Virus-Induced Cytoplasmic Aggregates and Inclusions Are Critical Cellular Regulatory and Antiviral Factors



Abstract
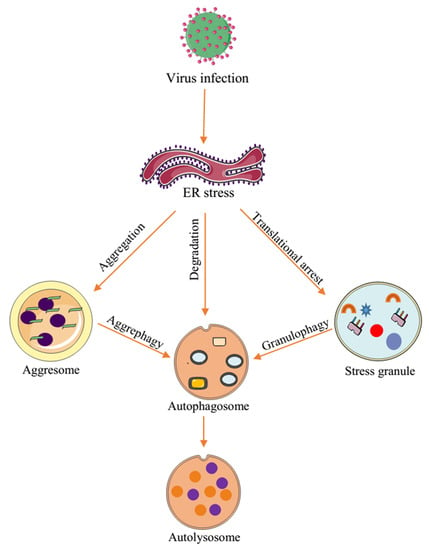
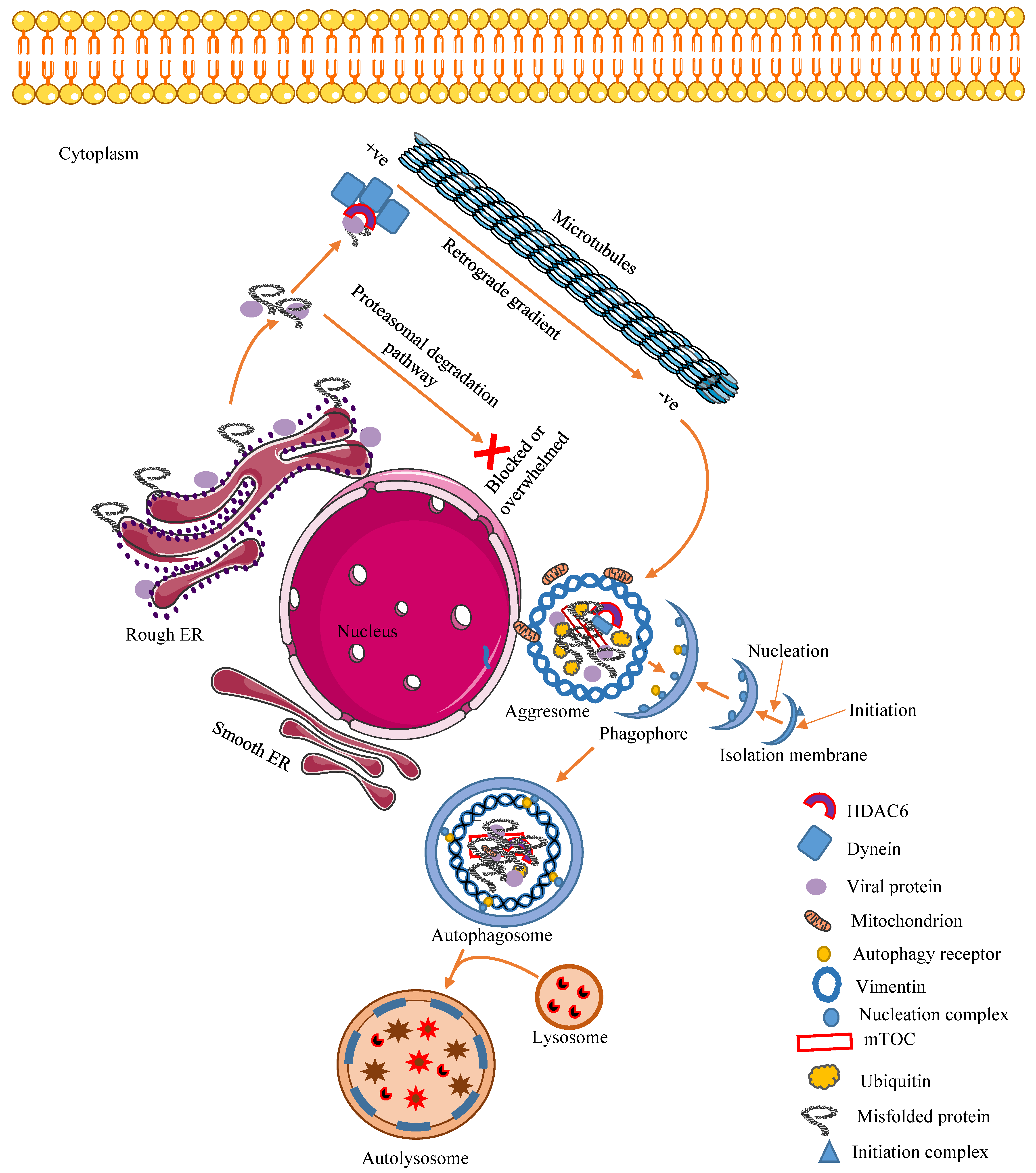
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. RNA Granules: Dynamic Modulators of Cellular Ribostasis and Antiviral Immunity
3. Aggresome: A Dynamic Modulator of Cellular Proteostasis and Antiviral Immunity
3.1. Mechanism of Aggresome Formation
3.2. Cellular Regulatory and Antiviral Effects of Aggresomes and Their Components
4. Autophagy: A Dynamic Modulator of Cellular Functions and Antiviral Immunity
Autophagy Regulates Cellular and Antiviral Responses to Virus Infection
5. Interplay between Virus-Induced Aggregates and Inclusions
6. Virus Exploitation of Cellular Inclusions and Aggregates
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Kaminskyy, V.; Zhivotovsky, B. To kill or be killed: How viruses interact with the cell death machinery. J. Intern. Med. 2010, 267, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Moshe, A.; Gorovits, R. Virus-induced aggregates in infected cells. Viruses 2012, 4, 2218–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wileman, T. Aggresomes and autophagy generate sites for virus replication. Science 2006, 312, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.G.; Loschi, M.; Desbats, M.A.; Boccaccio, G.L. RNA granules: The good, the bad and the ugly. Cell Signal. 2011, 23, 324–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, P.; Kedersha, N. Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Anderson, P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007, 431, 61–81. [Google Scholar] [CrossRef]
- Reineke, L.C.; Lloyd, R.E. Diversion of stress granules and P-bodies during viral infection. Virology 2013, 436, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Panas, M.D.; Ivanov, P.; Anderson, P. Mechanistic insights into mammalian stress granule dynamics. J. Cell Biol. 2016, 215, 313–323. [Google Scholar] [CrossRef]
- Gallois-Montbrun, S.; Kramer, B.; Swanson, C.M.; Byers, H.; Lynham, S.; Ward, M.; Malim, M.H. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J. Virol. 2007, 81, 2165–2178. [Google Scholar] [CrossRef] [Green Version]
- Valiente-Echeverria, F.; Melnychuk, L.; Mouland, A.J. Viral modulation of stress granules. Virus Res. 2012, 169, 430–437. [Google Scholar] [CrossRef]
- Lloyd, R.E. How do viruses interact with stress-associated RNA granules? PLoS Pathog. 2012, 8, e1002741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheth, U.; Parker, R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 2003, 300, 805–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makinen, K.; Lohmus, A.; Pollari, M. Plant RNA Regulatory Network and RNA Granules in Virus Infection. Front. Plant. Sci. 2017, 8, 2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.C.; Lloyd, R.E. Cytoplasmic RNA Granules and Viral Infection. Annu. Rev. Virol. 2014, 1, 147–170. [Google Scholar] [CrossRef] [Green Version]
- Walters, R.W.; Parker, R. Coupling of Ribostasis and Proteostasis: Hsp70 Proteins in mRNA Metabolism. Trends Biochem. Sci. 2015, 40, 552–559. [Google Scholar] [CrossRef] [Green Version]
- Mahboubi, H.; Stochaj, U. Cytoplasmic stress granules: Dynamic modulators of cell signaling and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 884–895. [Google Scholar] [CrossRef]
- Poblete-Duran, N.; Prades-Perez, Y.; Vera-Otarola, J.; Soto-Rifo, R.; Valiente-Echeverria, F. Who Regulates Whom? An Overview of RNA Granules and Viral Infections. Viruses 2016, 8, 180. [Google Scholar] [CrossRef]
- Beckham, C.J.; Parker, R. P bodies, stress granules, and viral life cycles. Cell Host Microbe 2008, 3, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Jogi, M.; Onomoto, K. Regulation of antiviral innate immune signaling by stress-induced RNA granules. J. Biochem. 2016, 159, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS ONE 2012, 7, e43031. [Google Scholar] [CrossRef]
- Bley, N.; Lederer, M.; Pfalz, B.; Reinke, C.; Fuchs, T.; Glass, M.; Moller, B.; Huttelmaier, S. Stress granules are dispensable for mRNA stabilization during cellular stress. Nucleic Acids Res. 2015, 43, e26. [Google Scholar] [CrossRef]
- Oh, S.W.; Onomoto, K.; Wakimoto, M.; Onoguchi, K.; Ishidate, F.; Fujiwara, T.; Yoneyama, M.; Kato, H.; Fujita, T. Leader-Containing Uncapped Viral Transcript Activates RIG-I in Antiviral Stress Granules. PLoS Pathog. 2016, 12, e1005444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Okabe, K.; Tani, T.; Funatsu, T. Dynamic association-dissociation and harboring of endogenous mRNAs in stress granules. J. Cell Sci. 2011, 124, 4087–4095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wileman, T. Aggresomes and pericentriolar sites of virus assembly: Cellular defense or viral design? Annu. Rev. Microbiol. 2007, 61, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Rozelle, D.K.; Filone, C.M.; Kedersha, N.; Connor, J.H. Activation of stress response pathways promotes formation of antiviral granules and restricts virus replication. Mol. Cell Biol. 2014, 34, 2003–2016. [Google Scholar] [CrossRef] [Green Version]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Diwaker, D.; Mishra, K.P.; Ganju, L. Effect of modulation of unfolded protein response pathway on dengue virus infection. Acta Biochim. Biophys. Sin. (Shanghai) 2015, 47, 960–968. [Google Scholar] [CrossRef] [Green Version]
- Olzmann, J.A.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination: A signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy 2008, 4, 85–87. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Statsyuk, A.V. An inhibitor of ubiquitin conjugation and aggresome formation. Chem. Sci. 2015, 6, 5235–5245. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Mata, R.; Gao, Y.S.; Sztul, E. Hassles with taking out the garbage: Aggravating aggresomes. Traffic 2002, 3, 388–396. [Google Scholar] [CrossRef]
- McConkey, D.J.; White, M.; Yan, W. HDAC inhibitor modulation of proteotoxicity as a therapeutic approach in cancer. Adv. Cancer Res. 2012, 116, 131–163. [Google Scholar] [CrossRef] [PubMed]
- Seibenhener, M.L.; Babu, J.R.; Geetha, T.; Wong, H.C.; Krishna, N.R.; Wooten, M.W. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell Biol. 2004, 24, 8055–8068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heir, R.; Ablasou, C.; Dumontier, E.; Elliott, M.; Fagotto-Kaufmann, C.; Bedford, F.K. The UBL domain of PLIC-1 regulates aggresome formation. EMBO Rep. 2006, 7, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Burnett, B.G.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin 3 regulates aggresome formation. Proc. Natl. Acad. Sci. USA 2005, 102, 4330–4335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigley, W.C.; Fabunmi, R.P.; Lee, M.G.; Marino, C.R.; Muallem, S.; DeMartino, G.N.; Thomas, P.J. Dynamic association of proteasomal machinery with the centrosome. J. Cell Biol. 1999, 145, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, K.; Jiang, Y.; He, Z.; Kitazato, K.; Wang, Y. Cellular defence or viral assist: The dilemma of HDAC6. J. Gen. Virol. 2017, 98, 322–337. [Google Scholar] [CrossRef]
- Serrador, J.M.; Cabrero, J.R.; Sancho, D.; Mittelbrunn, M.; Urzainqui, A.; Sanchez-Madrid, F. HDAC6 deacetylase activity links the tubulin cytoskeleton with immune synapse organization. Immunity 2004, 20, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Husain, M.; Cheung, C.Y. Histone deacetylase 6 inhibits influenza A virus release by downregulating the trafficking of viral components to the plasma membrane via its substrate, acetylated microtubules. J. Virol. 2014, 88, 11229–11239. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.P.; Tanaka, F.; Robitschek, J.; Sandoval, C.M.; Taye, A.; Markovic-Plese, S.; Fischbeck, K.H. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 2003, 12, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shevchenko, A.; Shevchenko, A.; Berk, A.J. Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J. Virol. 2005, 79, 14004–14016. [Google Scholar] [CrossRef] [Green Version]
- Dohner, K.; Nagel, C.H.; Sodeik, B. Viral stop-and-go along microtubules: Taking a ride with dynein and kinesins. Trends Microbiol. 2005, 13, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Meriin, A.B.; Wang, Y.; Sherman, M.Y. Isolation of aggresomes and other large aggregates. Curr. Protoc. Cell Biol. 2010, 48, 3–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, R.; Nanduri, P.; Rao, Y.; Panichelli, R.S.; Ito, A.; Yoshida, M.; Yao, T.P. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell 2013, 51, 819–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Salemi, L.M.; Almawi, A.W.; Lefebvre, K.J.; Schild-Poulter, C. Aggresome formation is regulated by RanBPM through an interaction with HDAC6. Biol. Open 2014, 3, 418–430. [Google Scholar] [CrossRef] [Green Version]
- Guidi, A.; Saccoccia, F.; Gennari, N.; Gimmelli, R.; Nizi, E.; Lalli, C.; Paonessa, G.; Papoff, G.; Bresciani, A.; Ruberti, G. Identification of novel multi-stage histone deacetylase (HDAC) inhibitors that impair Schistosoma mansoni viability and egg production. Parasites Vectors 2018, 11, 668. [Google Scholar] [CrossRef] [Green Version]
- Valera, M.S.; de Armas-Rillo, L.; Barroso-Gonzalez, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernandez, S.; Borel, S.; Garcia-Exposito, L.; Biard-Piechaczyk, M.; et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology 2015, 12, 53. [Google Scholar] [CrossRef] [Green Version]
- Dussart, S.; Courcoul, M.; Bessou, G.; Douaisi, M.; Duverger, Y.; Vigne, R.; Decroly, E. The Vif protein of human immunodeficiency virus type 1 is posttranslationally modified by ubiquitin. Biochem. Biophys. Res. Commun. 2004, 315, 66–72. [Google Scholar] [CrossRef]
- Mehle, A.; Strack, B.; Ancuta, P.; Zhang, C.; McPike, M.; Gabuzda, D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 2004, 279, 7792–7798. [Google Scholar] [CrossRef] [Green Version]
- Huo, L.; Li, D.; Sun, X.; Shi, X.; Karna, P.; Yang, W.; Liu, M.; Qiao, W.; Aneja, R.; Zhou, J. Regulation of Tat acetylation and transactivation activity by the microtubule-associated deacetylase HDAC6. J. Biol. Chem. 2011, 286, 9280–9286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusinzon, I.; Horvath, C.M. Positive and negative regulation of the innate antiviral response and beta interferon gene expression by deacetylation. Mol. Cell Biol. 2006, 26, 3106–3113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, S.; Fensterl, V.; Zhang, Y.; Veleeparambil, M.; Wetzel, J.L.; Sen, G.C. Inhibition of viral pathogenesis and promotion of the septic shock response to bacterial infection by IRF-3 are regulated by the acetylation and phosphorylation of its coactivators. MBio 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.J.; Lee, H.C.; Kim, J.H.; Park, S.Y.; Kim, T.H.; Lee, W.K.; Jang, D.J.; Yoon, J.E.; Choi, Y.I.; Kim, S.; et al. HDAC6 regulates cellular viral RNA sensing by deacetylation of RIG-I. EMBO J. 2016, 35, 429–442. [Google Scholar] [CrossRef]
- Kajitani, N.; Satsuka, A.; Yoshida, S.; Sakai, H. HPV18 E1^E4 is assembled into aggresome-like compartment and involved in sequestration of viral oncoproteins. Front. Microbiol. 2013, 4, 251. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef] [Green Version]
- Doring, T.; Zeyen, L.; Bartusch, C.; Prange, R. Hepatitis B Virus Subverts the Autophagy Elongation Complex Atg5-12/16L1 and Does Not Require Atg8/LC3 Lipidation for Viral Maturation. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Beatman, E.; Oyer, R.; Shives, K.D.; Hedman, K.; Brault, A.C.; Tyler, K.L.; Beckham, J.D. West Nile virus growth is independent of autophagy activation. Virology 2012, 433, 262–272. [Google Scholar] [CrossRef] [Green Version]
- Hafren, A.; Ustun, S.; Hochmuth, A.; Svenning, S.; Johansen, T.; Hofius, D. Turnip Mosaic Virus Counteracts Selective Autophagy of the Viral Silencing Suppressor HCpro. Plant. Physiol. 2018, 176, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Ge, X.; Xin, L.; Zhou, L.; Guo, X.; Yang, H. Nonstructural proteins 2C and 3D are involved in autophagy as induced by the encephalomyocarditis virus. Virol. J. 2014, 11, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozieres, A.; Viret, C.; Faure, M. Autophagy in Measles Virus Infection. Viruses 2017, 9, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, N.N.; Inagaki, F. Mechanisms of Autophagy. Annu. Rev. Biophys. 2015, 44, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Kohli, L.; Roth, K.A. Autophagy: Cerebral home cooking. Am. J. Pathol. 2010, 176, 1065–1071. [Google Scholar] [CrossRef]
- Fu, Q.; Shi, H.; Ni, W.; Shi, M.; Meng, L.; Zhang, H.; Ren, Y.; Guo, F.; Wang, P.; Qiao, J.; et al. Lentivirus-mediated Bos taurus bta-miR-29b overexpression interferes with bovine viral diarrhoea virus replication and viral infection-related autophagy by directly targeting ATG14 and ATG9A in Madin-Darby bovine kidney cells. J. Gen. Virol. 2015, 96, 85–94. [Google Scholar] [CrossRef]
- Sumpter, R., Jr.; Sirasanagandla, S.; Fernandez, A.F.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Pasztor, K.; Orosz, L.; Seprenyi, G.; Megyeri, K. Rubella virus perturbs autophagy. Med. Microbiol. Immunol. 2014, 203, 323–331. [Google Scholar] [CrossRef]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Kang, K.H.; Spector, S.A. Production of interferon alpha by human immunodeficiency virus type 1 in human plasmacytoid dendritic cells is dependent on induction of autophagy. J. Infect. Dis. 2012, 205, 1258–1267. [Google Scholar] [CrossRef]
- Lee, H.K.; Mattei, L.M.; Steinberg, B.E.; Alberts, P.; Lee, Y.H.; Chervonsky, A.; Mizushima, N.; Grinstein, S.; Iwasaki, A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 2010, 32, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Levine, B. Autophagy and viruses: Adversaries or allies? J. Innate Immun. 2013, 5, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Luo, H. Interplay between the cellular autophagy machinery and positive-stranded RNA viruses. Acta Biochim. Biophys. Sin. (Shanghai) 2012, 44, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Denizot, M.; Varbanov, M.; Espert, L.; Robert-Hebmann, V.; Sagnier, S.; Garcia, E.; Curriu, M.; Mamoun, R.; Blanco, J.; Biard-Piechaczyk, M. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 2008, 4, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Chiramel, A.I.; Brady, N.R.; Bartenschlager, R. Divergent roles of autophagy in virus infection. Cells 2013, 2, 83–104. [Google Scholar] [CrossRef] [Green Version]
- Dash, S.; Chava, S.; Aydin, Y.; Chandra, P.K.; Ferraris, P.; Chen, W.; Balart, L.A.; Wu, T.; Garry, R.F. Hepatitis C Virus Infection Induces Autophagy as a Prosurvival Mechanism to Alleviate Hepatic ER-Stress Response. Viruses 2016, 8, 150. [Google Scholar] [CrossRef]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investig. 2011, 121, 37–56. [Google Scholar] [CrossRef] [Green Version]
- Arimoto-Matsuzaki, K.; Saito, H.; Takekawa, M. TIA1 oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat. Commun. 2016, 7, 10252. [Google Scholar] [CrossRef] [Green Version]
- Joubert, P.E.; Meiffren, G.; Gregoire, I.P.; Pontini, G.; Richetta, C.; Flacher, M.; Azocar, O.; Vidalain, P.O.; Vidal, M.; Lotteau, V.; et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009, 6, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, N.; Kritsiligkou, P.; Grant, C.M. ER stress causes widespread protein aggregation and prion formation. J. Cell Biol. 2017, 216, 2295–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markmiller, S.; Fulzele, A.; Higgins, R.; Leonard, M.; Yeo, G.W.; Bennett, E.J. Active Protein Neddylation or Ubiquitylation Is Dispensable for Stress Granule Dynamics. Cell Rep. 2019, 27, 1356–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kedersha, N. RNA granules: Post-transcriptional and epigenetic modulators of gene expression. Nat. Rev. Mol. Cell Biol. 2009, 10, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef] [Green Version]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Kuo, S.H.; Lin, C.Y.; Fu, P.J.; Lin, Y.S.; Yeh, T.M.; Liu, H.S. Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Sci. Rep. 2018, 8, 489. [Google Scholar] [CrossRef]
- Mizushima, N. Physiological functions of autophagy. Curr Top. Microbiol. Immunol. 2009, 335, 71–84. [Google Scholar] [CrossRef]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Zhu, G.; Tang, Y.; Yan, J.; Han, S.; Yin, J.; Peng, B.; He, X.; Liu, W. HDAC6, A Novel Cargo for Autophagic Clearance of Stress Granules, Mediates the Repression of the Type I Interferon Response During Coxsackievirus A16 Infection. Front. Microbiol. 2020, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Monahan, Z.; Shewmaker, F.; Pandey, U.B. Stress granules at the intersection of autophagy and ALS. Brain Res. 2016, 1649, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamud, Y.; Qu, J.; Xue, Y.C.; Liu, H.; Deng, H.; Luo, H. CALCOCO2/NDP52 and SQSTM1/p62 differentially regulate coxsackievirus B3 propagation. Cell Death Differ. 2019, 26, 1062–1076. [Google Scholar] [CrossRef] [Green Version]
- Krick, R.; Bremer, S.; Welter, E.; Schlotterhose, P.; Muehe, Y.; Eskelinen, E.L.; Thumm, M. Cdc48/p97 and Shp1/p47 regulate autophagosome biogenesis in concert with ubiquitin-like Atg8. J. Cell Biol. 2010, 190, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Panas, M.D.; Schulte, T.; Thaa, B.; Sandalova, T.; Kedersha, N.; Achour, A.; McInerney, G.M. Viral and cellular proteins containing FGDF motifs bind G3BP to block stress granule formation. PLoS Pathog. 2015, 11, e1004659. [Google Scholar] [CrossRef] [Green Version]
- Valiente-Echeverria, F.; Melnychuk, L.; Vyboh, K.; Ajamian, L.; Gallouzi, I.E.; Bernard, N.; Mouland, A.J. eEF2 and Ras-GAP SH3 domain-binding protein (G3BP1) modulate stress granule assembly during HIV-1 infection. Nat. Commun. 2014, 5, 4819. [Google Scholar] [CrossRef] [Green Version]
- Nathans, R.; Chu, C.Y.; Serquina, A.K.; Lu, C.C.; Cao, H.; Rana, T.M. Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol. Cell 2009, 34, 696–709. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, J.D.; Tsai, W.C.; Lloyd, R.E. Multiple Poliovirus Proteins Repress Cytoplasmic RNA Granules. Viruses 2015, 7, 6127–6140. [Google Scholar] [CrossRef] [Green Version]
- Netherton, C.; Moffat, K.; Brooks, E.; Wileman, T. A guide to viral inclusions, membrane rearrangements, factories, and viroplasm produced during virus replication. Adv. Virus Res. 2007, 70, 101–182. [Google Scholar] [CrossRef]
- Muscolino, E.; Schmitz, R.; Loroch, S.; Caragliano, E.; Schneider, C.; Rizzato, M.; Kim, Y.H.; Krause, E.; Juranic Lisnic, V.; Sickmann, A.; et al. Herpesviruses induce aggregation and selective autophagy of host signalling proteins NEMO and RIPK1 as an immune-evasion mechanism. Nat. Microbiol. 2020, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Santarelli, R.; Farina, A.; Gonnella, R.; Lotti, L.V.; Faggioni, A.; Cirone, M. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J. Virol. 2014, 88, 12715–12726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemball, C.C.; Alirezaei, M.; Flynn, C.T.; Wood, M.R.; Harkins, S.; Kiosses, W.B.; Whitton, J.L. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 2010, 84, 12110–12124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennemann, N.J.; Coyne, C.B. Catch me if you can: The link between autophagy and viruses. PLoS Pathog. 2015, 11, e1004685. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [Green Version]
- Leidal, A.M.; Cyr, D.P.; Hill, R.J.; Lee, P.W.; McCormick, C. Subversion of autophagy by Kaposi’s sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe 2012, 11, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Lei, H.Y.; Liu, M.T.; Wang, J.R.; Chen, S.H.; Jiang-Shieh, Y.F.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008, 374, 240–248. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olasunkanmi, O.I.; Chen, S.; Mageto, J.; Zhong, Z. Virus-Induced Cytoplasmic Aggregates and Inclusions Are Critical Cellular Regulatory and Antiviral Factors. Viruses 2020, 12, 399. https://doi.org/10.3390/v12040399
Olasunkanmi OI, Chen S, Mageto J, Zhong Z. Virus-Induced Cytoplasmic Aggregates and Inclusions Are Critical Cellular Regulatory and Antiviral Factors. Viruses. 2020; 12(4):399. https://doi.org/10.3390/v12040399
Chicago/Turabian StyleOlasunkanmi, Oluwatayo Israel, Sijia Chen, James Mageto, and Zhaohua Zhong. 2020. "Virus-Induced Cytoplasmic Aggregates and Inclusions Are Critical Cellular Regulatory and Antiviral Factors" Viruses 12, no. 4: 399. https://doi.org/10.3390/v12040399
APA StyleOlasunkanmi, O. I., Chen, S., Mageto, J., & Zhong, Z. (2020). Virus-Induced Cytoplasmic Aggregates and Inclusions Are Critical Cellular Regulatory and Antiviral Factors. Viruses, 12(4), 399. https://doi.org/10.3390/v12040399