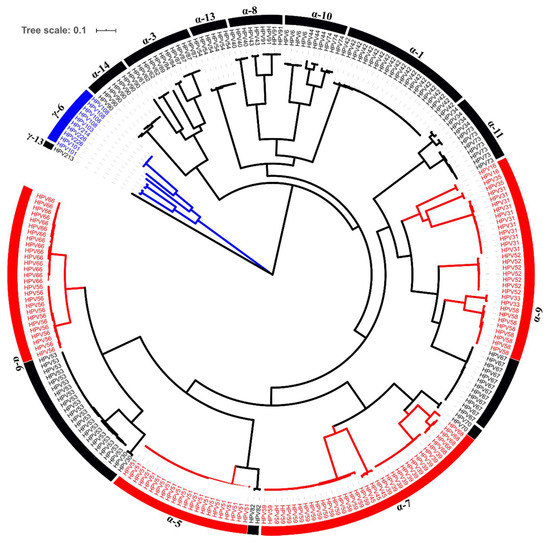
Figure 2.
Phylogenetic tree of 243 HPV isolates. The maximum likelihood tree was constructed using RAxML based on complete HPV genomes. High-risk HPV types are shown in red. Gamma-6 species are colored in blue.
Table 3.
Classification of high-risk complete HPV genomes sequenced in our study.
Table 4.
Classification of low-risk complete HPV genomes sequenced in our study.
Of a total 243 complete genomes, 49 (13 hr and 36 lr-HPV) were classified as belonging to 17 novel lineages and 14 novel sublineages of 20 HPV types including 4 hr and 16 lr-HPV genotypes (Table 3 and Table 4, Figure 3 and Figure 4 and Supplementary Figure S2). The remaining 194 complete genomes clustered with 46 existing lineages and 51 sublineages (Supplementary Figures S3 and S4).
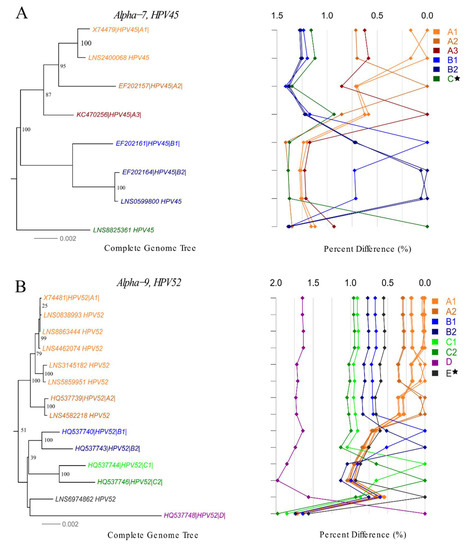
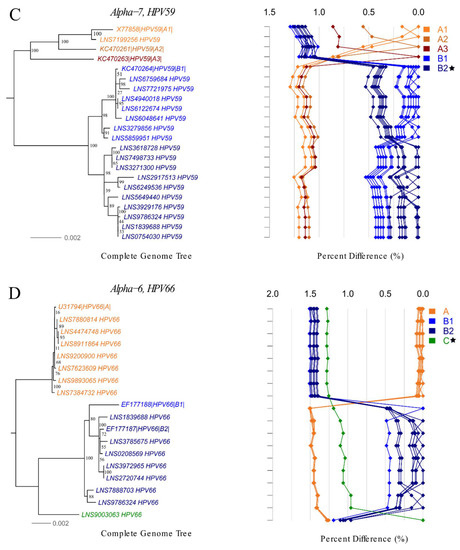
Figure 3.
Phylogenetic trees and pairwise distance comparisons for novel lineages/sublineages of high-risk HPV complete genome variants. Phylogenetic trees were inferred from a global alignment of the complete genome nucleotide sequences of the variants from following HPV genotypes using RAxML: (A) HPV45, (B) HPV52, (C) HPV59, (D) HPV66. A star ( ) indicates novel lineage/sublineage. The phylogenetic topology is shown on the left, pairwise differences for each isolate are shown on the right. Values for each isolate are connected by lines of different colors to distinguish each lineage and sublineage. Genomes from our collection are indicated with LNS identification number.
) indicates novel lineage/sublineage. The phylogenetic topology is shown on the left, pairwise differences for each isolate are shown on the right. Values for each isolate are connected by lines of different colors to distinguish each lineage and sublineage. Genomes from our collection are indicated with LNS identification number.
) indicates novel lineage/sublineage. The phylogenetic topology is shown on the left, pairwise differences for each isolate are shown on the right. Values for each isolate are connected by lines of different colors to distinguish each lineage and sublineage. Genomes from our collection are indicated with LNS identification number.
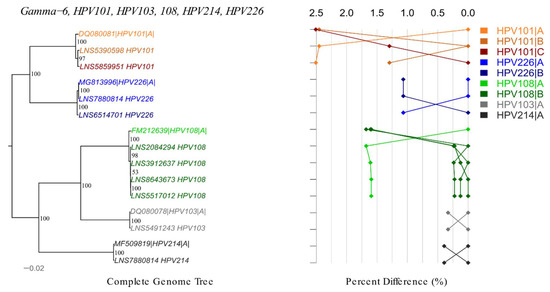
Figure 4.
Phylogenetic tree and pairwise distance comparisons for gamma-6 species. Phylogenetic trees were inferred from a global alignment of the complete genome nucleotide sequences of the variants from following HPV genotypes using RAxML: HPV101, HPV103, HPV108, HPV214, HPV226. The phylogenetic topology is shown on the left, pairwise differences for each isolate are shown on the right. Values for each isolate are connected by lines of different colors to distinguish each lineage and sublineage. Genomes from our collection are indicated with LNS identification number.
3.2.1. High-Risk HPV
In total, 118 complete hr-HPV genomes were assembled, spanning 24 existing lineages and 31 sublineages of 13 hr-HPV types (Supplementary Figure S3A–I). No complete genome of HPV18 could be obtained. We characterized novel lineages of HPV45, HPV52, HPV66 and a novel sublineage of HPV59 (Figure 3A–D). The novel lineage C of HPV45 differed by 0.93–1.35% and 1.38–1.39% from lineages A and B, respectively. The difference between the novel lineage E of HPV52 and other lineages ranged from 0.54% to 1.56%. Our analysis suggests that lineage B of HPV59 can be subdivided into sublineages B1 and B2. The pairwise distance between sublineages B1 and B2 varied from 0.27% to 0.53%. The novel lineage C of HPV66 differed by 1.25–1.41% and 0.96–1.32% from lineages A and B, respectively (Supplementary Table S5 and Figure 3A–D).
3.2.2. Low-Risk HPV
In total, the 114 genomes obtained of 21 lr-HPV types clustered with 22 existing lineages and 20 sublineages (Supplementary Figure S4A–H). According to the pairwise distance and the phylogenetic analysis, we identified 10 novel lineages (HPV34, HPV42, HPV43, HPV54, HPV73, HPV74, HPV84, and HPV90) and 13 sublineages (HPV40, HPV42, HPV43, HPV61, HPV62, HPV67, HPV84, HPV90, and HPV91) (Supplementary Figure S2A–M). The distance between new lineages with reference variants varied between 0.86% and 5.12%, while distance between new sublineages with the nearest variant varied between 0.38% and 0.83% (Supplementary Table S5).
3.2.3. Gamma HPV
We obtained 10 gamma-6 and one gamma-13 complete HPV genomes. In total, seven novel variants were identified including lineages A, B, C of HPV101 and lineages A, B of HPV108 and HPV226 (Figure 4). The difference amongst HPV101 lineages varied from 1.30% to 2.51%. The distance between A and B lineages was 1.59–1.68% for HPV108 and 1.07% for HPV226 (Supplementary Table S6). Variant of HPV213 gamma-13 genotype differed by 0.77% from reference genome.
4. Discussion
Our study showed a high diversity of human papillomavirus in women attending cervical cancer screening. We characterized 243 complete HPV genomes of 40 different genotypes and proposed the classification of novel lineages/sublineages for 4 hr-HPV and 16 lr-HPV genotypes. To the best of our knowledge, this is the first large scale study to present HPV diversity using unbiased NGS-RCA in healthy young women and comparing the analytical performance of a PCR-based genotyping assay with NGS-RCA.
Our findings suggest that the PCR-based the Anyplex assay is superior to the NGS in terms of sensitivity since only 16% of genotypes with low viral load by the Anyplex were concordantly identified by NGS. This is not surprising considering that Anyplex detection is based on type-specific primers, whereas RCA is a random amplification of circular DNA. Nevertheless, NGS detected an additional 110 genotypes compared to the Anyplex PCR. Further, our study suggests that concordance between the two methods increases with higher viral loads. In a recent clinical evaluation of the Anyplex assay, the detection of cervical intraepithelial neoplasia of grade 2 or worse (CIN2+) was associated with medium and high viral loads of the 14 high-risk types only, but not with low viral loads or low-risk genotypes [48]. In our study, only 3.5% participants were diagnosed with abnormal cytology, while we recovered 40% of complete genomes from these samples (Supplementary Table S7). As such, it would be important to validate the NGS approach in a clinical context using established protocol such as VALGENT [46].
In total, we detected 66 different HPV genotypes including 90 lineages/sublineages, of which 27 were novel and not previously described. Although, the diversity of hr-HPV genotypes has been well studied [4,13], we were able to identify novel variants of HPV45, HPV52, HPV59 and HPV66. Further, we proposed the classification of 10 novel lineages and 13 sublineages of lr-HPV which has been less studied. We were not able to obtain any complete HPV18 genomes (only five positive samples by both methods) as ~50% of the study population received HPV vaccine [20,49].
Several NGS-based approaches have been trialed, including RNA-baits, PCR-based and untargeted metagenomics [14,22,26,32,50,51,52]. Due to the high variability of HPV genomes across genera, species and even genotypes, designing primers or baits with acceptable sensitivity in a cost-effective manner is a major challenge. In our study, we found the untargeted rolling-circle amplification approach, initially implemented for HPV sequencing by Meiring [53], to be a very useful tool for characterizing HPV variants and detection. Identification of new variants could be important for adopting PCR-based assays, as we observed substantial discordance in HPV type-specific genotyping of some hr and lr HPVs.
Previous studies applied an RCA-based approach in immune-suppressed individuals, where high viral load would facilitate HPV detection and complete genome recovery. Pastrana et al. reported the discovery of 83 novel HPV types using RCA-NGS with the prior physical enrichment of viral DNA from immunodeficient patients [32]. This method was successfully used for HPV detection and characterization by researchers in South Africa and Brazil in HIV positive populations [14,53]. Results of the study by Wang et al. suggest that RCA-based sequencing is more accurate than the long-PCR template cloning and deep sequencing of HPV58 [54].
In our study, we obtained 243 HPV complete genomes, which increases the number of publicly available complete HPV genomes on GenBank (~2283 complete genomes on April 2020) by approximately 10%. Our dataset provides important genomic insights on HPV variants where currently only a few complete genomes were available, such as HPV42, HPV59, HPV66, HPV67, HPV90, and HPV91.
Interestingly, we also obtained 11 complete HPV genomes of gamma species, with a population prevalence of 3.7% in our population. Our findings are in line with the previous research which found gamma-6 species in cervical samples confirming their mucosal tropism [26,55,56,57,58]. Although gamma-6 pathogenicity is not well described, HPV108 could induce dysplasia in organotypic keratinocytes [57]. In our study, gamma-6 genotypes were present independently only in four samples, whereas other 23 samples were co-infected with other alpha HPVs. As gamma-6 HPV genotypes lack the E6 oncogene, they may have a lower oncogenic potential [56].
One of the limitations of our study was the absence of negative and positive control during the sequencing, in order to account for the cross contamination and index hopping. Therefore, our detection criteria might produce some false positive results. Nevertheless, only 3% of the samples were defined as HPV positive by NGS with at least one concordantly mapping read pair to the reference genome. Moreover, when considering more stringent detection criteria for HPV positivity by NGS (mapping coverage of 1000 bp), the agreement between methods did not change significantly (Supplementary Table S8). The NGS detection threshold of concordant read pairs might have a higher impact on genotyping results, rather than HPV positivity. We accounted for potential PCR or sequencing errors during the assembly process by stringent quality controls and we excluded three genomes from the analysis due to the presence of chimeric contigs.
The major advantage of the NGS method is that it requires no prior knowledge of which HPV genotypes are present in the samples. Overall, we were able to detect 51 different HPV genotypes including 26, which are not detectable by the Anyplex assay. As the majority of these HPV types belong to IARC Group 3 (not classifiable as to its carcinogenicity to humans) [5], our findings currently do not have direct clinical implications in the context of cervical cancer screening. Nevertheless, this approach is certainly useful from a population and evolutionary biology perspective and may have potential to address new disease etiology in the future.
Supplementary Materials
The following are available online at https://www.mdpi.com/1999-4915/12/12/1437/s1, Figure S1A. Flow chart illustrating generation of blind assembly input data. Bar chart illustrating the generation of the blind assembly input data. The bar labels show the average single read count over 744 samples. For HG (Human Genome) we used genome version GRCh38. The average input size per sample for a blind genome assembly is 93,675 reads, which corresponds on average to ~14% of the raw sample size. Figure S1B. Flow chart illustrating generation of bowtie assembly input data. Bar chart illustrating the generation of the bowtie assembly input data. The lower graph shows the generation of the genome assembly input data only considering HPV positive samples. Figure S2A–M. Phylogenetic trees and pairwise distance comparisons for novel lineages/sublineages of low-risk HPV complete genome variants. Phylogenetic trees were inferred from a global alignment of the complete genome nucleotide sequences of the variants from following low risk HPV genotypes using RAxML: (A) HPV34, (B) HPV40, (C) HPV42, (D) HPV43, (E) HPV54, (F) HPV61, (G) HPV62, (H) HPV67, (I) HPV73, (J) HPV74, (K) HPV84, (L) HPV90, (M) HPV91. A star () indicates novel lineage/sublineage. The phylogenetic trees are shown on the left, pairwise differences for each isolate are shown on the right. Values for each isolate are connected by lines of different colors to distinguish each lineage and sublineage. Genomes from our collection are indicated with LNS identification number. Figure S3A–H. Phylogenetic trees and pairwise distance comparisons of high-risk HPV complete genome variants. Phylogenetic trees were inferred from a global alignment of the complete genome nucleotide sequences of the variants from following high risk HPV genotypes using RAxML: (A) HPV16, (B) HPV31, (C) HPV33, (D) HPV35, (E) HPV39, (F) HPV51, (G) HPV56, (H) HPV58, (I) 68. The phylogenetic trees are shown on the left, pairwise differences for each isolate are shown on the right. Values for each isolate are connected by lines of different colors to distinguish each lineage and sublineage. Genomes from our collection are indicated with LNS identification number. Figure S4A–H. Phylogenetic trees and pairwise distance comparisons of low-risk complete genome variants. Phylogenetic trees were inferred from a global alignment of the complete genome nucleotide sequences of the variants from following low risk HPV genotypes using RAxML: (A) HPV6, (B) HPV30, (C) HPV44, (D) HPV53, (E) HPV70, (F) HPV82, (G) HPV87, (H) HPV89. Phylogenetic trees are shown on the left, pairwise differences for each isolate are shown on the right. Values for each isolate are connected by lines of different colors to distinguish each lineage and sublineage. Genomes from our collection are indicated with LNS identification number.
) indicates novel lineage/sublineage. The phylogenetic trees are shown on the left, pairwise differences for each isolate are shown on the right. Values for each isolate are connected by lines of different colors to distinguish each lineage and sublineage. Genomes from our collection are indicated with LNS identification number. Figure S3A–H. Phylogenetic trees and pairwise distance comparisons of high-risk HPV complete genome variants. Phylogenetic trees were inferred from a global alignment of the complete genome nucleotide sequences of the variants from following high risk HPV genotypes using RAxML: (A) HPV16, (B) HPV31, (C) HPV33, (D) HPV35, (E) HPV39, (F) HPV51, (G) HPV56, (H) HPV58, (I) 68. The phylogenetic trees are shown on the left, pairwise differences for each isolate are shown on the right. Values for each isolate are connected by lines of different colors to distinguish each lineage and sublineage. Genomes from our collection are indicated with LNS identification number. Figure S4A–H. Phylogenetic trees and pairwise distance comparisons of low-risk complete genome variants. Phylogenetic trees were inferred from a global alignment of the complete genome nucleotide sequences of the variants from following low risk HPV genotypes using RAxML: (A) HPV6, (B) HPV30, (C) HPV44, (D) HPV53, (E) HPV70, (F) HPV82, (G) HPV87, (H) HPV89. Phylogenetic trees are shown on the left, pairwise differences for each isolate are shown on the right. Values for each isolate are connected by lines of different colors to distinguish each lineage and sublineage. Genomes from our collection are indicated with LNS identification number.Author Contributions
A.L. and J.M. designed and conducted the study. A.L., A.W.-B. and J.M. designed NGS pipelines. A.W.-B. implemented the NGS pipelines and contributed to methods part of the manuscript. A.L. analyzed sequence data, performed the statistical analysis, interpreted findings and drafted the manuscript. I.K. and Z.C. contributed to generating figures. J.T. performed HPV genotyping and next generation sequencing. M.F.’s lab performed cytological testing. P.P. led recruitment at the Planning Familial. M.A., F.M., and S.W., advised and critically reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Fonds National de la Recherche Luxembourg [FNR grant no 14/BM/7932582 and PRIDE17/11823097]. M.A. was supported by the RISCC Network [grant no. 8478459], funded by the Horizon 2020 Programme for Research and Innovation of the European Commission (Brussels, Belgium).
Acknowledgments
We thank doctors at the Planning Familial (Sandrine Casarotto, Ulla Heckert-Hauck, Christine Kalonji, Jacqueline Klein-Zimmermann, Brigitte Marchand, Anne-Laure Ricard-Michaux) as well the private gynecologists (Annick Aerts, Jean-Marc Billion, Pascale George, Margareta Kirsch, Isabelle Meyer) for study recruitment and sample collection. We thank Seegene for providing laboratory equipment for the duration of the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Einstein, M.H.; Schiller, J.T.; Viscidi, R.P.; Strickler, H.D.; Coursaget, P.; Tan, T.; Halsey, N.; Jenkins, D. Clinician’s guide to human papillomavirus immunology: Knowns and unknowns. Lancet Infect. Dis. 2009, 9, 347–356. [Google Scholar] [CrossRef]
- de Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef]
- Chan, P.K.; Zhang, C.; Park, J.S.; Smith-McCune, K.K.; Palefsky, J.M.; Giovannelli, L.; Coutlee, F.; Hibbitts, S.; Konno, R.; Settheetham-Ishida, W.; et al. Geographical distribution and oncogenic risk association of human papillomavirus type 58 E6 and E7 sequence variations. Int. J. Cancer 2013, 132, 2528–2536. [Google Scholar] [CrossRef]
- Chen, Z.; Schiffman, M.; Herrero, R.; Desalle, R.; Anastos, K.; Segondy, M.; Sahasrabuddhe, V.V.; Gravitt, P.E.; Hsing, A.W.; Burk, R.D. Evolution and taxonomic classification of human papillomavirus 16 (HPV16)-related variant genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. PLoS ONE 2011, 6, e20183. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; Ghissassi, F.E.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Arbyn, M.; Tommasino, M.; Depuydt, C.; Dillner, J. Are 20 human papillomavirus types causing cervical cancer? J. Pathol. 2014, 234, 431–435. [Google Scholar] [CrossRef]
- Xi, L.F.; Kiviat, N.B.; Hildesheim, A.; Galloway, D.A.; Wheeler, C.M.; Ho, J.; Koutsky, L.A. Human papillomavirus type 16 and 18 variants: Race-related distribution and persistence. J. Natl. Cancer Inst. 2006, 98, 1045–1052. [Google Scholar] [CrossRef]
- Xi, L.F.; Schiffman, M.; Koutsky, L.A.; Hulbert, A.; Lee, S.K.; Defilippis, V.; Shen, Z.; Kiviat, N.B. Association of human papillomavirus type 31 variants with risk of cervical intraepithelial neoplasia grades 2–3. Int. J. Cancer 2012, 131, 2300–2307. [Google Scholar] [CrossRef]
- Mirabello, L.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, Z.; Wentzensen, N.; Zhang, X.; Yu, K.; Yang, Q.; Mitchell, J.; et al. HPV16 Sublineage Associations With Histology-Specific Cancer Risk Using HPV Whole-Genome Sequences in 3200 Women. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Godínez, J.M.; Heideman, D.A.M.; Gheit, T.; Alemany, L.; Snijders, P.J.F.; Tommasino, M.; Meijer, C.J.L.M.; de Sanjosé, S.; Bosch, F.X.; Bravo, I.G. Differential presence of Papillomavirus variants in cervical cancer: An analysis for HPV33, HPV45 and HPV58. Infect. Genet. Evol. 2013, 13, 96–104. [Google Scholar] [CrossRef]
- Nicolás-Párraga, S.; Gandini, C.; Pimenoff, V.N.; Alemany, L.; de Sanjosé, S.; Bosch, F.X.; Bravo, I.G.; The RIS HPV TT and HPV VVAP study groups. HPV16 variants distribution in invasive cancers of the cervix, vulva, vagina, penis, and anus. Cancer Med. 2016, 5, 2909–2919. [Google Scholar] [CrossRef] [PubMed]
- Xi, L.F.; Schiffman, M.; Koutsky, L.A.; Hughes, J.P.; Winer, R.L.; Mao, C.; Hulbert, A.; Lee, S.K.; Shen, Z.; Kiviat, N.B. Lineages of oncogenic human papillomavirus types other than type 16 and 18 and risk for cervical intraepithelial neoplasia. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Schiffman, M.; Herrero, R.; DeSalle, R.; Anastos, K.; Segondy, M.; Sahasrabuddhe, V.V.; Gravitt, P.E.; Hsing, A.W.; Chan, P.K.S.; et al. Classification and evolution of human papillomavirus genome variants: Alpha-5 (HPV26, 51, 69, 82), Alpha-6 (HPV30, 53, 56, 66), Alpha-11 (HPV34, 73), Alpha-13 (HPV54) and Alpha-3 (HPV61). Virology 2018, 516, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, J.D.; Alves, B.M.; Prellwitz, I.M.; Furtado, C.; Meyrelles, A.R.; Machado, E.S.; Seuanez, H.N.; Soares, M.A.; Soares, E.A. Identification of novel human papillomavirus lineages and sublineages in HIV/HPV-coinfected pregnant women by next-generation sequencing. Virology 2016, 493, 202–208. [Google Scholar] [CrossRef]
- Jelen, M.M.; Chen, Z.; Kocjan, B.J.; Hošnjak, L.; Burt, F.J.; Chan, P.K.S.; Chouhy, D.; Combrinck, C.E.; Estrade, C.; Fiander, A.; et al. Global Genomic Diversity of Human Papillomavirus 11 Based on 433 Isolates and 78 Complete Genome Sequences. J. Virol. 2016, 90, 5503–5513. [Google Scholar] [CrossRef]
- Poljak, M.; Valenčak, A.O.; Domjanič, G.G.; Xu, L.; Arbyn, M. Commercially available molecular tests for human papillomaviruses: A global overview. Clin. Microbiol. Infect. 2020. [Google Scholar] [CrossRef]
- Ostrbenk, A.; Xu, L.; Arbyn, M.; Poljak, M. Clinical and Analytical Evaluation of the Anyplex II HPV HR Detection Assay within the VALGENT-3 Framework. J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef]
- Hesselink, A.T.; Sahli, R.; Berkhof, J.; Snijders, P.J.; van der Salm, M.L.; Agard, D.; Bleeker, M.C.; Heideman, D.A. Clinical validation of Anyplex II HPV HR Detection according to the guidelines for HPV test requirements for cervical cancer screening. J. Clin. Virol. 2016, 76, 36–39. [Google Scholar] [CrossRef]
- Latsuzbaia, A.; Tapp, J.; Nguyen, T.; Fischer, M.; Arbyn, M.; Weyers, S.; Mossong, J. Analytical performance evaluation of Anyplex II HPV28 and Euroarray HPV for genotyping of cervical samples. Diagn. Microbiol. Infect. Dis. 2016, 85, 318–322. [Google Scholar] [CrossRef]
- Latsuzbaia, A.; Arbyn, M.; Tapp, J.; Fischer, M.; Weyers, S.; Pesch, P.; Mossong, J. Effectiveness of bivalent and quadrivalent human papillomavirus vaccination in Luxembourg. Cancer Epidemiol. 2019, 63, 101593. [Google Scholar] [CrossRef]
- Arroyo, L.S.; Smelov, V.; Bzhalava, D.; Eklund, C.; Hultin, E.; Dillner, J. Next generation sequencing for human papillomavirus genotyping. J. Clin. Virol. 2013, 58, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Muhr, L.S.A.; Bzhalava, D.; Lagheden, C.; Eklund, C.; Johansson, H.; Forslund, O.; Dillner, J.; Hultin, E. Does human papillomavirus-negative condylomata exist? Virology 2015, 485, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Galati, L.; Brancaccio, R.N.; Robitaille, A.; Cuenin, C.; Luzi, F.; Fiorucci, G.; Chiantore, M.V.; Marascio, N.; Matera, G.; Liberto, M.C.; et al. Detection of human papillomaviruses in paired healthy skin and actinic keratosis by next generation sequencing. Papillomavirus Res. 2020, 9, 100196. [Google Scholar] [CrossRef] [PubMed]
- Murahwa, A.T.; Meiring, T.L.; Mbulawa, Z.Z.A.; Williamson, A.-L. Complete Genome Sequences of Four Novel Human Gammapapillomavirus Types, HPV-219, HPV-220, HPV-221, and HPV-222, Isolated from Penile Skin Swabs from South African Men. Genome Announc. 2018, 6, e00584-18. [Google Scholar] [CrossRef] [PubMed]
- Kocjan, B.J.; Steyer, A.; Sagadin, M.; Hosnjak, L.; Poljak, M. Novel human papillomavirus type 174 from a cutaneous squamous cell carcinoma. Genome Announc. 2013, 1, e00445-13. [Google Scholar] [CrossRef]
- Latsuzbaia, A.; Arbyn, M.; Dutta, S.; Fischer, M.; Gheit, T.; Tapp, J.; Tommasino, M.; Weyers, S.; Mossong, J. Complete Genome Sequence of a Novel Human Gammapapillomavirus Isolated from a Cervical Swab in Luxembourg. Genome Announc. 2018, 6. [Google Scholar] [CrossRef]
- Van Doorslaer, K.; Li, Z.; Xirasagar, S.; Maes, P.; Kaminsky, D.; Liou, D.; Sun, Q.; Kaur, R.; Huyen, Y.; McBride, A.A. The Papillomavirus Episteme: A major update to the papillomavirus sequence database. Nucleic Acids Res. 2017, 45, D499–D506. [Google Scholar] [CrossRef]
- Mühr, L.S.A.; Eklund, C.; Dillner, J. Towards quality and order in human papillomavirus research. Virology 2018, 519, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.R.; Cai, Y.C.; Giesler, T.L.; Farchaus, J.W.; Sundaram, S.T.; Ortiz-Rivera, M.; Hosta, L.P.; Hewitt, P.L.; Mamone, J.A.; Palaniappan, C.; et al. TempliPhi, phi29 DNA polymerase based rolling circle amplification of templates for DNA sequencing. Biotechniques 2002, 32, 44–47. [Google Scholar] [CrossRef]
- Rector, A.; Tachezy, R.; Van Ranst, M. A sequence-independent strategy for detection and cloning of circular DNA virus genomes by using multiply primed rolling-circle amplification. J. Virol. 2004, 78, 4993–4998. [Google Scholar] [CrossRef] [PubMed]
- Murahwa, A.T.; Meiring, T.L.; Mbulawa, Z.Z.A.; Williamson, A.L. Discovery, characterisation and genomic variation of six novel Gammapapillomavirus types from penile swabs in South Africa. Papillomavirus Res. 2019, 7, 102–111. [Google Scholar] [CrossRef]
- Pastrana, D.V.; Peretti, A.; Welch, N.L.; Borgogna, C.; Olivero, C.; Badolato, R.; Notarangelo, L.D.; Gariglio, M.; FitzGerald, P.C.; McIntosh, C.E.; et al. Metagenomic Discovery of 83 New Human Papillomavirus Types in Patients with Immunodeficiency. mSphere 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, R.N.; Robitaille, A.; Dutta, S.; Rollison, D.E.; Fischer, N.; Grundhoff, A.; Tommasino, M.; Gheit, T. Complete Genome Sequence of a Novel Human Gammapapillomavirus Isolated from Skin. Genome Announc. 2017, 5, e00833-17. [Google Scholar] [CrossRef] [PubMed]
- Latsuzbaia, A.; Hebette, G.; Fischer, M.; Arbyn, M.; Weyers, S.; Vielh, P.; Schmitt, F.; Mossong, J. Introduction of liquid-based cytology and human papillomavirus testing in cervical cancer screening in Luxembourg. Diagn. Cytopathol. 2017, 45, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Brestovac, B.; Wong, M.E.; Costantino, P.S.; Groth, D. A rapid DNA extraction method suitable for human papillomavirus detection. J. Med. Virol. 2014, 86, 653–657. [Google Scholar] [CrossRef]
- Jung, S.; Lee, B.; Lee, K.N.; Kim, Y.; Oh, E.J. Clinical Validation of Anyplex II HPV HR Detection Test for Cervical Cancer Screening in Korea. Arch. Pathol. Lab. Med. 2016, 140, 276–280. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 15 November 2018).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Shean, R.C.; Makhsous, N.; Stoddard, G.D.; Lin, M.J.; Greninger, A.L. VAPiD: A lightweight cross-platform viral annotation pipeline and identification tool to facilitate virus genome submissions to NCBI GenBank. BMC Bioinform. 2019, 20, 48. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef]
- Arbyn, M.; Depuydt, C.; Benoy, I.; Bogers, J.; Cuschieri, K.; Schmitt, M.; Pawlita, M.; Geraets, D.; Heard, I.; Gheit, T.; et al. VALGENT: A protocol for clinical validation of human papillomavirus assays. J. Clin. Virol. 2016, 76, S14–S21. [Google Scholar] [CrossRef] [PubMed]
- Landis, J.R.; Koch, G.G. The measurement of observer agreement for categorical data. Biometrics 1977, 33, 159–174. [Google Scholar] [CrossRef]
- Baasland, I.; Romundstad, P.R.; Eide, M.L.; Jonassen, C.M. Clinical performance of Anyplex II HPV28 by human papillomavirus type and viral load in a referral population. PLoS ONE 2019, 14, e0210997. [Google Scholar] [CrossRef]
- Latsuzbaia, A.; Arbyn, M.; Weyers, S.; Mossong, J. Human papillomavirus vaccination coverage in Luxembourg—Implications of lowering and restricting target age groups. Vaccine 2018, 36, 2411–2416. [Google Scholar] [CrossRef]
- Park, Y.; Lee, E.; Choi, J.; Jeong, S.; Kim, H.S. Comparison of the Abbott RealTime High-Risk Human Papillomavirus (HPV), Roche Cobas HPV, and Hybrid Capture 2 assays to direct sequencing and genotyping of HPV DNA. J. Clin. Microbiol. 2012, 50, 2359–2365. [Google Scholar] [CrossRef]
- Li, T.; Unger, E.R.; Batra, D.; Sheth, M.; Steinau, M.; Jasinski, J.; Jones, J.; Rajeevan, M.S. Universal Human Papillomavirus Typing Assay: Whole-Genome Sequencing following Target Enrichment. J. Clin. Microbiol. 2017, 55, 811–823. [Google Scholar] [CrossRef]
- Li, T.; Unger, E.R.; Rajeevan, M.S. Universal human papillomavirus typing by whole genome sequencing following target enrichment: Evaluation of assay reproducibility and limit of detection. BMC Genom. 2019, 20, 231. [Google Scholar] [CrossRef]
- Meiring, T.L.; Salimo, A.T.; Coetzee, B.; Maree, H.J.; Moodley, J.; Hitzeroth, I.I.; Freeborough, M.J.; Rybicki, E.P.; Williamson, A.L. Next-generation sequencing of cervical DNA detects human papillomavirus types not detected by commercial kits. Virol. J. 2012, 9, 164. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Ni, T.; Xie, X.; Zhu, J.; Zheng, Z.-M. Genome sequencing accuracy by RCA-seq versus long PCR template cloning and sequencing in identification of human papillomavirus type 58. Cell Biosci. 2014, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Schiffman, M.; Herrero, R.; Desalle, R.; Burk, R.D. Human papillomavirus (HPV) types 101 and 103 isolated from cervicovaginal cells lack an E6 open reading frame (ORF) and are related to gamma-papillomaviruses. Virology 2007, 360, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; McBride, A.A. Molecular archeological evidence in support of the repeated loss of a papillomavirus gene. Sci. Rep. 2016, 6, 33028. [Google Scholar] [CrossRef] [PubMed]
- Nobre, R.J.; Herraez-Hernandez, E.; Fei, J.W.; Langbein, L.; Kaden, S.; Grone, H.J.; de Villiers, E.M. E7 oncoprotein of novel human papillomavirus type 108 lacking the E6 gene induces dysplasia in organotypic keratinocyte cultures. J. Virol. 2009, 83, 2907–2916. [Google Scholar] [CrossRef]
- Ameur, A.; Meiring, T.L.; Bunikis, I.; Haggqvist, S.; Lindau, C.; Lindberg, J.H.; Gustavsson, I.; Mbulawa, Z.Z.; Williamson, A.L.; Gyllensten, U. Comprehensive profiling of the vaginal microbiome in HIV positive women using massive parallel semiconductor sequencing. Sci. Rep. 2014, 4, 4398. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).