PERK-Mediated Unfolded Protein Response Signaling Restricts Replication of the Tick-Borne Flavivirus Langat Virus

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Viruses
2.2. Antibodies
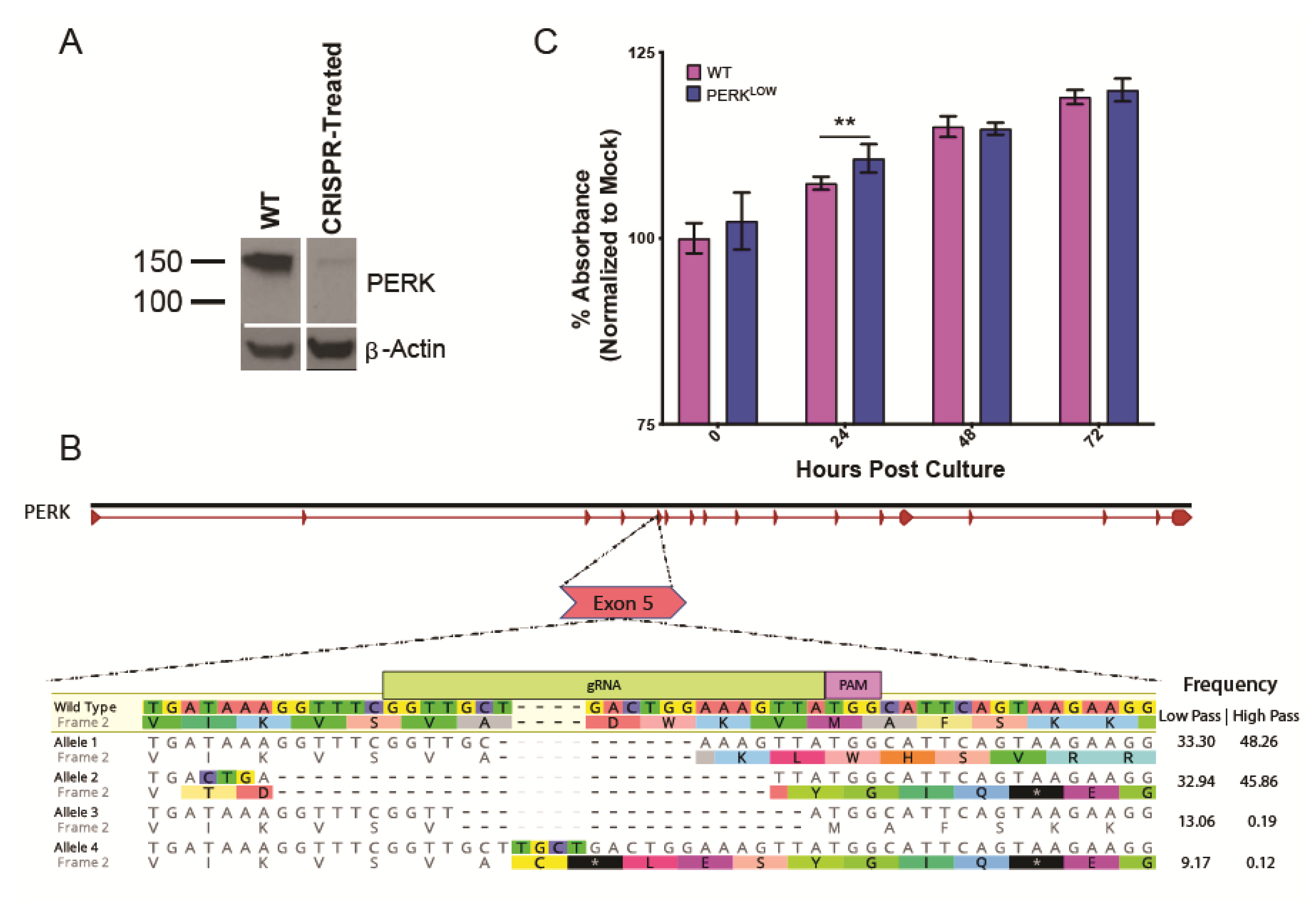
2.3. Generation of CRISPR Knockdown Cell Line
2.4. Cell Viability
2.5. Virus Quantification
2.6. Intracellular Genome Quantification
2.7. Protein Analysis
2.8. Immunofluorescence (IF) and Image Analysis
3. Results
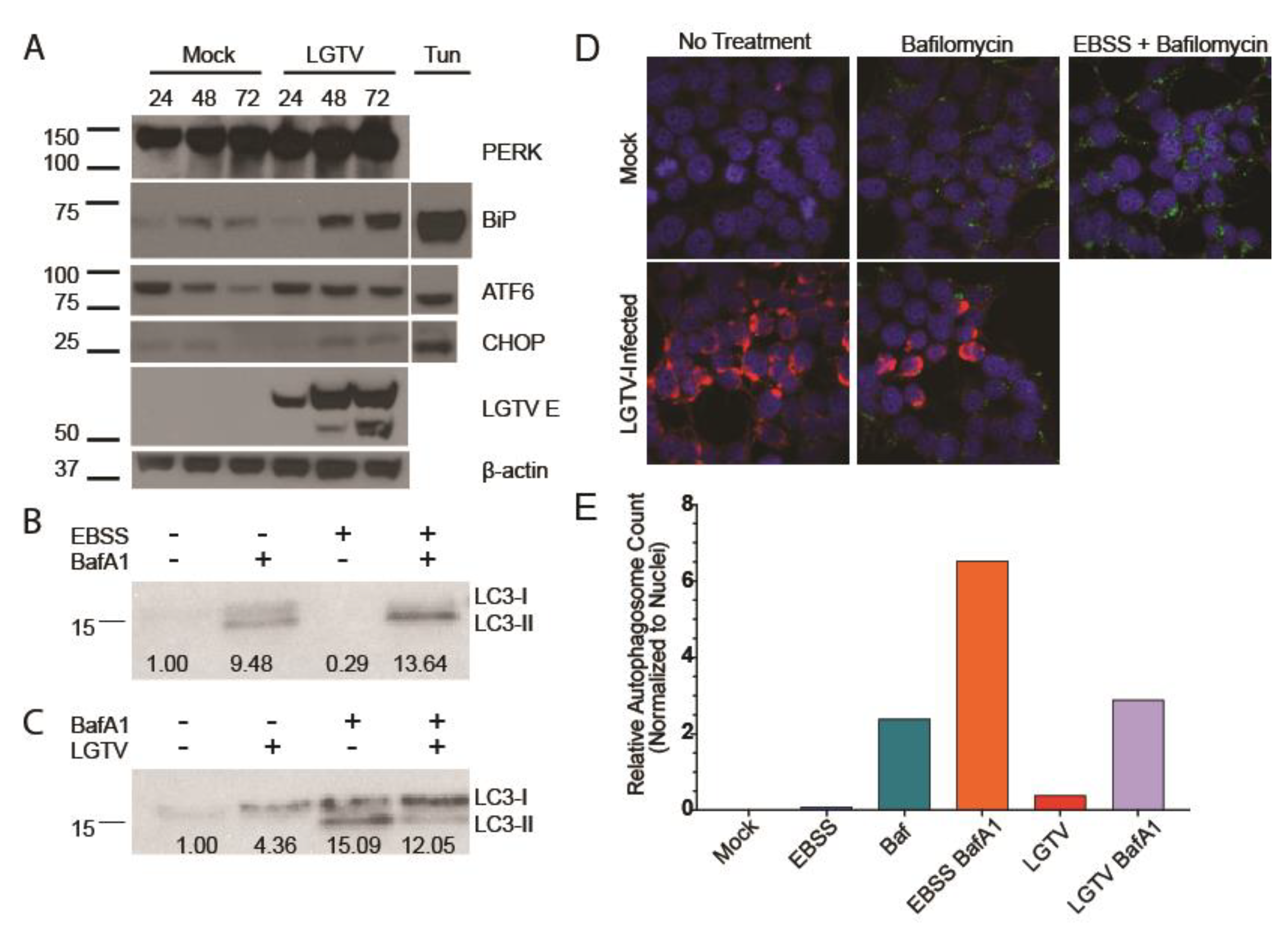
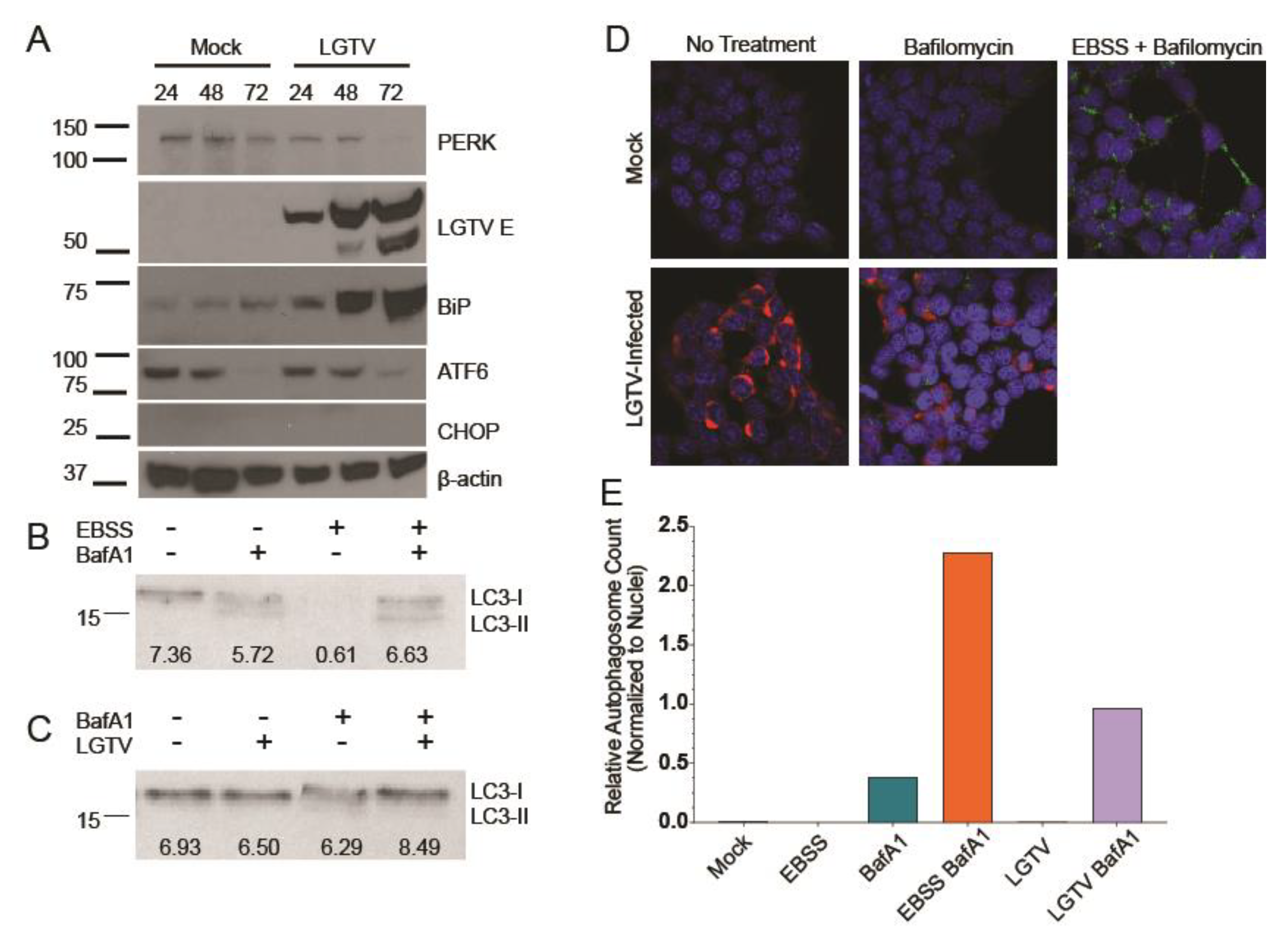
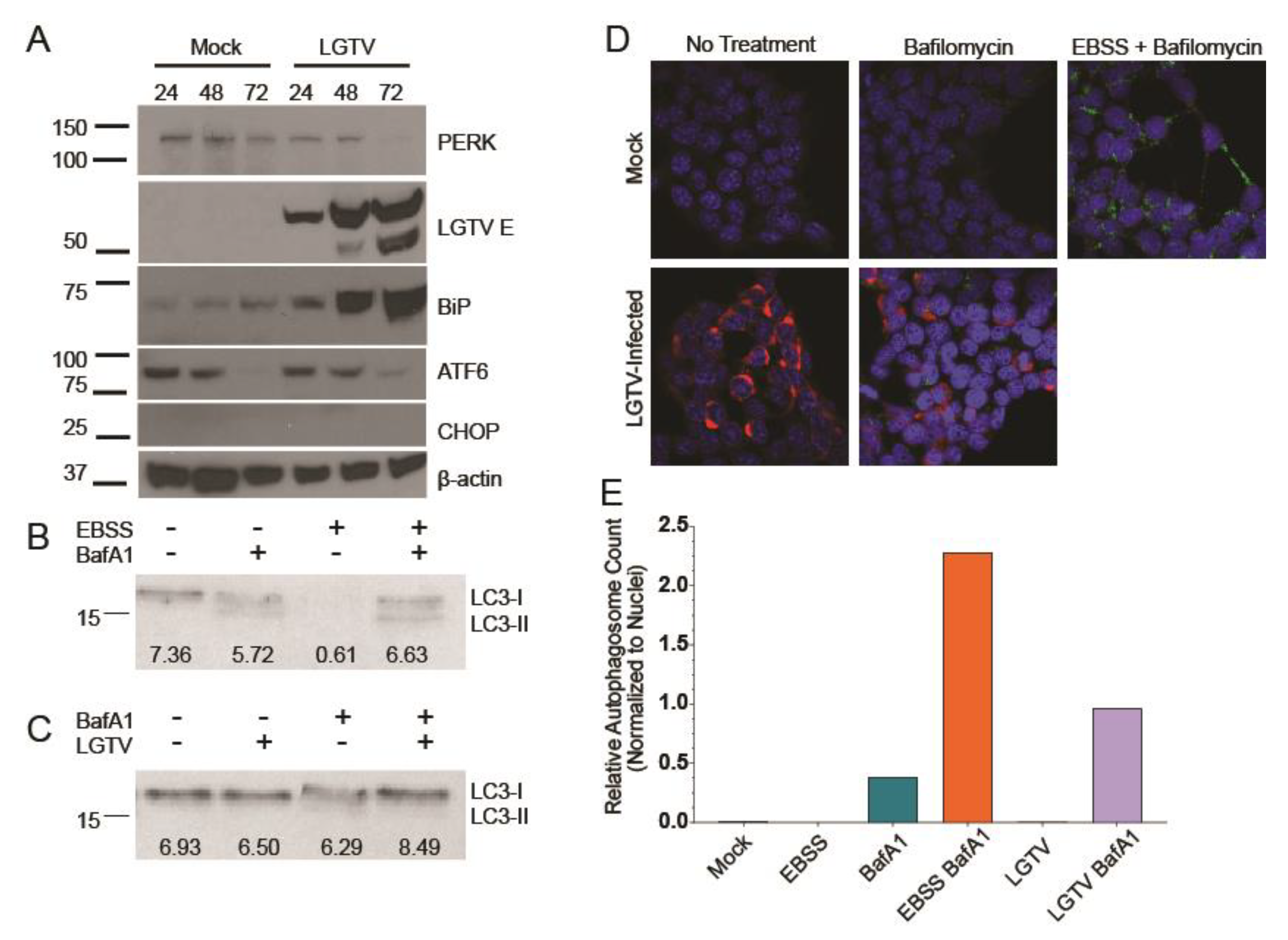
3.1. LGTV Infection Activates the UPR
3.2. LGTV Infection Alters Autophagic Flux
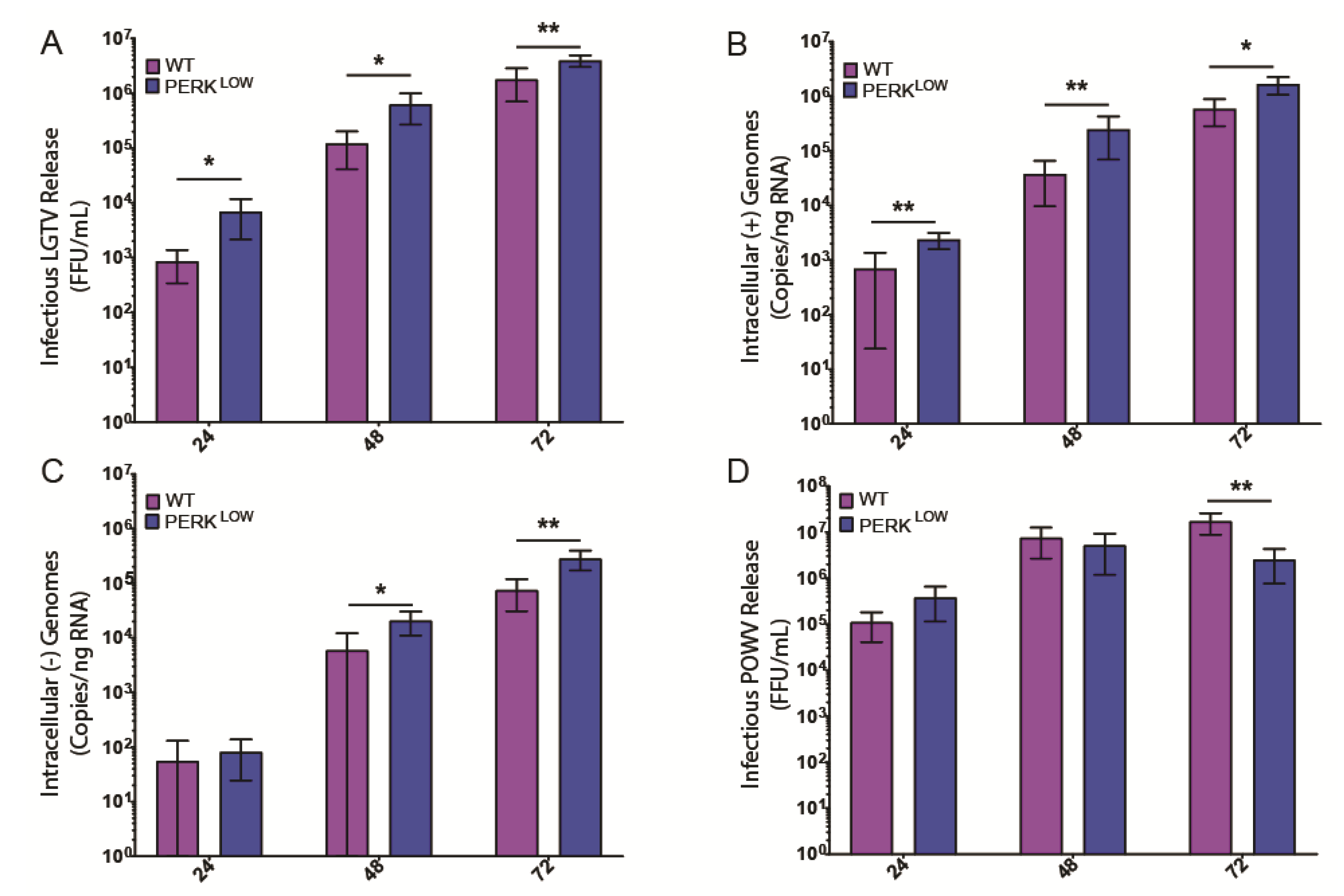
3.3. PERK Has an Antiviral Effect on LGTV Replication
3.4. PERK-Mediated CHOP Expression Is Important to Control LGTV Infection
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Fernandez-Garcia, M.D.; Mazzon, M.; Jacobs, M.; Amara, A. Pathogenesis of flavivirus infections: using and abusing the host cell. Cell Host Microbe 2009, 5, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.F.; Armstrong, P.M. Prevalence and genetic characterization of Powassan virus strains infecting Ixodes scapularis in Connecticut. Am. J. Trop. Med. Hyg. 2012, 87, 754–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, A.P., 2nd; Peters, R.J.; Prusinski, M.A.; Falco, R.C.; Ostfeld, R.S.; Kramer, L.D. Isolation of deer tick virus (Powassan virus, lineage II) from Ixodes scapularis and detection of antibody in vertebrate hosts sampled in the Hudson Valley, New York State. Parasit Vectors 2013, 6, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebel, G.D. Update on Powassan virus: emergence of a North American tick-borne flavivirus. Annu. Rev. Entomol. 2010, 55, 95–110. [Google Scholar] [CrossRef]
- Paules, C.I.; Marston, H.D.; Bloom, M.E.; Fauci, A.S. Tickborne Diseases—Confronting a Growing Threat. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef]
- Bogovic, P.; Strle, F. Tick-borne encephalitis: A review of epidemiology, clinical characteristics, and management. World J. Clin. Cases 2015, 3, 430–441. [Google Scholar] [CrossRef]
- Rieille, N.; Bressanelli, S.; Freire, C.C.; Arcioni, S.; Gern, L.; Peter, O.; Voordouw, M.J. Prevalence and phylogenetic analysis of tick-borne encephalitis virus (TBEV) in field-collected ticks (Ixodes ricinus) in southern Switzerland. Parasit Vectors 2014, 7, 443. [Google Scholar] [CrossRef] [Green Version]
- Ogden, N.H.; Radojevic, M.; Wu, X.; Duvvuri, V.R.; Leighton, P.A.; Wu, J. Estimated effects of projected climate change on the basic reproductive number of the Lyme disease vector Ixodes scapularis. Environ. Health Perspect 2014, 122, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Piantadosi, A.; Rubin, D.B.; McQuillen, D.P.; Hsu, L.; Lederer, P.A.; Ashbaugh, C.D.; Duffalo, C.; Duncan, R.; Thon, J.; Bhattacharyya, S.; et al. Emerging Cases of Powassan Virus Encephalitis in New England: Clinical Presentation, Imaging, and Review of the Literature. Clin. Infect. Dis. 2016, 62, 707–713. [Google Scholar] [CrossRef] [Green Version]
- Maffioli, C.; Grandgirard, D.; Engler, O.; Leib, S.L. A tick-borne encephalitis model in infant rats infected with langat virus. J. Neuropathol Exp. Neurol. 2014, 73, 1107–1115. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.S.; Walter, P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 1996, 87, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Rose, M.D.; Misra, L.M.; Vogel, J.P. KAR2, a karyogamy gene, is the yeast homolog of the mammalian BiP/GRP78 gene. Cell 1989, 57, 1211–1221. [Google Scholar] [CrossRef]
- Ryoo, H.D.; Domingos, P.M.; Kang, M.J.; Steller, H. Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J. 2007, 26, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewy, T.G.; Grabowski, J.M.; Bloom, M.E. BiP: Master Regulator of the Unfolded Protein Response and Crucial Factor in Flavivirus Biology. Yale J. Biol. Med. 2017, 90, 291–300. [Google Scholar]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Luo, S.; Baumeister, P.; Yang, S.; Abcouwer, S.F.; Lee, A.S. Induction of Grp78/BiP by translational block: activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J. Biol. Chem. 2003, 278, 37375–37385. [Google Scholar] [CrossRef] [Green Version]
- Hurtley, S.M.; Bole, D.G.; Hoover-Litty, H.; Helenius, A.; Copeland, C.S. Interactions of misfolded influenza virus hemagglutinin with binding protein (BiP). J. Cell Biol. 1989, 108, 2117–2126. [Google Scholar] [CrossRef]
- Baltzis, D.; Qu, L.K.; Papadopoulou, S.; Blais, J.D.; Bell, J.C.; Sonenberg, N.; Koromilas, A.E. Resistance to vesicular stomatitis virus infection requires a functional cross talk between the eukaryotic translation initiation factor 2alpha kinases PERK and PKR. J. Virol. 2004, 78, 12747–12761. [Google Scholar] [CrossRef] [Green Version]
- Tardif, K.D.; Waris, G.; Siddiqui, A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 2005, 13, 159–163. [Google Scholar] [CrossRef]
- Jordan, R.; Wang, L.; Graczyk, T.M.; Block, T.M.; Romano, P.R. Replication of a cytopathic strain of bovine viral diarrhea virus activates PERK and induces endoplasmic reticulum stress-mediated apoptosis of MDBK cells. J. Virol. 2002, 76, 9588–9599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isler, J.A.; Skalet, A.H.; Alwine, J.C. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 2005, 79, 6890–6899. [Google Scholar] [CrossRef] [Green Version]
- Wati, S.; Soo, M.L.; Zilm, P.; Li, P.; Paton, A.W.; Burrell, C.J.; Beard, M.; Carr, J.M. Dengue virus infection induces upregulation of GRP78, which acts to chaperone viral antigen production. J. Virol. 2009, 83, 12871–12880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.P.; Chang, C.M.; Hung, C.Y.; Tsai, M.C.; Schuyler, S.C.; Wang, R.Y. Japanese encephalitis virus co-opts the ER-stress response protein GRP78 for viral infectivity. Virol. J. 2011, 8, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medigeshi, G.R.; Lancaster, A.M.; Hirsch, A.J.; Briese, T.; Lipkin, W.I.; Defilippis, V.; Fruh, K.; Mason, P.W.; Nikolich-Zugich, J.; Nelson, J.A. West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 2007, 81, 10849–10860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena, J.; Harris, E. Dengue virus modulates the unfolded protein response in a time-dependent manner. J. Biol. Chem. 2011, 286, 14226–14236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrose, R.L.; Mackenzie, J.M. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J. Virol. 2011, 85, 2723–2732. [Google Scholar] [CrossRef] [Green Version]
- Datan, E.; Roy, S.G.; Germain, G.; Zali, N.; McLean, J.E.; Golshan, G.; Harbajan, S.; Lockshin, R.A.; Zakeri, Z. Dengue-induced autophagy, virus replication and protection from cell death require ER stress (PERK) pathway activation. Cell Death Dis. 2016, 7, e2127. [Google Scholar] [CrossRef] [Green Version]
- Carletti, T.; Zakaria, M.K.; Faoro, V.; Reale, L.; Kazungu, Y.; Licastro, D.; Marcello, A. Viral priming of cell intrinsic innate antiviral signaling by the unfolded protein response. Nat. Commun. 2019, 10, 3889. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Achazi, K.; Niedrig, M. Tick-borne encephalitis virus triggers inositol-requiring enzyme 1 (IRE1) and transcription factor 6 (ATF6) pathways of unfolded protein response. Virus Res. 2013, 178, 471–477. [Google Scholar] [CrossRef] [Green Version]
- Boyle, K.B.; Randow, F. The role of “eat-me” signals and autophagy cargo receptors in innate immunity. Curr. Opin. Microbiol. 2013, 16, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection - a double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Parnell, L.A.; Diamond, M.S.; Mysorekar, I.U. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J. Exp. Med. 2017, 214, 2303–2313. [Google Scholar] [CrossRef]
- Tasaki, T.; Nukuzuma, S.; Takegami, T. Impaired Japanese encephalitis virus replication in p62/SQSTM1 deficient mouse embryonic fibroblasts. Microbiol. Immunol. 2016, 60, 708–711. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Lei, H.Y.; Liu, M.T.; Wang, J.R.; Chen, S.H.; Jiang-Shieh, Y.F.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008, 374, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Metz, P.; Chiramel, A.; Chatel-Chaix, L.; Alvisi, G.; Bankhead, P.; Mora-Rodriguez, R.; Long, G.; Hamacher-Brady, A.; Brady, N.R.; Bartenschlager, R. Dengue Virus Inhibition of Autophagic Flux and Dependency of Viral Replication on Proteasomal Degradation of the Autophagy Receptor p62. J. Virol. 2015, 89, 8026–8041. [Google Scholar] [CrossRef] [Green Version]
- Beatman, E.; Oyer, R.; Shives, K.D.; Hedman, K.; Brault, A.C.; Tyler, K.L.; Beckham, J.D. West Nile virus growth is independent of autophagy activation. Virology 2012, 433, 262–272. [Google Scholar] [CrossRef] [Green Version]
- Vandergaast, R.; Fredericksen, B.L. West Nile virus (WNV) replication is independent of autophagy in mammalian cells. PLoS ONE 2012, 7, e45800. [Google Scholar] [CrossRef] [PubMed]
- Mlera, L.; Meade-White, K.; Saturday, G.; Scott, D.; Bloom, M.E. Modeling Powassan virus infection in Peromyscus leucopus, a natural host. PLoS Negl. Trop. Dis. 2017, 11, e0005346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offerdahl, D.K.; Dorward, D.W.; Hansen, B.T.; Bloom, M.E. A three-dimensional comparison of tick-borne flavivirus infection in mammalian and tick cell lines. PLoS ONE 2012, 7, e47912. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, J.M.; Perera, R.; Roumani, A.M.; Hedrick, V.E.; Inerowicz, H.D.; Hill, C.A.; Kuhn, R.J. Changes in the Proteome of Langat-Infected Ixodes scapularis ISE6 Cells: Metabolic Pathways Associated with Flavivirus Infection. PLoS Negl. Trop. Dis. 2016, 10, e0004180. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, J.M.; Tsetsarkin, K.A.; Long, D.; Scott, D.P.; Rosenke, R.; Schwan, T.G.; Mlera, L.; Offerdahl, D.K.; Pletnev, A.G.; Bloom, M.E. Flavivirus Infection of Ixodes scapularis (Black-Legged Tick) Ex Vivo Organotypic Cultures and Applications for Disease Control. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlera, L.; Offerdahl, D.K.; Martens, C.; Porcella, S.F.; Melik, W.; Bloom, M.E. Development of a Model System for Tick-Borne Flavivirus Persistence in HEK 293T Cells. MBio 2015, 6, e00614. [Google Scholar] [CrossRef] [Green Version]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef] [Green Version]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Xu, Y.; Xu, L.; Maccauro, G.; Rossi, B.; Chen, Y.; Li, H.; Zhang, J.; Sun, H.; Yang, Y.; et al. Regulation of autophagy via PERK-eIF2alpha effectively relieve the radiation myelitis induced by iodine-125. PLoS ONE 2013, 8, e76819. [Google Scholar] [CrossRef]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, E.; Kouroku, Y.; Isoai, A.; Kumagai, H.; Misutani, A.; Matsuda, C.; Hayashi, Y.K.; Momoi, T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 2007, 16, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Best, S.M.; Morris, K.L.; Shannon, J.G.; Robertson, S.J.; Mitzel, D.N.; Park, G.S.; Boer, E.; Wolfinbarger, J.B.; Bloom, M.E. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J. Virol. 2005, 79, 12828–12839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, M.S. Virus and host determinants of West Nile virus pathogenesis. PLoS Pathog. 2009, 5, e1000452. [Google Scholar] [CrossRef] [Green Version]
- Blazquez, A.B.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.C.; Martin-Acebes, M.A. Stress responses in flavivirus-infected cells: Activation of unfolded protein response and autophagy. Front. Microbiol. 2014, 5, 266. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, L.; Naik, I.; Braunstein, Z.; Zhong, J.; Ren, B. Transcription Factor C/EBP Homologous Protein in Health and Diseases. Front. Immunol. 2017, 8, 1612. [Google Scholar] [CrossRef]
- Williams, B.R. Signal integration via PKR. Sci. STKE 2001, 2001. [Google Scholar] [CrossRef]
- Han, A.P.; Yu, C.; Lu, L.; Fujiwara, Y.; Browne, C.; Chin, G.; Fleming, M.; Leboulch, P.; Orkin, S.H.; Chen, J.J. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001, 20, 6909–6918. [Google Scholar] [CrossRef] [Green Version]
- Kimball, S.R. Regulation of translation initiation by amino acids in eukaryotic cells. Prog. Mol. Subcell Biol. 2001, 26, 155–184. [Google Scholar]
- Elbahesh, H.; Scherbik, S.V.; Brinton, M.A. West Nile virus infection does not induce PKR activation in rodent cells. Virology 2011, 421, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.C.; Yu, C.Y.; Liang, J.J.; Lin, E.; Liao, C.L.; Lin, Y.L. Blocking double-stranded RNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J. Virol. 2012, 86, 10347–10358. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, J.M.; Offerdahl, D.K.; Bloom, M.E. The Use of Ex Vivo Organ Cultures in Tick-Borne Virus Research. ACS Infect. Dis. 2018, 4, 247–256. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewy, T.G.; Offerdahl, D.K.; Grabowski, J.M.; Kellman, E.; Mlera, L.; Chiramel, A.; Bloom, M.E. PERK-Mediated Unfolded Protein Response Signaling Restricts Replication of the Tick-Borne Flavivirus Langat Virus. Viruses 2020, 12, 328. https://doi.org/10.3390/v12030328
Lewy TG, Offerdahl DK, Grabowski JM, Kellman E, Mlera L, Chiramel A, Bloom ME. PERK-Mediated Unfolded Protein Response Signaling Restricts Replication of the Tick-Borne Flavivirus Langat Virus. Viruses. 2020; 12(3):328. https://doi.org/10.3390/v12030328
Chicago/Turabian StyleLewy, Tyler G., Danielle K. Offerdahl, Jeffrey M. Grabowski, Eliza Kellman, Luwanika Mlera, Abhilash Chiramel, and Marshall E. Bloom. 2020. "PERK-Mediated Unfolded Protein Response Signaling Restricts Replication of the Tick-Borne Flavivirus Langat Virus" Viruses 12, no. 3: 328. https://doi.org/10.3390/v12030328
APA StyleLewy, T. G., Offerdahl, D. K., Grabowski, J. M., Kellman, E., Mlera, L., Chiramel, A., & Bloom, M. E. (2020). PERK-Mediated Unfolded Protein Response Signaling Restricts Replication of the Tick-Borne Flavivirus Langat Virus. Viruses, 12(3), 328. https://doi.org/10.3390/v12030328