Comparison of CRISPR and Marker-Based Methods for the Engineering of Phage T7

 , ,
, ,  , and
, and 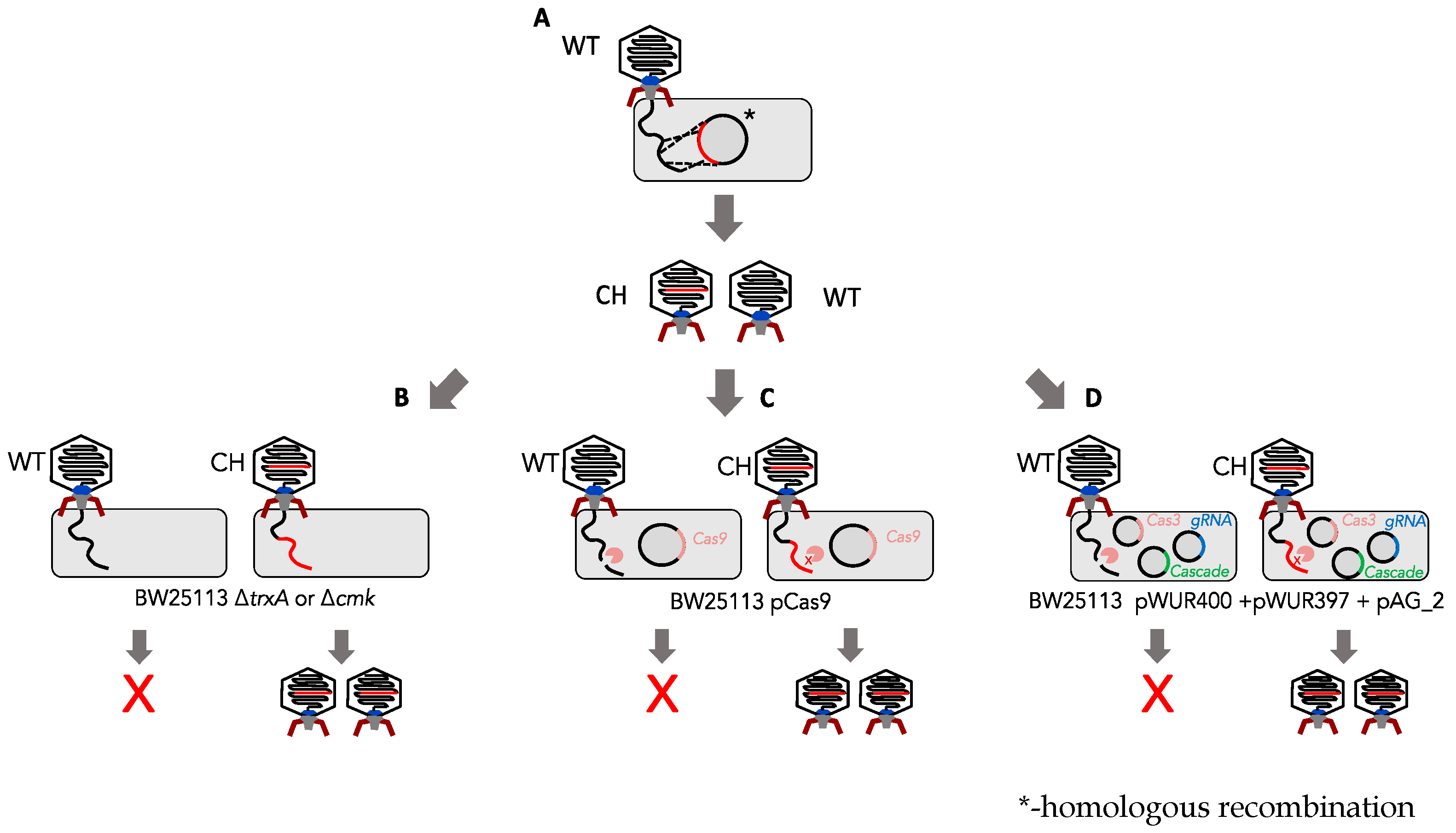{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Phages
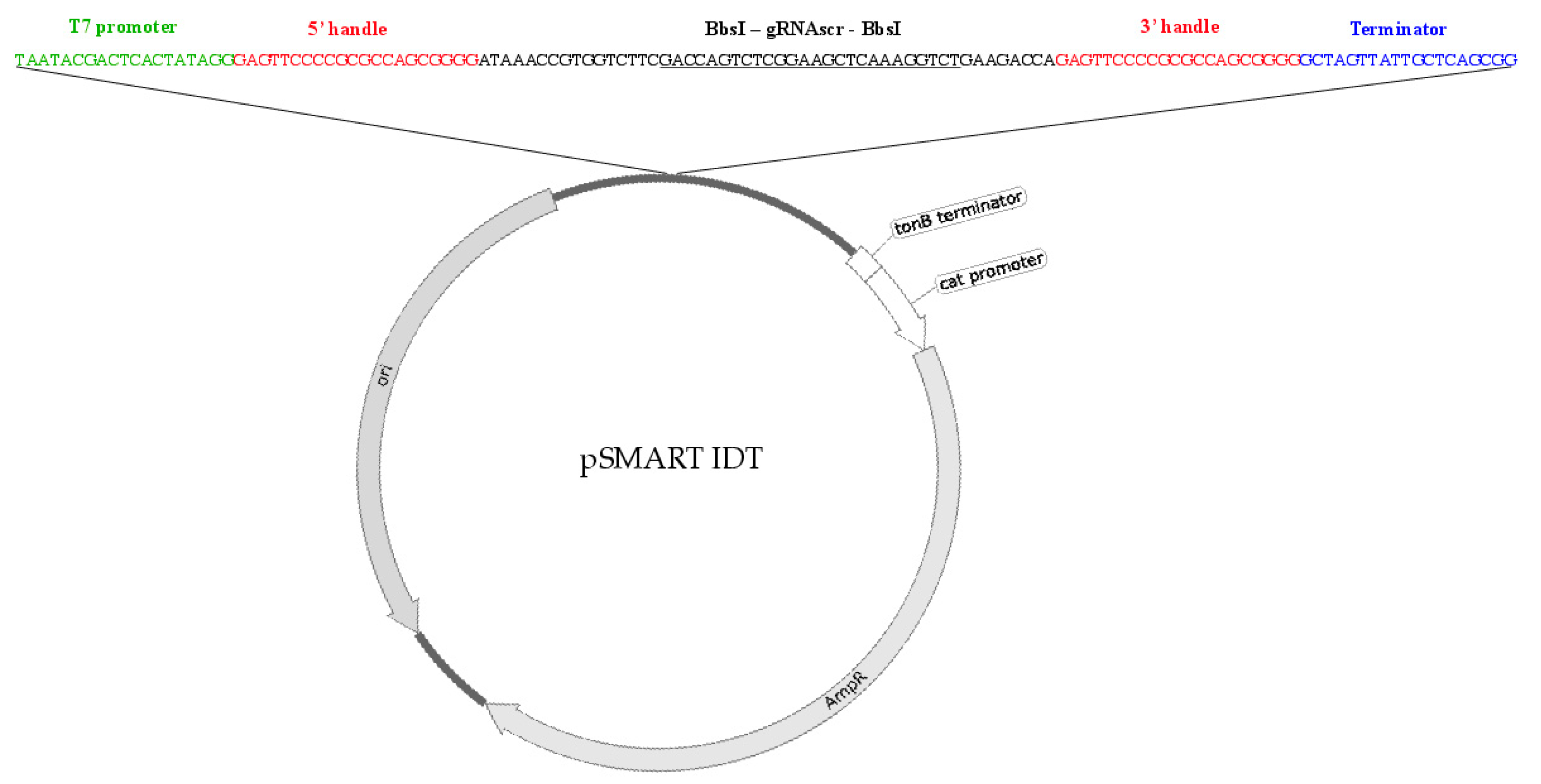
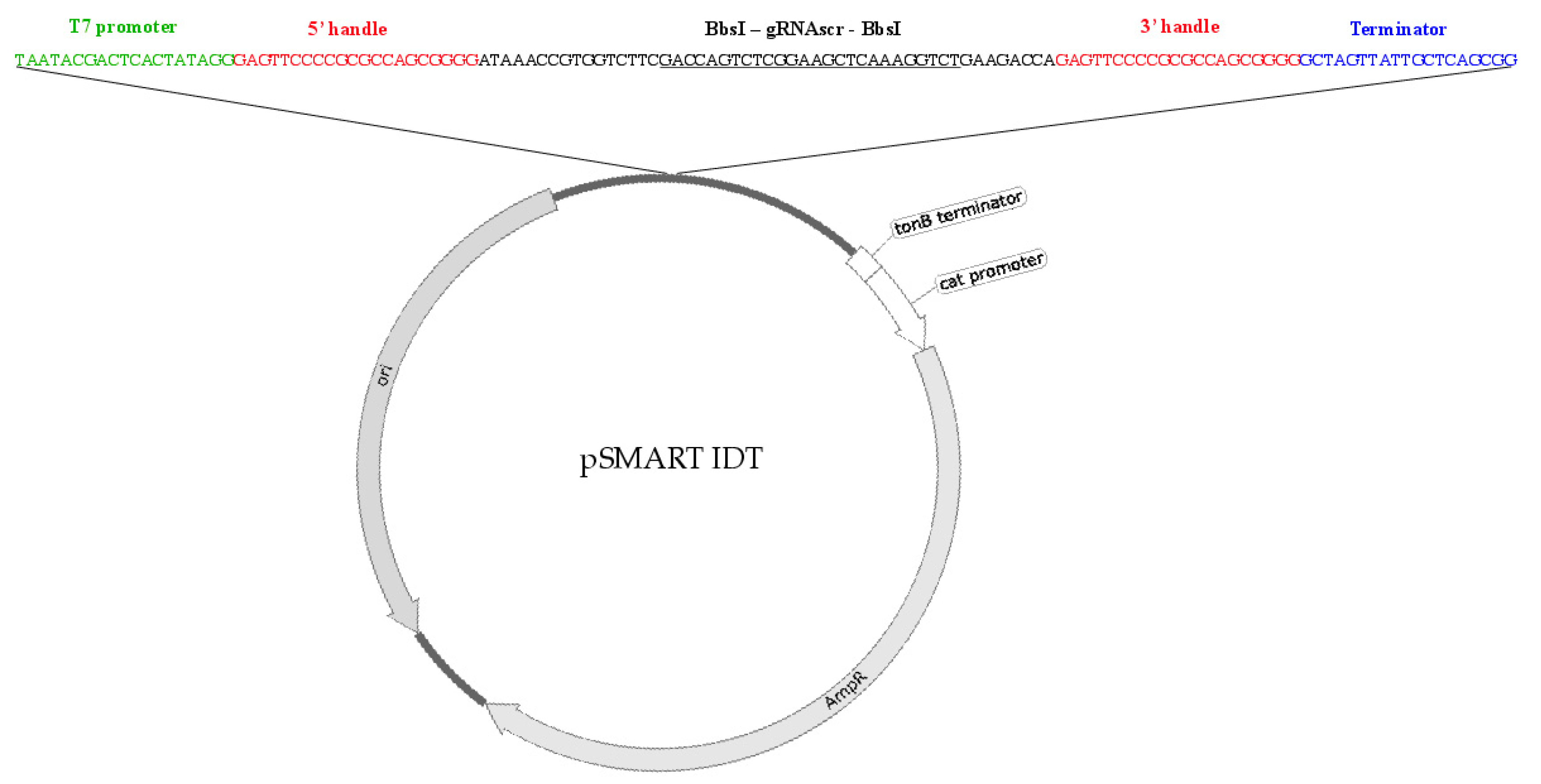
2.2. Type I CRISPR gRNA Delivery Vector Component/Vector Design
2.3. Type I CRISPR gRNA Design
2.4. Type II CRISPR gRNA Design
2.5. Golden Gate Cloning
2.6. PCR
2.7. Gibson Assembly
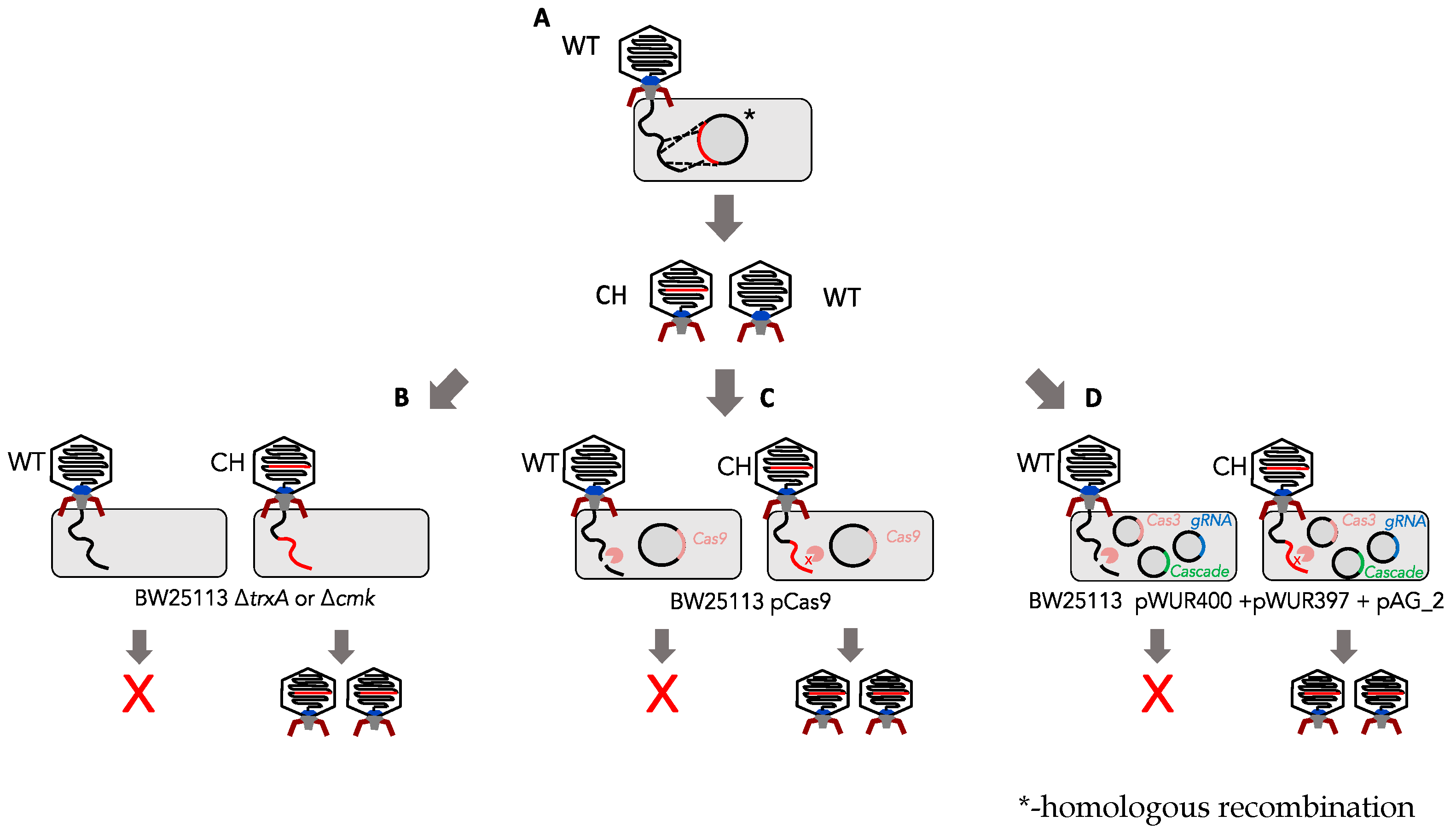
2.8. Homologous Recombination and In Trans Complementation
2.9. Confirmation of Phage Mutants
2.10. Type I and Type II CRISPR Screening Assays
3. Results
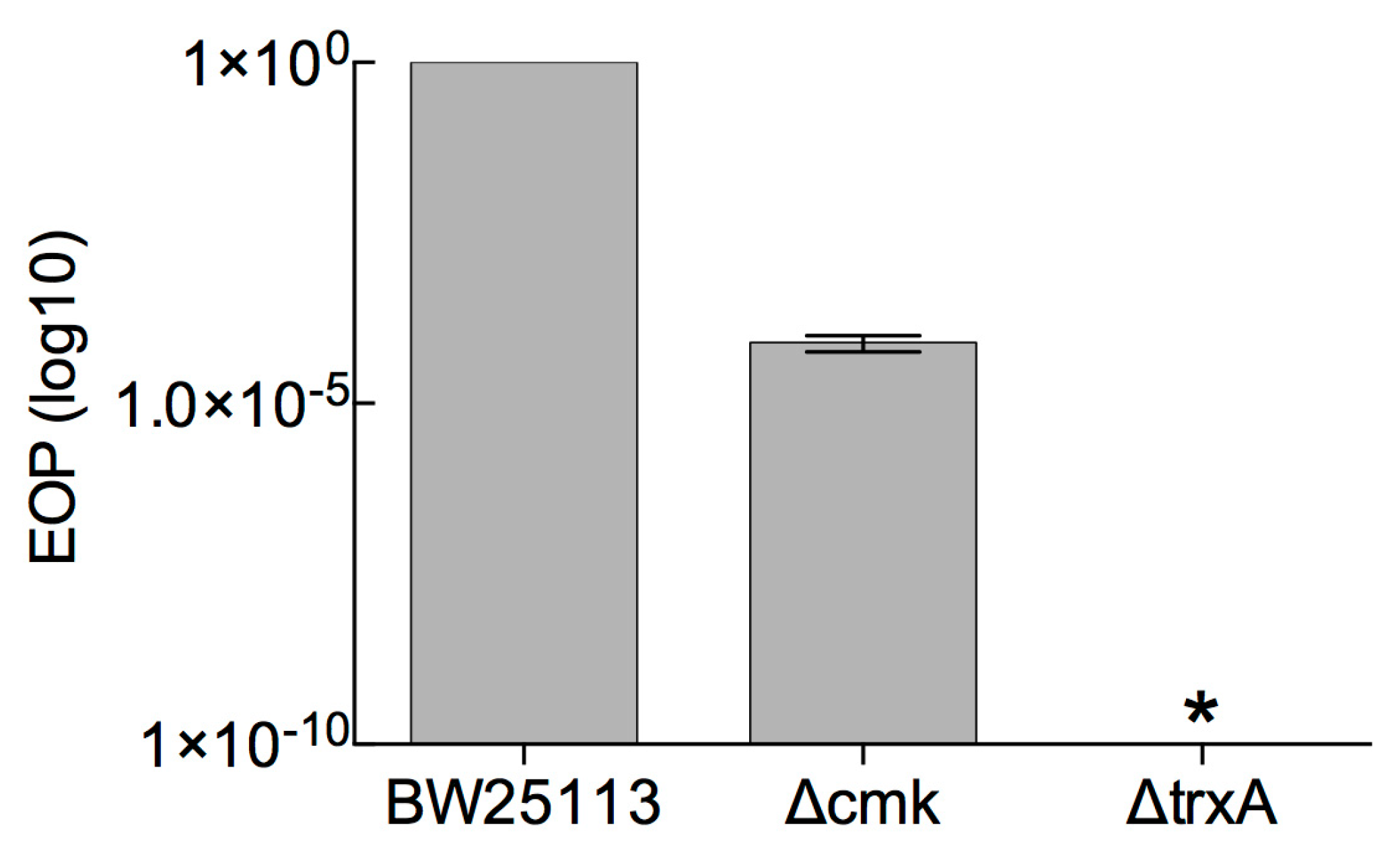
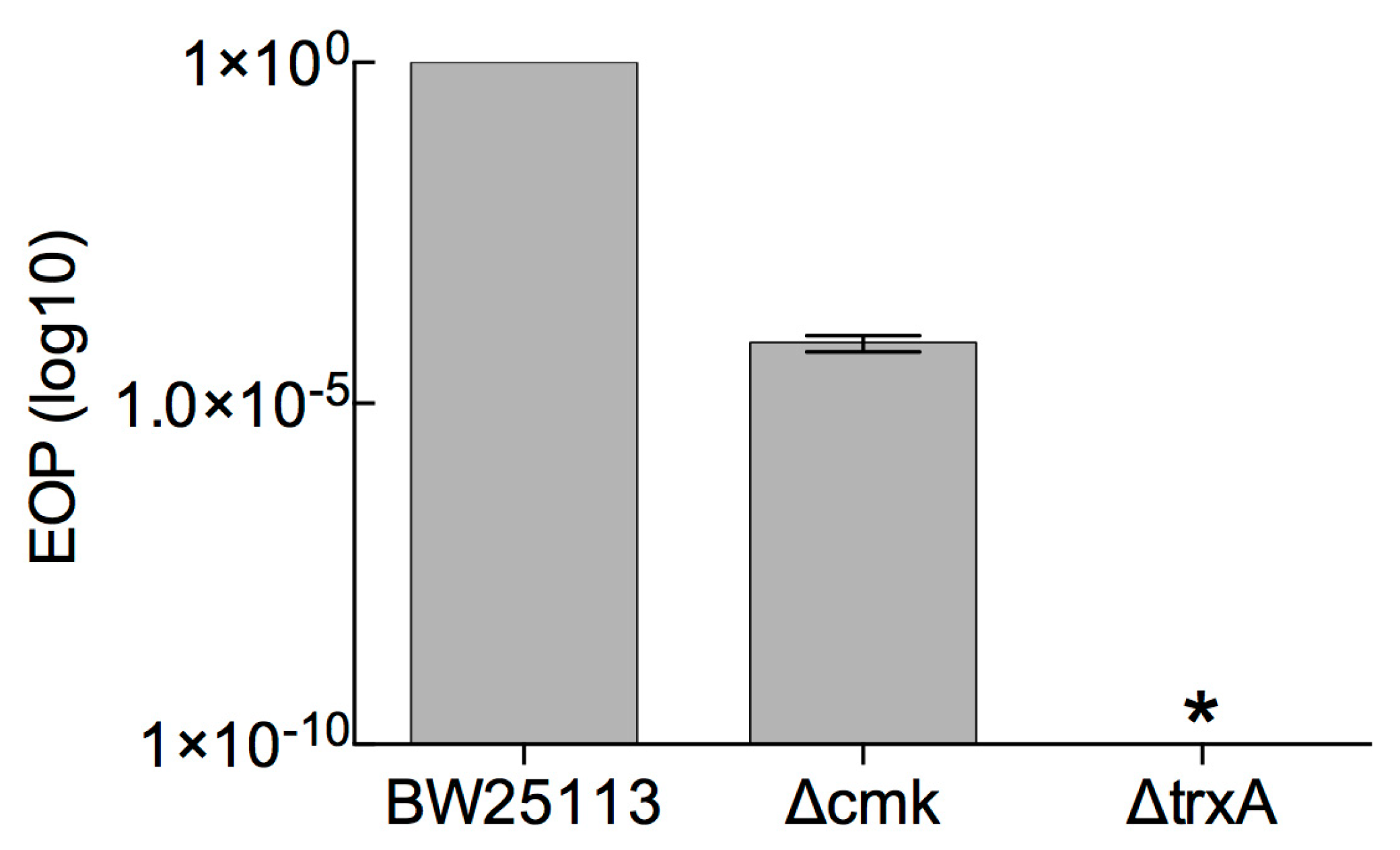
3.1. Determination of Marker-Based (cmk and trxA) Selection Efficiency against Phage T7
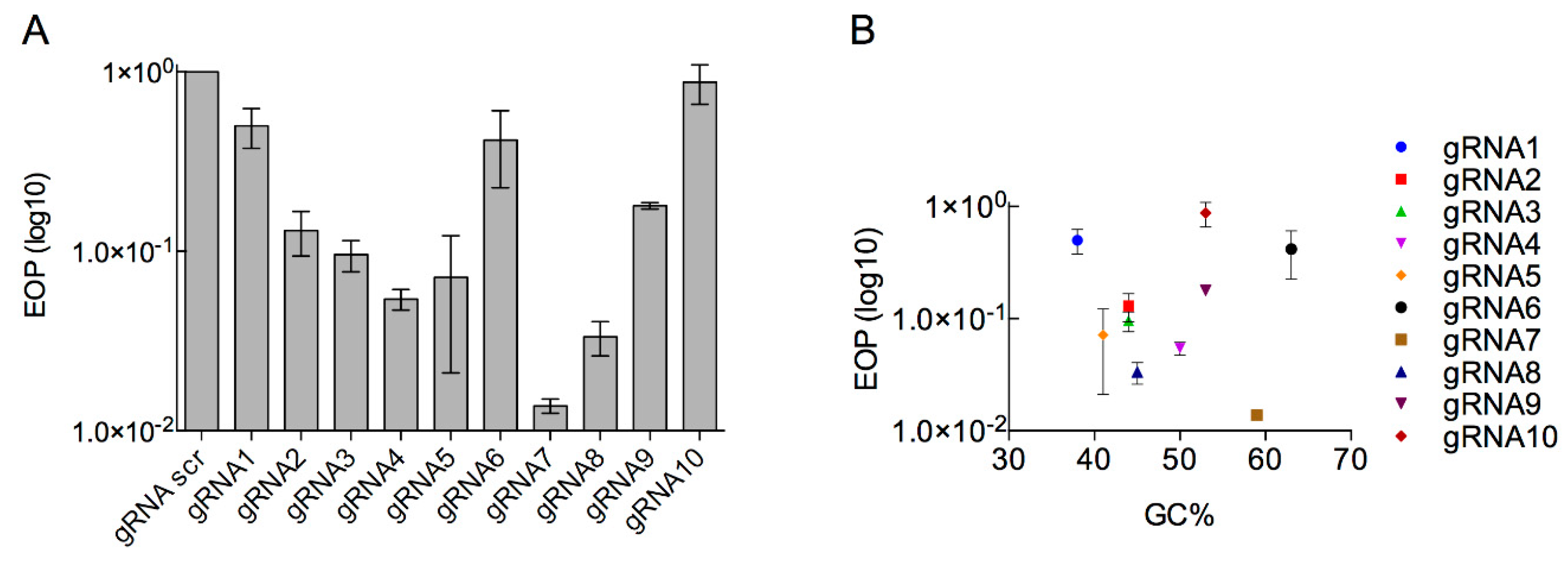
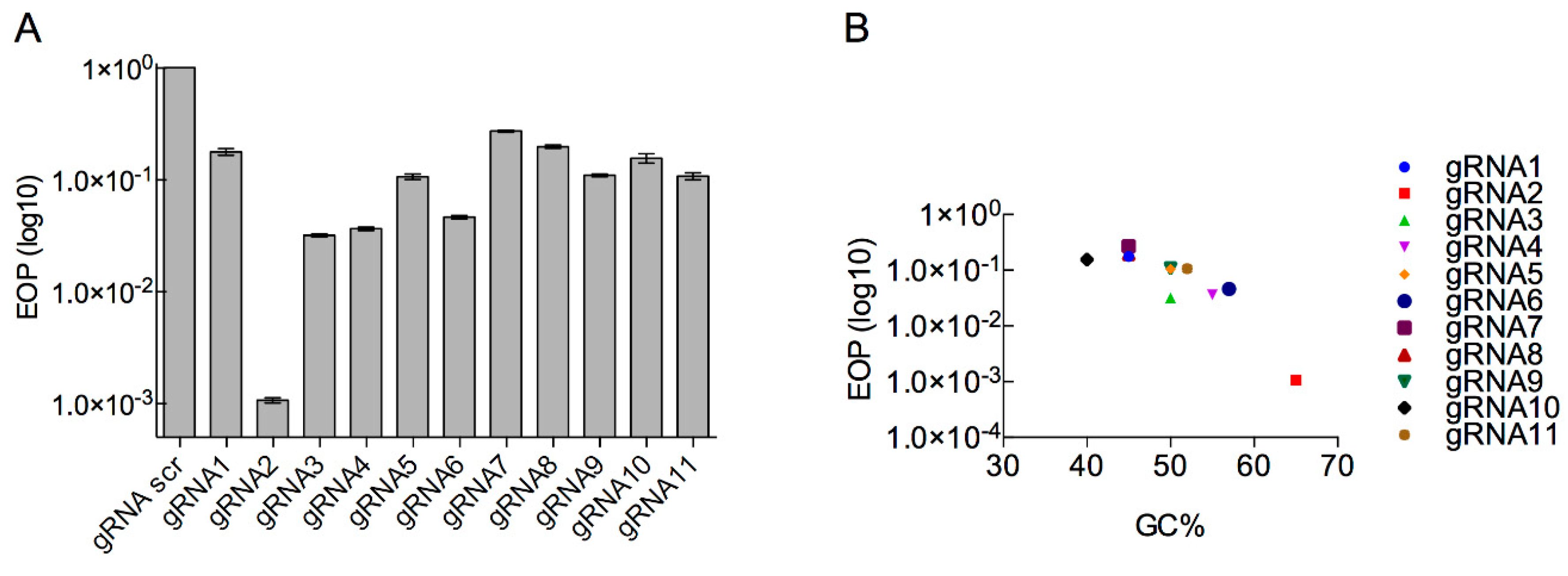
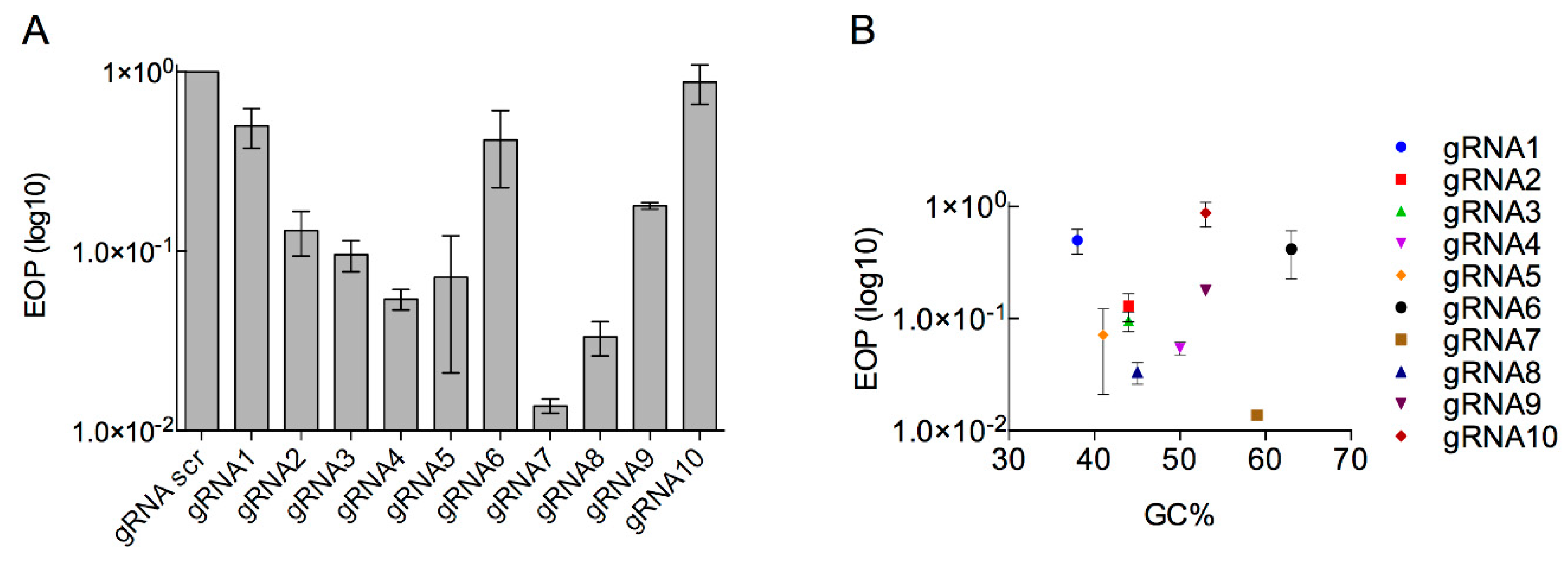
3.2. Determination of the Efficiency of Type I CRISPR for Selecting against Wild-Type Phage T7
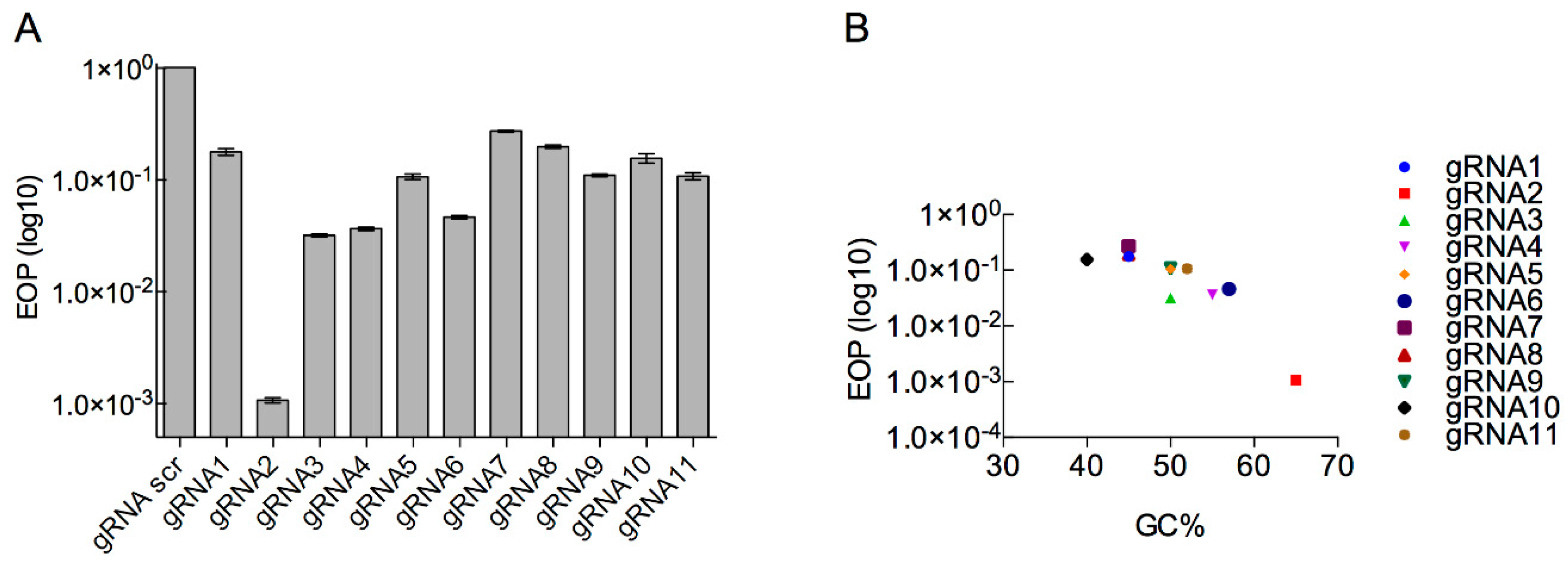
3.3. Determination of Type II CRISPR Efficiency as a Method for Engineered Phage T7 Selection
3.4. Generation of T7 Tail Fibre Mutants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.W.; Harper, D.; et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G. Bacteriophage Therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Kȩsik-Szeloch, A.; Drulis-Kawa, Z.; Weber-Da̧browska, B.; Kassner, J.; Majkowska-Skrobek, G.; Augustyniak, D.; Łusiak-Szelachowska, M.; Zaczek, M.; Górski, A.; Kropinski, A.M. Characterising the biology of novel lytic bacteriophages infecting multidrug resistant Klebsiella pneumoniae. Virol. J. 2013, 10, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, H.; Pinto, G.; Hendrix, H.; Noben, J.P.; Gawor, J.; Kropinski, A.M.; Łobocka, M.; Lavigne, R.; Azeredo, J. A lytic Providencia rettgeri virus of potential therapeutic value is a deepbranching member of the T5virus genus. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratton, C.W. Dead bugs don’t mutate: Susceptibility issues in the emergence of bacterial resistance. Emerg. Infect. Dis. 2003, 9, 10–16. [Google Scholar] [CrossRef]
- Erez, Z.; Steinberger-Levy, I.; Shamir, M.; Doron, S.; Stokar-Avihail, A.; Peleg, Y.; Melamed, S.; Leavitt, A.; Savidor, A.; Albeck, S.; et al. Communication between viruses guides lysis-lysogeny decisions. Nature 2017, 541, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Burns, N.; James, C.E.; Harrison, E. Polylysogeny magnifies competitiveness of a bacterial pathogen in vivo. Evol. Appl. 2015, 8, 346–351. [Google Scholar] [CrossRef]
- Clokie, M.R.J.; Kropinski, A.M. Bacteriophages: Methods and Protocols Volume 1: Isolation, Characterization, and Interactions; Springer: Berlin/Heidelberg, Germany, 2009; ISBN 978-1-60327-164-6. [Google Scholar]
- Krylov, V.N. Phage Therapy in Terms of Bacteriophage Genetics: Hopes, Prospects, Safety, Limitations. Russ. J. Genet. 2001, 37, 715–730. [Google Scholar] [CrossRef]
- Qadir, M.I.; Mobeen, T.; Masood, A. Phage therapy: Progress in pharmacokinetics. Braz. J. Pharm. Sci. 2018. [Google Scholar] [CrossRef]
- Abedon, S.; Thomas-Abedon, C. Phage Therapy Pharmacology. Curr. Pharm. Biotechnol. 2010, 11, 28–47. [Google Scholar] [CrossRef]
- Lucchesi, P.; Carraway, M.; Marinus, M.G. Analysis of forward mutations induced by N-methyl-N’-nitrosoguanidine in the bacteriophage P22 mnt repressor gene. J. Bacteriol. 1986, 166, 34–37. [Google Scholar] [CrossRef] [Green Version]
- Court, D.L.; Sawitzke, J.A.; Thomason, L.C. Genetic Engineering Using Homologous Recombination. Annu. Rev. Genet. 2002, 36, 361–388. [Google Scholar] [CrossRef] [PubMed]
- Qimron, U.; Marintcheva, B.; Tabor, S.; Richardson, C.C. Genomewide screens for Escherichia coli genes affecting growth of T7 bacteriophage. Proc. Natl. Acad. Sci. USA 2006, 103, 19039–19044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yosef, I.; Goren, M.G.; Globus, R.; Molshanski-Mor, S.; Qimron, U. Extending the Host Range of Bacteriophage Particles for DNA Transduction. Mol. Cell 2017, 66, 721–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiro, R.; Molshanski-Mor, S.; Yosef, I.; Milam, S.L.; Erickson, H.P.; Qimron, U. Gene product 0.4 increases bacteriophage T7 competitiveness by inhibiting host cell division. Proc. Natl. Acad. Sci. USA 2013, 110, 19549–19554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auster, O.; Globus, R.; Yosef, I.; Qimron, U. Optimizing DNA transduction by selection of mutations that evade bacterial defense systems. RNA Biol. 2019, 16, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, A.; Oldfield, L.M.; Julia, C.; Marinelli, L.J.; Piuri, M.; Swigon, Z.; Kessel, V.; Hatfull, G.F. BRED: A Simple and Powerful Tool for Constructing Mutant and Recombinant Bacteriophage Genomes. PLoS ONE 2008, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Fehér, T.; Karcagi, I.; Blattner, F.R.; Pósfai, G. Bacteriophage recombineering in the lytic state using the lambda red recombinases. Microb. Biotechnol. 2012, 5, 466–476. [Google Scholar] [CrossRef] [Green Version]
- Van Kessel, J.C.; Hatfull, G.F. Mycobacterial recombineering. Methods Mol. Biol. 2008, 435, 203–215. [Google Scholar]
- Marinelli, L.J.; Hatfull, G.F.; Piuri, M. Recombineering: A powerful tool for modification of bacteriophage genomes. Bacteriophage 2012, 2, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Jaschke, P.R.; Lieberman, E.K.; Rodriguez, J.; Sierra, A.; Endy, D. A fully decompressed synthetic bacteriophage øX174 genome assembled and archived in yeast. Virology 2012, 434, 278–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 2015, 1, 187–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiro, R.; Shitrit, D.; Qimron, U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014, 11, 42–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Box, A.M.; McGuffie, M.J.; O’Hara, B.J.; Seed, K.D. Functional analysis of bacteriophage immunity through a Type I-E CRISPR-Cas system in Vibrio cholerae and its application in bacteriophage genome engineering. J. Bacteriol. 2016, 198, 578–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemay, M.L.; Tremblay, D.M.; Moineau, S. Genome Engineering of Virulent Lactococcal Phages Using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1351–1358. [Google Scholar] [CrossRef]
- Shen, J.; Zhou, J.; Chen, G.-Q.; Xiu, Z.-L. Efficient genome engineering of a virulent Klebsiella bacteriophage using CRISPR-Cas9. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Tao, P.; Wu, X.; Tang, W.C.; Zhu, J.; Rao, V. Engineering of Bacteriophage T4 Genome Using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Yaung, S.J.; Esvelt, K.M.; Church, G.M. CRISPR/Cas9-mediated phage resistance is not impeded by the DNA modifications of phage T4. PLoS ONE 2014, 9, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Hoshiga, F.; Yoshizaki, K.; Takao, N.; Miyanaga, K.; Tanji, Y. Modification of T2 phage infectivity toward Escherichia coli O157:H7 via using CRISPR/Cas9. FEMS Microbiol. Lett. 2019. [Google Scholar] [CrossRef]
- Bari, S.M.N.; Walker, F.C.; Cater, K.; Aslan, B.; Hatoum-Aslan, A. Strategies for Editing Virulent Staphylococcal Phages Using CRISPR-Cas10. ACS Synth. Biol. 2017, 6, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- Bryson, A.L.; Hwang, Y.; Sherrill-Mix, S.; Wu, G.D.; Lewis, J.D.; Black, L.; Clark, T.A.; Bushman, F.D. Covalent modification of bacteriophage T4 DNA inhibits CRISPRCas9. MBio 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Grishin, N.V.; Shabalina, S.A.; Wolf, Y.I.; Koonin, E.V. A putative RNA-interference-based immune system in prokaryotes: Computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct 2006, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouns, S.J.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.H.; Snijders, A.P.L.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; van der Oost, J. Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michniewski, S.; Redgwell, T.; Grigonyte, A.; Rihtman, B.; Aguilo-Ferretjans, M.; Christie-Oleza, J.; Jameson, E.; Scanlan, D.J.; Millard, A.D. Riding the wave of genomics to investigate aquatic coliphage diversity and activity. Environ. Microbiol. 2019, 21, 2112–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.A.; Erdmann, S.; Mojica, F.J.M.; Garrett, R.A. Protospacer recognition motifs: Mixed identities and functional diversity. RNA Biol. 2013, 10, 891–899. [Google Scholar] [CrossRef] [Green Version]
- Engler, C.; Kandzia, R.; Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PLoS ONE 2008, 3, e3647. [Google Scholar] [CrossRef] [Green Version]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Rihtman, B.; Meaden, S.; Clokie, M.R.; Koskella, B.; Millard, A.D. Assessing Illumina technology for the high-throughput sequencing of bacteriophage genomes. PeerJ 2016, 4, e2055. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, N.A.; Fass, J.N. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33) [Software]. Available online: https://github.com/najoshi/sickle (accessed on 12 January 2020).
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labuhn, M.; Adams, F.F.; Ng, M.; Knoess, S.; Schambach, A.; Charpentier, E.M.; Schwarzer, A.; Mateo, J.L.; Klusmann, J.H.; Heckl, D. Refined sgRNA efficacy prediction improves largeand small-scale CRISPR-Cas9 applications. Nucleic Acids Res. 2018, 46, 1375–1385. [Google Scholar] [CrossRef] [Green Version]
- Bin Moon, S.; Lee, J.M.; Kang, J.G.; Lee, N.E.; Ha, D.I.; Kim, D.Y.; Kim, S.H.; Yoo, K.; Kim, D.; Ko, J.H.; et al. Highly efficient genome editing by CRISPR-Cpf1 using CRISPR RNA with a uridinylate-rich 3′-overhang. Nat. Commun. 2018, 9, 3651. [Google Scholar] [CrossRef]
- Luo, M.L.; Mullis, A.S.; Leenay, R.T.; Beisel, C.L. Repurposing endogenous type i CRISPR-Cas systems for programmable gene repression. Nucleic Acids Res. 2015, 43, 674–681. [Google Scholar] [CrossRef] [Green Version]
- Majsec, K.; Bolt, E.L.; Ivančić-Baće, I. Cas3 is a limiting factor for CRISPR-Cas immunity in Escherichia coli cells lacking H-NS. BMC Microbiol. 2016, 16. [Google Scholar] [CrossRef] [Green Version]
- Ueguchi, C.; Kakeda, M.; Mizuno, T. Autoregulatory expression of the Escherichia coli hns gene encoding a nucleoid protein: H-NS functions as a repressor of its own transcription. MGG Mol. Gen. Genet. 1993, 236, 171–178. [Google Scholar] [CrossRef]
- Liu, M.; Deora, R.; Simons, R.W.; Doulatov, S.; Hodes, A.; Dai, L.; Mandhana, N.; Zimmerly, S.; Miller, J.F.; Liu, M.; et al. Tropism switching in Bordetella bacteriophage defines a family of diversity-generating retroelements. Nature 2004, 431, 476–481. [Google Scholar]
- Zhao, X.; Wu, W.; Qi, Z.; Cui, Y.; Yan, F.; Guo, Z.; Wang, Z.; Wang, H.; Deng, H.; Xue, Y.; et al. The complete genome sequence and proteomics of Yersinia pestis phage Yep-phi. J. Gen. Virol. 2011, 92, 216–221. [Google Scholar] [CrossRef]
- Liu, M.; Gingery, M.; Doulatov, S.R.; Liu, Y.; Hodes, A.; Baker, S.; Davis, P.; Simmonds, M.; Churcher, C.; Mungall, K.; et al. Genomic and Genetic Analysis of Bordetella Bacteriophages Encoding Reverse Transcriptase-Mediated Tropism-Switching Cassettes. J. Bacteriol. 2004, 186, 1503–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Cui, Y.; Yan, Y.; Du, Z.; Tan, Y.; Yang, H.; Bi, Y.; Zhang, P.; Zhou, L.; Zhou, D.; et al. Outer Membrane Proteins Ail and OmpF of Yersinia pestis Are Involved in the Adsorption of T7-Related Bacteriophage Yep-phi. J. Virol. 2013, 87, 12260–12269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, L.; Suthers, P.F.; Yin, J. Effects of Escherichia coli physiology on growth of phage T7 in vivo and in silico. J. Bacteriol. 2002, 184, 1888–1894. [Google Scholar] [CrossRef] [Green Version]
- Wilson, L.O.W.; O’Brien, A.R.; Bauer, D.C. The current state and future of CRISPR-Cas9 gRNA design tools. Front. Pharmacol. 2018, 9, 749. [Google Scholar] [CrossRef]
- Park, J.; Kim, J.S.; Bae, S. Cas-Database: Web-based genome-wide guide RNA library design for gene knockout screens using CRISPR-Cas9. Bioinformatics 2016, 32, 2017–2023. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martel, B.; Moineau, S. CRISPR-Cas: An efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 2014, 42, 9504–9513. [Google Scholar] [CrossRef] [Green Version]
- Marraffini, L.A. The CRISPR-Cas System of Streptococcus Pyogenes: Function and Applications; University of Oklahoma Health Sciences Center: Oklahoma, OK, USA, 2016. [Google Scholar]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigonyte, A.M.; Harrison, C.; MacDonald, P.R.; Montero-Blay, A.; Tridgett, M.; Duncan, J.; Sagona, A.P.; Constantinidou, C.; Jaramillo, A.; Millard, A. Comparison of CRISPR and Marker-Based Methods for the Engineering of Phage T7. Viruses 2020, 12, 193. https://doi.org/10.3390/v12020193
Grigonyte AM, Harrison C, MacDonald PR, Montero-Blay A, Tridgett M, Duncan J, Sagona AP, Constantinidou C, Jaramillo A, Millard A. Comparison of CRISPR and Marker-Based Methods for the Engineering of Phage T7. Viruses. 2020; 12(2):193. https://doi.org/10.3390/v12020193
Chicago/Turabian StyleGrigonyte, Aurelija M., Christian Harrison, Paul R. MacDonald, Ariadna Montero-Blay, Matthew Tridgett, John Duncan, Antonia P. Sagona, Chrystala Constantinidou, Alfonso Jaramillo, and Andrew Millard. 2020. "Comparison of CRISPR and Marker-Based Methods for the Engineering of Phage T7" Viruses 12, no. 2: 193. https://doi.org/10.3390/v12020193
APA StyleGrigonyte, A. M., Harrison, C., MacDonald, P. R., Montero-Blay, A., Tridgett, M., Duncan, J., Sagona, A. P., Constantinidou, C., Jaramillo, A., & Millard, A. (2020). Comparison of CRISPR and Marker-Based Methods for the Engineering of Phage T7. Viruses, 12(2), 193. https://doi.org/10.3390/v12020193