Suppression of a Subset of Interferon-Induced Genes by Human Papillomavirus Type 16 E7 via a Cyclin Dependent Kinase 8-Dependent Mechanism

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning of E7 Mutants
2.2. Cell Culture and Creation of Cell Lines
2.3. Cellular Growth Rates
2.4. RNA Extraction, RT-qPCR, and Western Blotting
2.5. RNA Sequencing
2.6. siRNA Transfection
2.7. Immunoprecipitation
2.8. Chromatin Immunoprecipitation
2.9. Statistics
3. Results
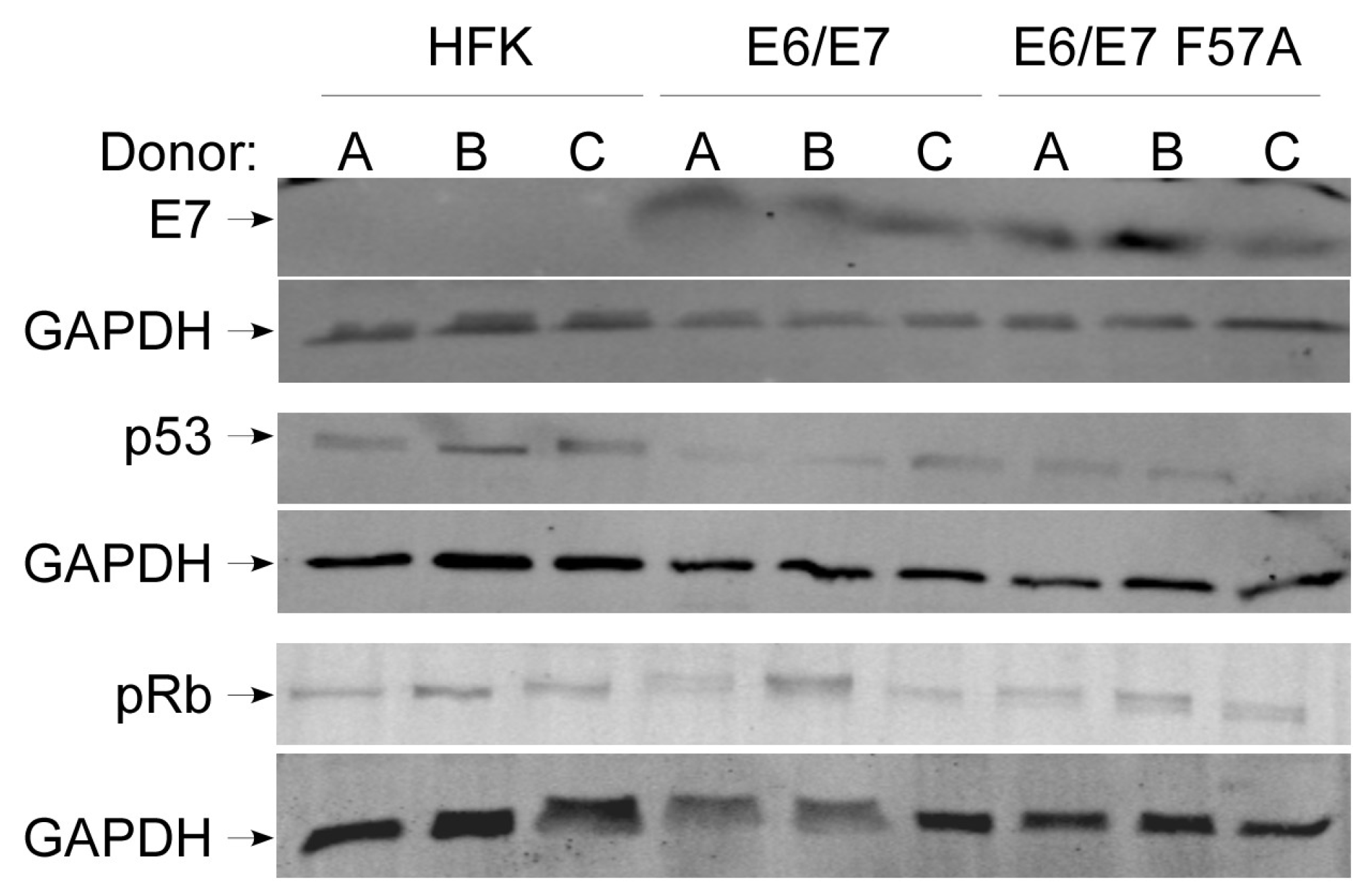
3.1. The CR3 of E7 Contributes to Episomal Maintenance of the HPV16 Genome
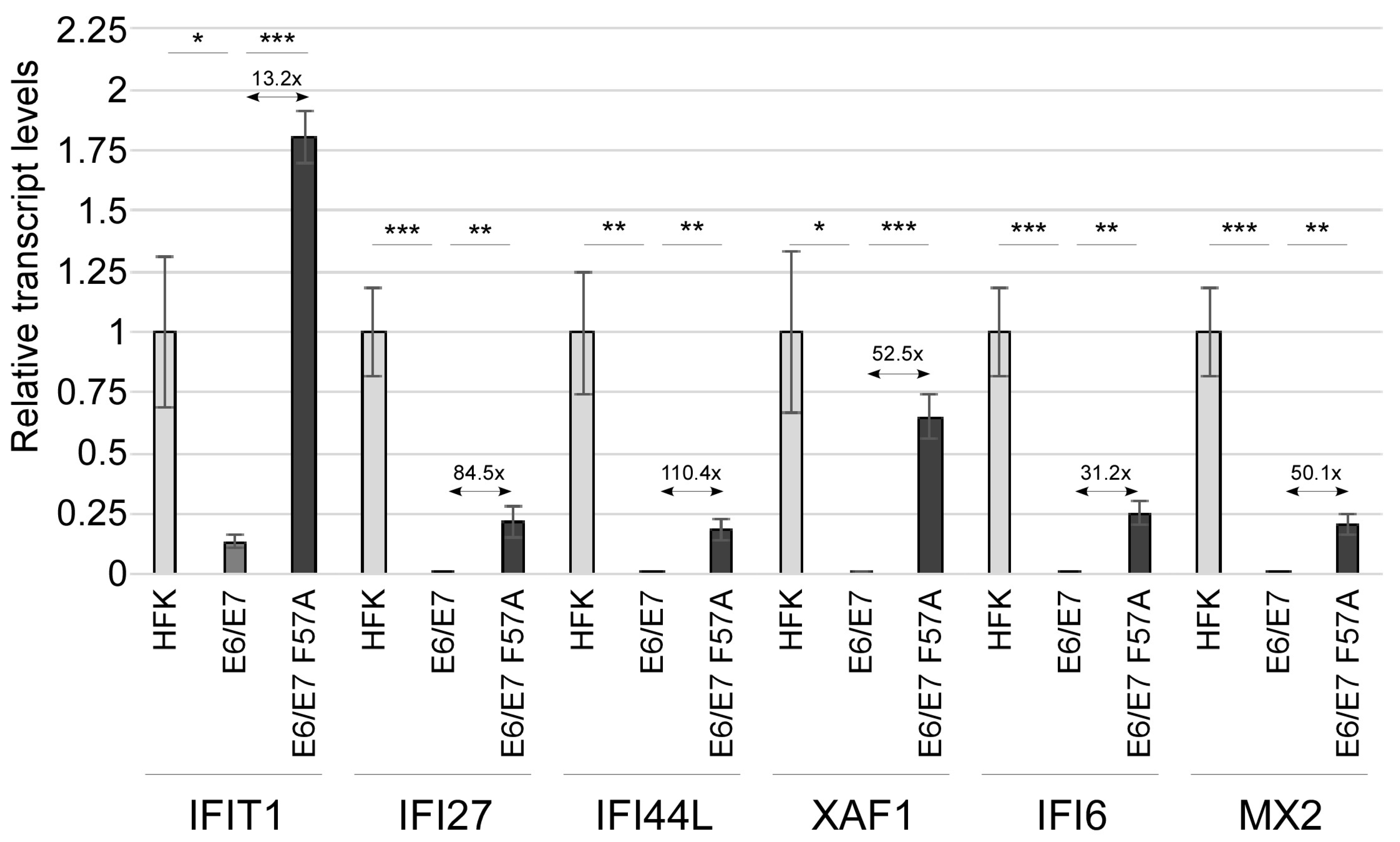
3.2. E7 F57A Is Defective in E7-Mediated Suppression of ISGs
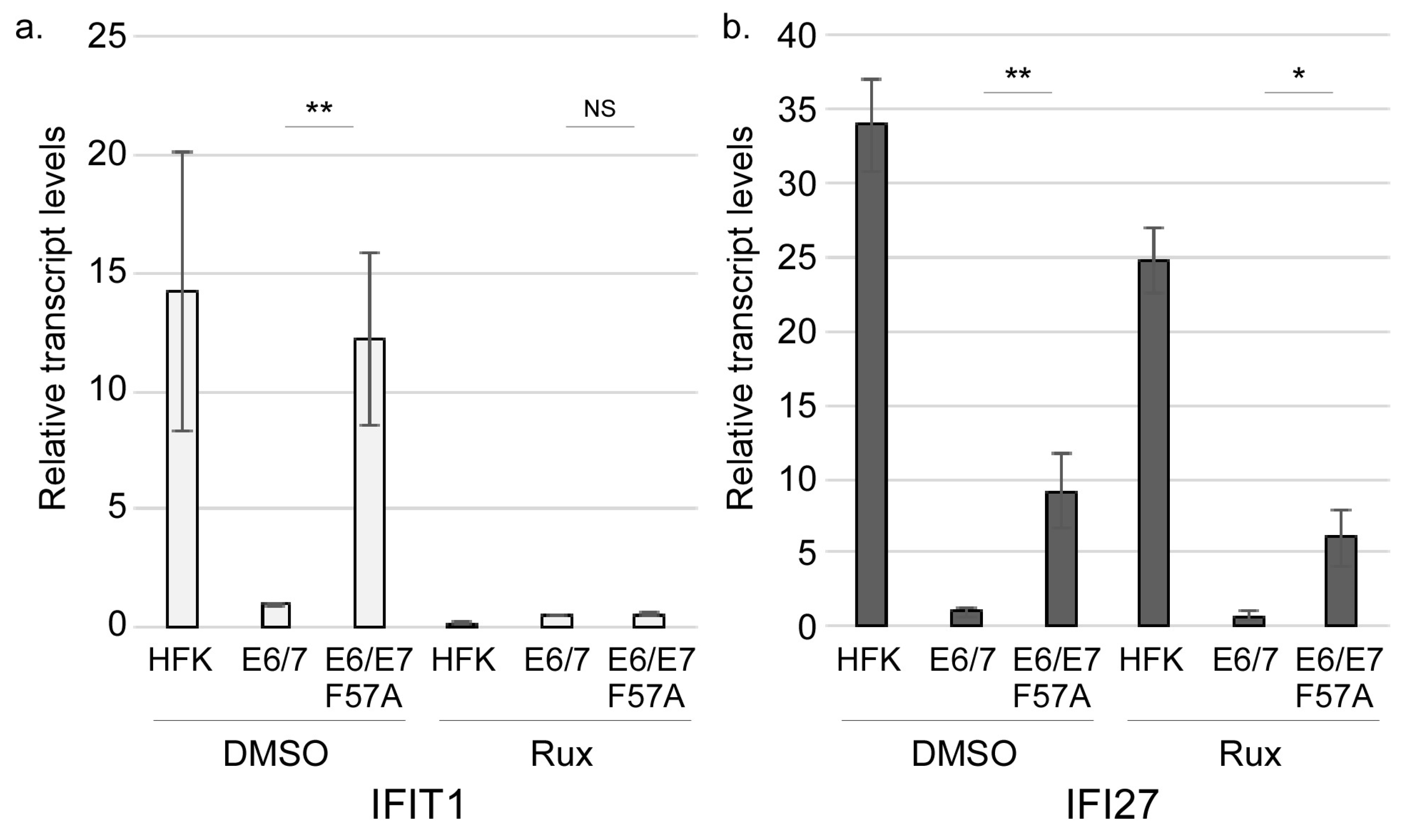
3.3. E7 May Function Downstream of JAK Activation to Suppress Interferon-Stimulated Gene Expression
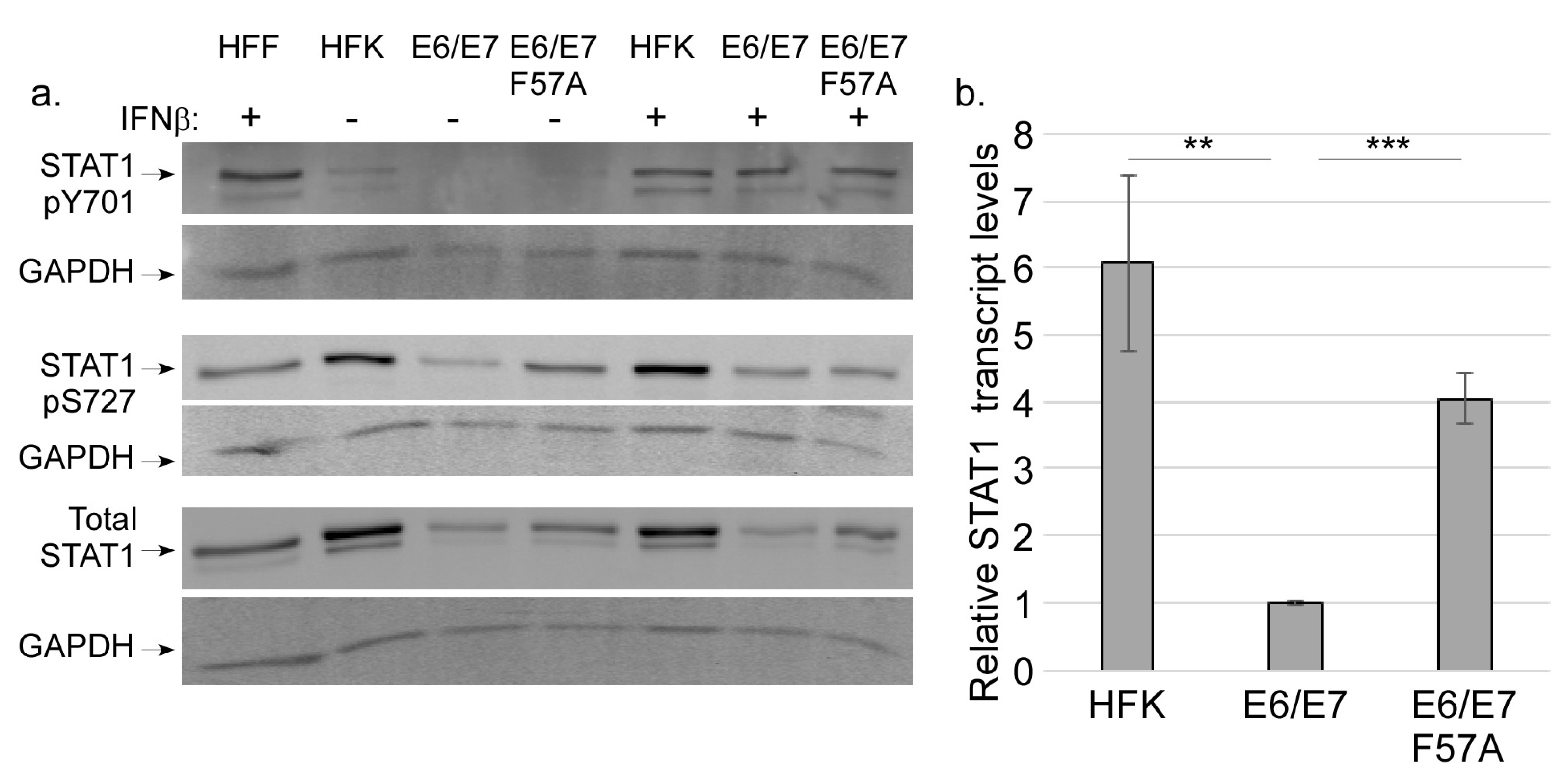
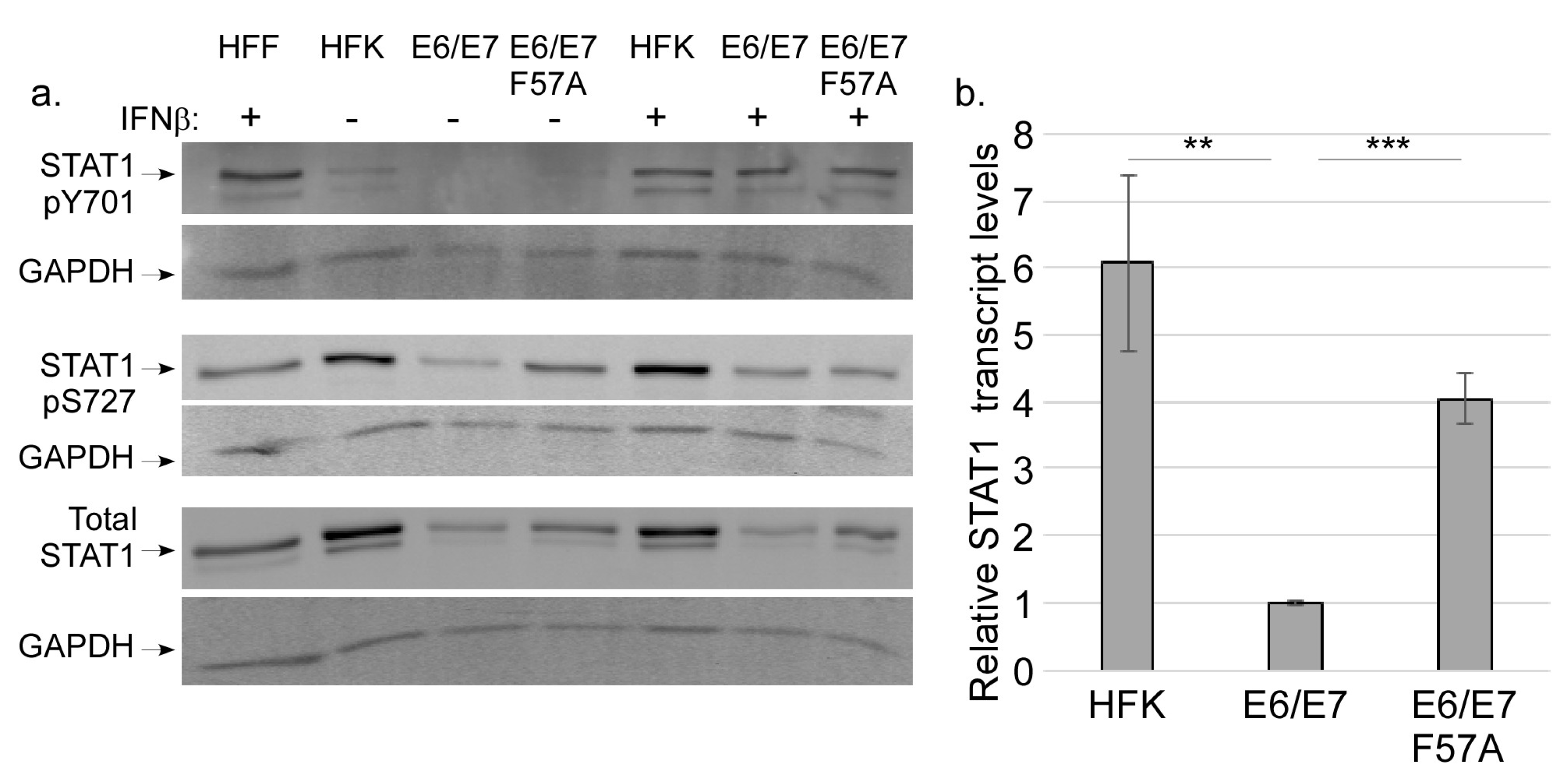
3.4. E7 Does not Regulate STAT Activation
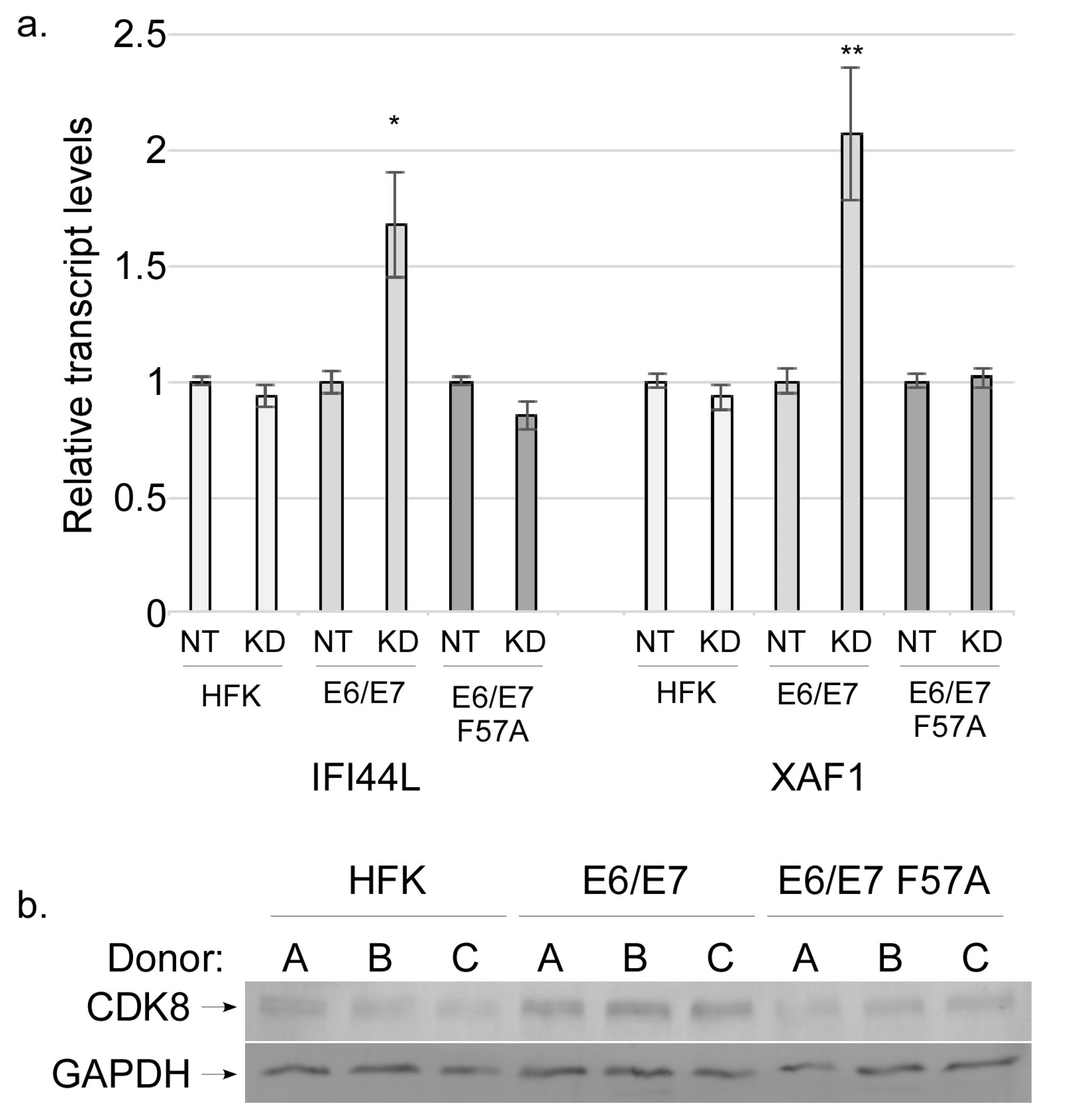
3.5. CDK8 Associates with E7 and Contributes to ISG Suppression
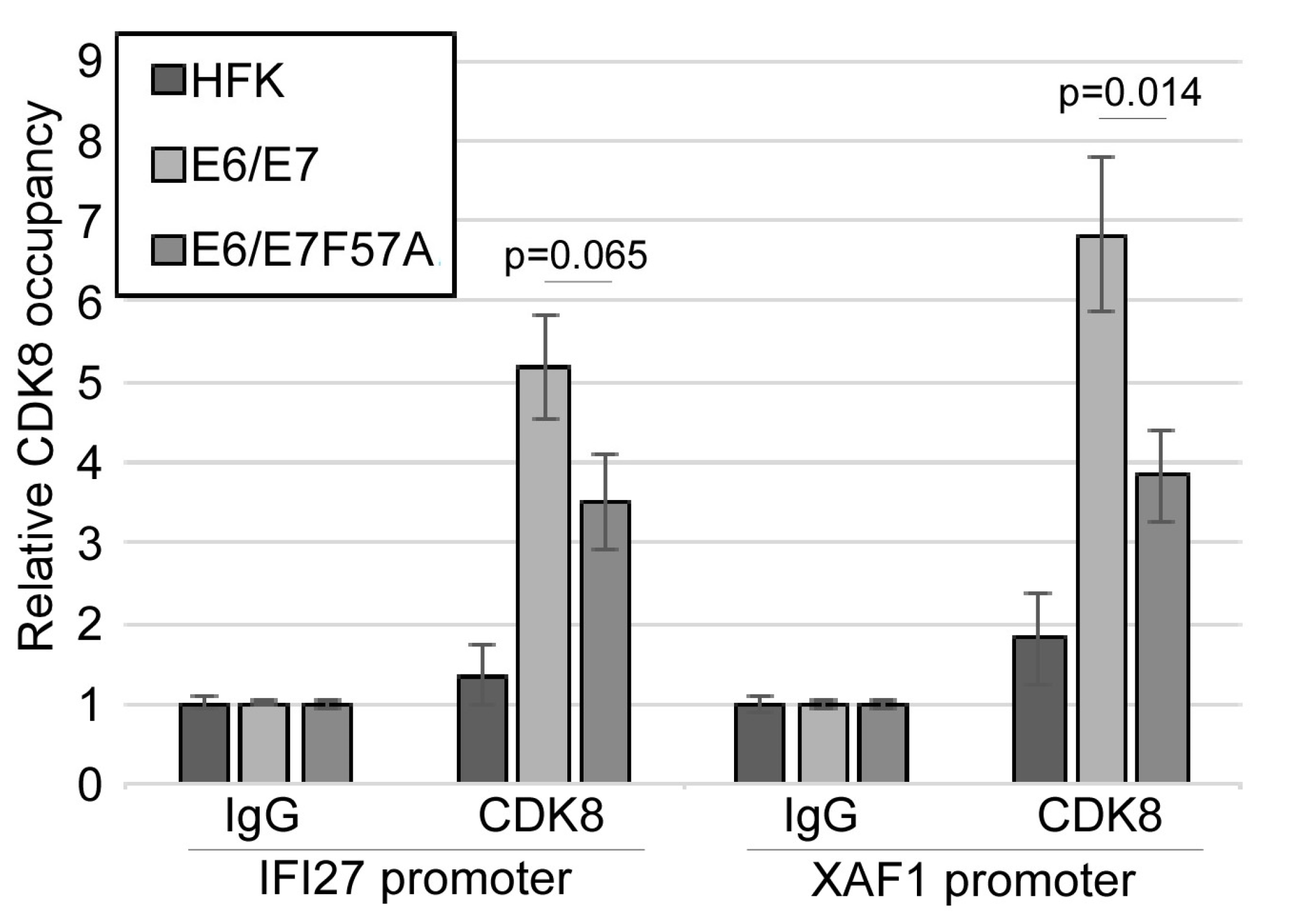
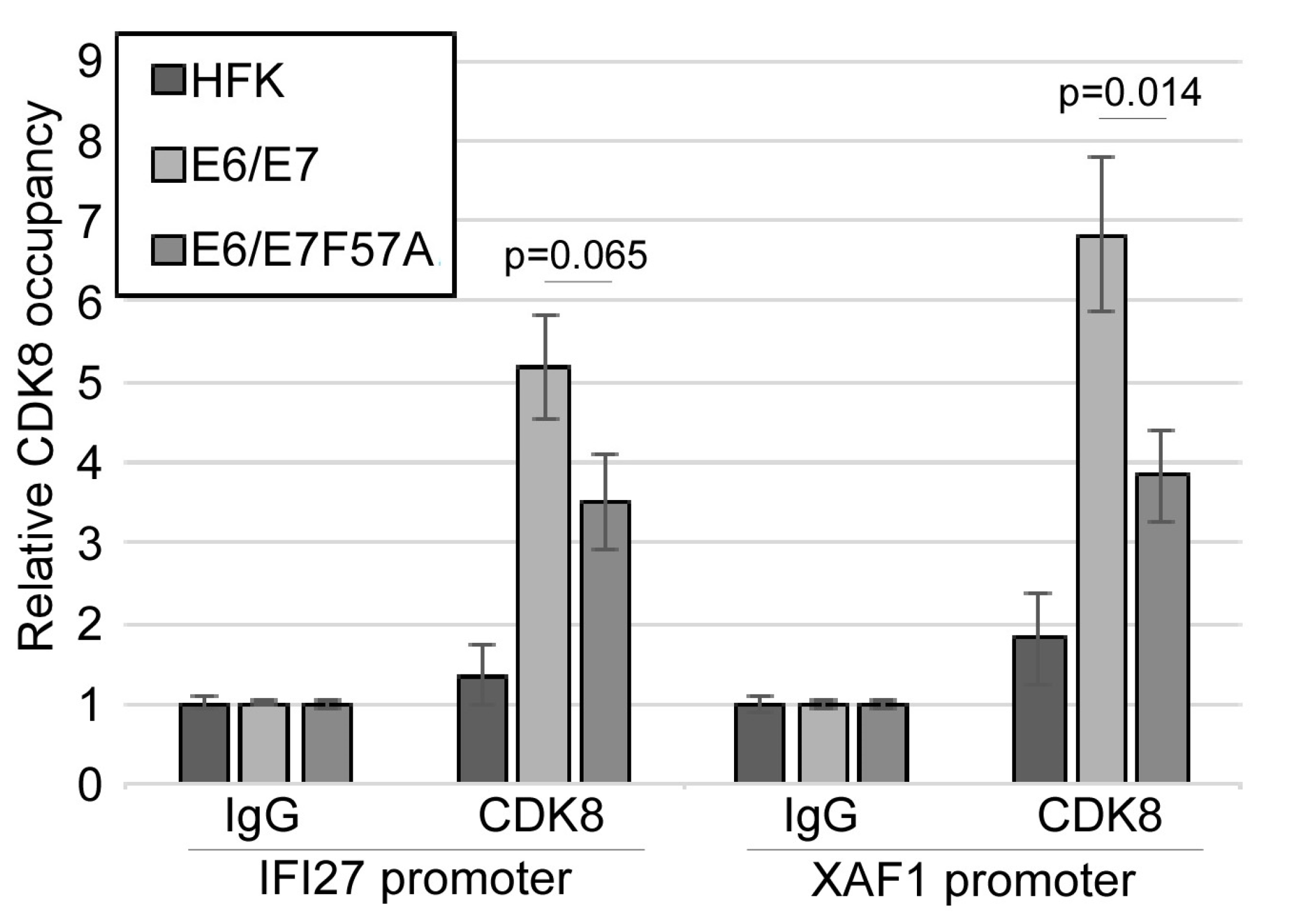
3.6. CDK8 Occupies the Promoters of Interferon-Stimulated Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moscicki, A.B.; Schiffman, M.; Burchell, A.; Albero, G.; Giuliano, A.R.; Goodman, M.T.; Kjaer, S.K.; Palefsky, J. Updating the natural history of human papillomavirus and anogenital cancers. Vaccine 2012, 30, F24–F33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef] [Green Version]
- Clifford, G.M.; Smith, J.S.; Aguado, T.; Franceschi, S. Comparison of HPV type distribution in high-grade cervical lesions and cervical cancer: A meta-analysis. Br. J. Cancer 2003, 89, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, A.A. Mechanisms and strategies of papillomavirus replication. Biol. Chem. 2017, 398, 919–927. [Google Scholar] [CrossRef]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, C. Mechanisms by which HPV Induces a Replication Competent Environment in Differentiating Keratinocytes. Viruses 2017, 9, 261. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.; Laimins, L.A. Persistence of human papillomavirus infection: Keys to malignant progression. Trends Microbiol. 2011, 19, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Gielen, V.; Schmitt, D.; Thivolet, J. HLA class I antigen (heavy and light chain) expression by Langerhans cells and keratinocytes of the normal human epidermis: Ultrastructural quantitation using immunogold labelling procedure. Arch. Dermatol. Res. 1988, 280, 131–136. [Google Scholar] [CrossRef]
- Kubo, A.; Nagao, K.; Yokouchi, M.; Sasaki, H.; Amagai, M. External antigen uptake by Langerhans cells with reorganization of epidermal tight junction barriers. J. Exp. Med. 2009, 206, 2937–2946. [Google Scholar] [CrossRef] [Green Version]
- Woodby, B.L.; Songock, W.K.; Scott, M.L.; Raikhy, G.; Bodily, J.M. Induction of Interferon Kappa in Human Papillomavirus 16 Infection by Transforming Growth Factor Beta-Induced Promoter Demethylation. J. Virol. 2018, 92, e01714-17. [Google Scholar] [CrossRef] [Green Version]
- Halbert, C.L.; Demers, G.W.; Galloway, D.A. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J. Virol. 1991, 65, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Munger, K.; Basile, J.R.; Duensing, S.; Eichten, A.; Gonzalez, S.L.; Grace, M.; Zacny, V.L. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene 2001, 20, 7888–7898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vousden, K.H.; Vojtesek, B.; Fisher, C.; Lane, D. HPV-16 E7 or adenovirus E1A can overcome the growth arrest of cells immortalized with a temperature-sensitive p53. Oncogene 1993, 8, 1697–1702. [Google Scholar] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westrich, J.A.; Warren, C.J.; Pyeon, D. Evasion of host immune defenses by human papillomavirus. Virus. Res. 2017, 231, 21–33. [Google Scholar] [CrossRef]
- Songock, W.K.; Kim, S.M.; Bodily, J.M. The human papillomavirus E7 oncoprotein as a regulator of transcription. Virus. Res. 2017, 231, 56–75. [Google Scholar] [CrossRef] [Green Version]
- Bodily, J.M.; Mehta, K.P.; Laimins, L.A. Human papillomavirus E7 enhances hypoxia-inducible factor 1-mediated transcription by inhibiting binding of histone deacetylases. Cancer Res. 2011, 71, 1187–1195. [Google Scholar] [CrossRef] [Green Version]
- Bernat, A.; Avvakumov, N.; Mymryk, J.S.; Banks, L. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene 2003, 22, 7871–7881. [Google Scholar] [CrossRef] [Green Version]
- Brehm, A.; Nielsen, S.J.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999, 18, 2449–2458. [Google Scholar] [CrossRef] [Green Version]
- Burgers, W.A.; Blanchon, L.; Pradhan, S.; de Launoit, Y.; Kouzarides, T.; Fuks, F. Viral oncoproteins target the DNA methyltransferases. Oncogene 2007, 26, 1650–1655. [Google Scholar] [CrossRef] [Green Version]
- Massimi, P.; Pim, D.; Banks, L. Human papillomavirus type 16 E7 binds to the conserved carboxy-terminal region of the TATA box binding protein and this contributes to E7 transforming activity. J. Gen. Virol. 1997, 78, 2607–2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Kim, E.J.; Kwon, H.J.; Hwang, E.S.; Namkoong, S.E.; Um, S.J. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin-Drubin, M.E.; Huh, K.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with E2F6. J. Virol. 2008, 82, 8695–8705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar]
- Chellappan, S.; Kraus, V.B.; Kroger, B.; Munger, K.; Howley, P.M.; Phelps, W.C.; Nevins, J.R. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc. Natl. Acad. Sci. USA 1992, 89, 4549–4553. [Google Scholar] [CrossRef] [Green Version]
- Munger, K.; Werness, B.A.; Dyson, N.; Phelps, W.C.; Harlow, E.; Howley, P.M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989, 8, 4099–4105. [Google Scholar] [CrossRef]
- LaFleur, D.W.; Nardelli, B.; Tsareva, T.; Mather, D.; Feng, P.; Semenuk, M.; Taylor, K.; Buergin, M.; Chinchilla, D.; Roshke, V.; et al. Interferon-kappa, a novel type I interferon expressed in human keratinocytes. J. Biol. Chem. 2001, 276, 39765–39771. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, S.A.; Salditt-Georgieff, M.; Darnell, J.E., Jr. Tyrosine-phosphorylated Stat1 and Stat2 plus a 48-kDa protein all contact DNA in forming interferon-stimulated-gene factor 3. Proc. Natl. Acad. Sci. USA 1995, 92, 3829–3833. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xu, L.; Su, J.; Peppelenbosch, M.P.; Pan, Q. Transcriptional Regulation of Antiviral Interferon-Stimulated Genes. Trends Microbiol. 2017, 25, 573–584. [Google Scholar] [CrossRef]
- Bancerek, J.; Poss, Z.C.; Steinparzer, I.; Sedlyarov, V.; Pfaffenwimmer, T.; Mikulic, I.; Dolken, L.; Strobl, B.; Muller, M.; Taatjes, D.J.; et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 2013, 38, 250–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilz, A.; Ramsauer, K.; Heidari, H.; Leitges, M.; Kovarik, P.; Decker, T. Phosphorylation of the Stat1 transactivating domain is required for the response to type I interferons. EMBO Rep. 2003, 4, 368–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadzak, I.; Schiff, M.; Gattermeier, I.; Glinitzer, R.; Sauer, I.; Saalmuller, A.; Yang, E.; Schaljo, B.; Kovarik, P. Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 8944–8949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinparzer, I.; Sedlyarov, V.; Rubin, J.D.; Eislmayr, K.; Galbraith, M.D.; Levandowski, C.B.; Vcelkova, T.; Sneezum, L.; Wascher, F.; Amman, F.; et al. Transcriptional Responses to IFN-gamma Require Mediator Kinase-Dependent Pause Release and Mechanistically Distinct CDK8 and CDK19 Functions. Mol. Cell 2019, 76, 485–499. [Google Scholar] [CrossRef]
- Uddin, S.; Sassano, A.; Deb, D.K.; Verma, A.; Majchrzak, B.; Rahman, A.; Malik, A.B.; Fish, E.N.; Platanias, L.C. Protein kinase C-delta (PKC-delta ) is activated by type I interferons and mediates phosphorylation of Stat1 on serine 727. J. Biol. Chem. 2002, 277, 14408–14416. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Antonsson, A.; Payne, E.; Hengst, K.; McMillan, N.A. The human papillomavirus type 16 E7 protein binds human interferon regulatory factor-9 via a novel PEST domain required for transformation. J. Interferon Cytokine Res. 2006, 26, 455–461. [Google Scholar] [CrossRef]
- Barnard, P.; McMillan, N.A. The human papillomavirus E7 oncoprotein abrogates signaling mediated by interferon-alpha. Virology 1999, 259, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Mehta, K.P.; Laimins, L.A. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J. Virol. 2011, 85, 9486–9494. [Google Scholar] [CrossRef] [Green Version]
- Nees, M.; Geoghegan, J.M.; Hyman, T.; Frank, S.; Miller, L.; Woodworth, C.D. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J. Virol. 2001, 75, 4283–4296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perea, S.; Lopezocejo, O.; Vongabain, A.; Arana, M. Human papillomavirus type-16 (HPV-16) major transforming proteins functionally interact with interferon signaling mechanisms. Int. J. Oncol. 1997, 11, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.H.; Bortnik, V.; McMillan, N.A.; Idris, A. cGAS-STING responses are dampened in high-risk HPV type 16 positive head and neck squamous cell carcinoma cells. Microb. Pathog. 2019, 132, 162–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, S.J.; Rhyu, J.W.; Kim, E.J.; Jeon, K.C.; Hwang, E.S.; Park, J.S. Abrogation of IRF-1 response by high-risk HPV E7 protein in vivo. Cancer Lett. 2002, 179, 205–212. [Google Scholar] [CrossRef]
- Bodily, J.M.; Mehta, K.P.; Cruz, L.; Meyers, C.; Laimins, L.A. The E7 open reading frame acts in cis and in trans to mediate differentiation-dependent activities in the human papillomavirus type 16 life cycle. J. Virol. 2011, 85, 8852–8862. [Google Scholar] [CrossRef] [Green Version]
- Edmonds, C.; Vousden, K.H. A point mutational analysis of human papillomavirus type 16 E7 protein. J. Virol. 1989, 63, 2650–2656. [Google Scholar] [CrossRef] [Green Version]
- Todorovic, B.; Massimi, P.; Hung, K.; Shaw, G.S.; Banks, L.; Mymryk, J.S. Systematic analysis of the amino acid residues of human papillomavirus type 16 E7 conserved region 3 involved in dimerization and transformation. J. Virol. 2011, 85, 10048–10057. [Google Scholar] [CrossRef] [Green Version]
- Fant, C.B.; Taatjes, D.J. Regulatory functions of the Mediator kinases CDK8 and CDK19. Transcription 2019, 10, 76–90. [Google Scholar] [CrossRef]
- Meyer, K.D.; Donner, A.J.; Knuesel, M.T.; York, A.G.; Espinosa, J.M.; Taatjes, D.J. Cooperative activity of cdk8 and GCN5L within Mediator directs tandem phosphoacetylation of histone H3. EMBO J. 2008, 27, 1447–1457. [Google Scholar] [CrossRef] [Green Version]
- Nemet, J.; Jelicic, B.; Rubelj, I.; Sopta, M. The two faces of Cdk8, a positive/negative regulator of transcription. Biochimie 2014, 97, 22–27. [Google Scholar] [CrossRef]
- Knuesel, M.T.; Meyer, K.D.; Bernecky, C.; Taatjes, D.J. The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. 2009, 23, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donner, A.J.; Szostek, S.; Hoover, J.M.; Espinosa, J.M. CDK8 is a stimulus-specific positive coregulator of p53 target genes. Mol. Cell 2007, 27, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galbraith, M.D.; Allen, M.A.; Bensard, C.L.; Wang, X.; Schwinn, M.K.; Qin, B.; Long, H.W.; Daniels, D.L.; Hahn, W.C.; Dowell, R.D.; et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 2013, 153, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Ding, N.; Zhou, H.; Esteve, P.O.; Chin, H.G.; Kim, S.; Xu, X.; Joseph, S.M.; Friez, M.J.; Schwartz, C.E.; Pradhan, S.; et al. Mediator links epigenetic silencing of neuronal gene expression with x-linked mental retardation. Mol. Cell 2008, 31, 347–359. [Google Scholar] [CrossRef] [Green Version]
- Bodily, J.M.; Hennigan, C.; Wrobel, G.A.; Rodriguez, C.M. Regulation of the human papillomavirus type 16 late promoter by E7 and the cell cycle. Virology 2013, 443, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, M.L.; Woodby, B.L.; Ulicny, J.; Raikhy, G.; Orr, A.W.; Songock, W.K.; Bodily, J.M. Human papillomavirus type 16 E5 inhibits interferon signaling and supports episomal viral maintenance. J. Virol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Laimins, L.A. Differentiation of HPV-containing cells using organotypic "raft" culture or methylcellulose. Methods Mol. Med. 2005, 119, 157–169. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, M.C.; Frattini, M.G.; Grossman, S.R.; Laimins, L.A. Human papillomavirus type 18 E7 protein requires intact Cys-X-X-Cys motifs for zinc binding, dimerization, and transformation but not for Rb binding. J. Virol. 1993, 67, 3142–3150. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Clements, A.; Zhao, K.; Marmorstein, R. Structure of the human Papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. J. Biol. Chem. 2006, 281, 578–586. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.E.; Pena, L.; Sen, G.C.; Park, J.K.; Laimins, L.A. Long-term effect of interferon on keratinocytes that maintain human papillomavirus type 31. J. Virol. 2002, 76, 8864–8874. [Google Scholar] [CrossRef] [Green Version]
- Herdman, M.T.; Pett, M.R.; Roberts, I.; Alazawi, W.O.; Teschendorff, A.E.; Zhang, X.Y.; Stanley, M.A.; Coleman, N. Interferon-beta treatment of cervical keratinocytes naturally infected with human papillomavirus 16 episomes promotes rapid reduction in episome numbers and emergence of latent integrants. Carcinogenesis 2006, 27, 2341–2353. [Google Scholar] [CrossRef] [Green Version]
- Pett, M.; Coleman, N. Integration of high-risk human papillomavirus: A key event in cervical carcinogenesis? J. Pathol. 2007, 212, 356–367. [Google Scholar] [CrossRef]
- Bordignon, V.; Di Domenico, E.G.; Trento, E.; D’Agosto, G.; Cavallo, I.; Pontone, M.; Pimpinelli, F.; Mariani, L.; Ensoli, F. How Human Papillomavirus Replication and Immune Evasion Strategies Take Advantage of the Host DNA Damage Repair Machinery. Viruses 2017, 9, 390. [Google Scholar] [CrossRef] [Green Version]
- Cheon, H.; Stark, G.R. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc. Natl. Acad. Sci. USA 2009, 106, 9373–9378. [Google Scholar] [CrossRef] [Green Version]
- Staab, J.; Herrmann-Lingen, C.; Meyer, T. CDK8 as the STAT1 serine 727 kinase? JAK-STAT 2013, 2, e24275. [Google Scholar] [CrossRef] [Green Version]
- Songock, W.K.; Scott, M.L.; Bodily, J.M. Regulation of the human papillomavirus type 16 late promoter by transcriptional elongation. Virology 2017, 507, 179–191. [Google Scholar] [CrossRef]
- Akoulitchev, S.; Chuikov, S.; Reinberg, D. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature 2000, 407, 102–106. [Google Scholar] [CrossRef]
- Donner, A.J.; Ebmeier, C.C.; Taatjes, D.J.; Espinosa, J.M. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat. Struct. Mol. Biol. 2010, 17, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Galbraith, M.D.; Donner, A.J.; Espinosa, J.M. CDK8: A positive regulator of transcription. Transcription 2010, 1, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taatjes, D.J. The human Mediator complex: A versatile, genome-wide regulator of transcription. Trends Biochem. Sci. 2010, 35, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borden, E.C.; Sen, G.C.; Uze, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef]
- Sung, P.S.; Cheon, H.; Cho, C.H.; Hong, S.H.; Park, D.Y.; Seo, H.I.; Park, S.H.; Yoon, S.K.; Stark, G.R.; Shin, E.C. Roles of unphosphorylated ISGF3 in HCV infection and interferon responsiveness. Proc. Natl. Acad. Sci. USA 2015, 112, 10443–10448. [Google Scholar] [CrossRef] [Green Version]
- Kovarik, P.; Stoiber, D.; Eyers, P.A.; Menghini, R.; Neininger, A.; Gaestel, M.; Cohen, P.; Decker, T. Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-gamma uses a different signaling pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 13956–13961. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef]
- Poss, Z.C.; Ebmeier, C.C.; Odell, A.T.; Tangpeerachaikul, A.; Lee, T.; Pelish, H.E.; Shair, M.D.; Dowell, R.D.; Old, W.M.; Taatjes, D.J. Identification of Mediator Kinase Substrates in Human Cells using Cortistatin A and Quantitative Phosphoproteomics. Cell Rep. 2016, 15, 436–450. [Google Scholar] [CrossRef] [Green Version]
- Alarcon, C.; Zaromytidou, A.I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Feng, D.; Wang, Q.; Abdulla, A.; Xie, X.J.; Zhou, J.; Sun, Y.; Yang, E.S.; Liu, L.P.; Vaitheesvaran, B.; et al. Regulation of lipogenesis by cyclin-dependent kinase 8-mediated control of SREBP-1. J. Clin. Investg. 2012, 122, 2417–2427. [Google Scholar] [CrossRef]
- Boyer, T.G.; Martin, M.E.; Lees, E.; Ricciardi, R.P.; Berk, A.J. Mammalian Srb/Mediator complex is targeted by adenovirus E1A protein. Nature 1999, 399, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.L.; Cantin, G.T.; Wang, G.; Shevchenko, A.; Shevchenko, A.; Berk, A.J. Transcription control by E1A and MAP kinase pathway via Sur2 mediator subunit. Science 2002, 296, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Vijayalingam, S.; Chinnadurai, G. Adenovirus L-E1A activates transcription through mediator complex-dependent recruitment of the super elongation complex. J. Virol. 2013, 87, 3425–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Berk, A.J. In vivo association of adenovirus large E1A protein with the human mediator complex in adenovirus-infected and -transformed cells. J. Virol. 2002, 76, 9186–9193. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Number of Samples | Episomal | Integrated | Fraction Episomal | p Value (Wt v Mutant) |
|---|---|---|---|---|---|
| HPV16 (wild type) | 51 | 40 | 11 | 0.78 | |
| M84S | 8 | 4 | 4 | 0.50 | 0.18 |
| G85A | 6 | 3 | 3 | 0.50 | 0.15 |
| Y52A | 10 | 5 | 5 | 0.50 | 0.11 |
| ED80-81KK | 5 | 2 | 3 | 0.40 | 0.09 |
| D62K | 7 | 3 | 4 | 0.43 | 0.07 |
| N53D | 12 | 6 | 6 | 0.50 | 0.07 |
| S63D | 9 | 3 | 6 | 0.33 | <0.05 |
| QKP96-98EEA | 11 | 3 | 8 | 0.27 | <0.01 |
| V55T | 13 | 4 | 9 | 0.31 | <0.01 |
| T64D | 7 | 1 | 6 | 0.14 | <0.01 |
| R77E | 7 | 0 | 7 | 0.00 | <0.001 |
| R66E | 13 | 2 | 11 | 0.15 | <0.0001 |
| F57A | 34 | 6 | 28 | 0.18 | <0.0001 |
| Pathway Name | # Entities Found | # Entities Total | Entities FDR |
|---|---|---|---|
| Interferon alpha/beta signaling | 10 | 184 | <0.0001 |
| Interferon Signaling | 10 | 392 | <0.0001 |
| Cytokine Signaling in Immune system | 11 | 1055 | <0.001 |
| Transport of fatty acids | 2 | 18 | 0.01 |
| Transport of vitamins, nucleosides, and related molecules | 3 | 100 | 0.02 |
| Upregulated | Downregulated | ||
|---|---|---|---|
| Gene ID | Fold Change | Gene ID | Fold Change |
| SLC44A5 | 5713.3 | ARL2-SNX15 | −4881.7 |
| GPAT2 | 3410.2 | AC013489.1 | −4599.3 |
| SLC27A6 | 170.4 | AC139530.2 | −4185.4 |
| VAV1 | 147.7 | UGT1A3 | −4061.3 |
| IFI27 | 49.3 | AC097658.1 | −441.7 |
| KRBOX1 | 47.7 | USP32P2 | −102.9 |
| IFI44L | 34.3 | MTND1P23 | −69.8 |
| XAF1 | 29.8 | SHC1P1 | −59.0 |
| IFI6 | 23.7 | FOXF2 | -49.8 |
| MX2 | 22.6 | MAMLD1 | −43.6 |
| FYB1 | 19.5 | FAM103A2P | −30.7 |
| FAM26E | 19.1 | SPINK7 | −30.1 |
| BST2 | 17.7 | RAB4B-EGLN2 | −30.0 |
| AL163051.1 | 10.6 | SPRR4 | −28.9 |
| AC009268.2 | 8.1 | AC008556.1 | −26.6 |
| AL136295.5 | 7.6 | VASH2 | −21.7 |
| AL450992.2 | 7.4 | SORD2P | −18.7 |
| PART1 | 3.4 | LYPD2 | −17.7 |
| AC010326.4 | 3.1 | SEPT7P7 | −17.2 |
| AC022400.7 | 2.5 | PKNOX2 | −14.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rice, S.; Kim, S.-m.; Rodriguez, C.; Songock, W.; Raikhy, G.; Lopez, R.; Henderson, L.; Yusufji, A.; Bodily, J. Suppression of a Subset of Interferon-Induced Genes by Human Papillomavirus Type 16 E7 via a Cyclin Dependent Kinase 8-Dependent Mechanism. Viruses 2020, 12, 311. https://doi.org/10.3390/v12030311
Rice S, Kim S-m, Rodriguez C, Songock W, Raikhy G, Lopez R, Henderson L, Yusufji A, Bodily J. Suppression of a Subset of Interferon-Induced Genes by Human Papillomavirus Type 16 E7 via a Cyclin Dependent Kinase 8-Dependent Mechanism. Viruses. 2020; 12(3):311. https://doi.org/10.3390/v12030311
Chicago/Turabian StyleRice, Sadie, Seong-man Kim, Cynthia Rodriguez, William Songock, Gaurav Raikhy, Rebecca Lopez, Lauren Henderson, Arjun Yusufji, and Jason Bodily. 2020. "Suppression of a Subset of Interferon-Induced Genes by Human Papillomavirus Type 16 E7 via a Cyclin Dependent Kinase 8-Dependent Mechanism" Viruses 12, no. 3: 311. https://doi.org/10.3390/v12030311
APA StyleRice, S., Kim, S.-m., Rodriguez, C., Songock, W., Raikhy, G., Lopez, R., Henderson, L., Yusufji, A., & Bodily, J. (2020). Suppression of a Subset of Interferon-Induced Genes by Human Papillomavirus Type 16 E7 via a Cyclin Dependent Kinase 8-Dependent Mechanism. Viruses, 12(3), 311. https://doi.org/10.3390/v12030311