Host Protective Immune Responses against Influenza A Virus Infection


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
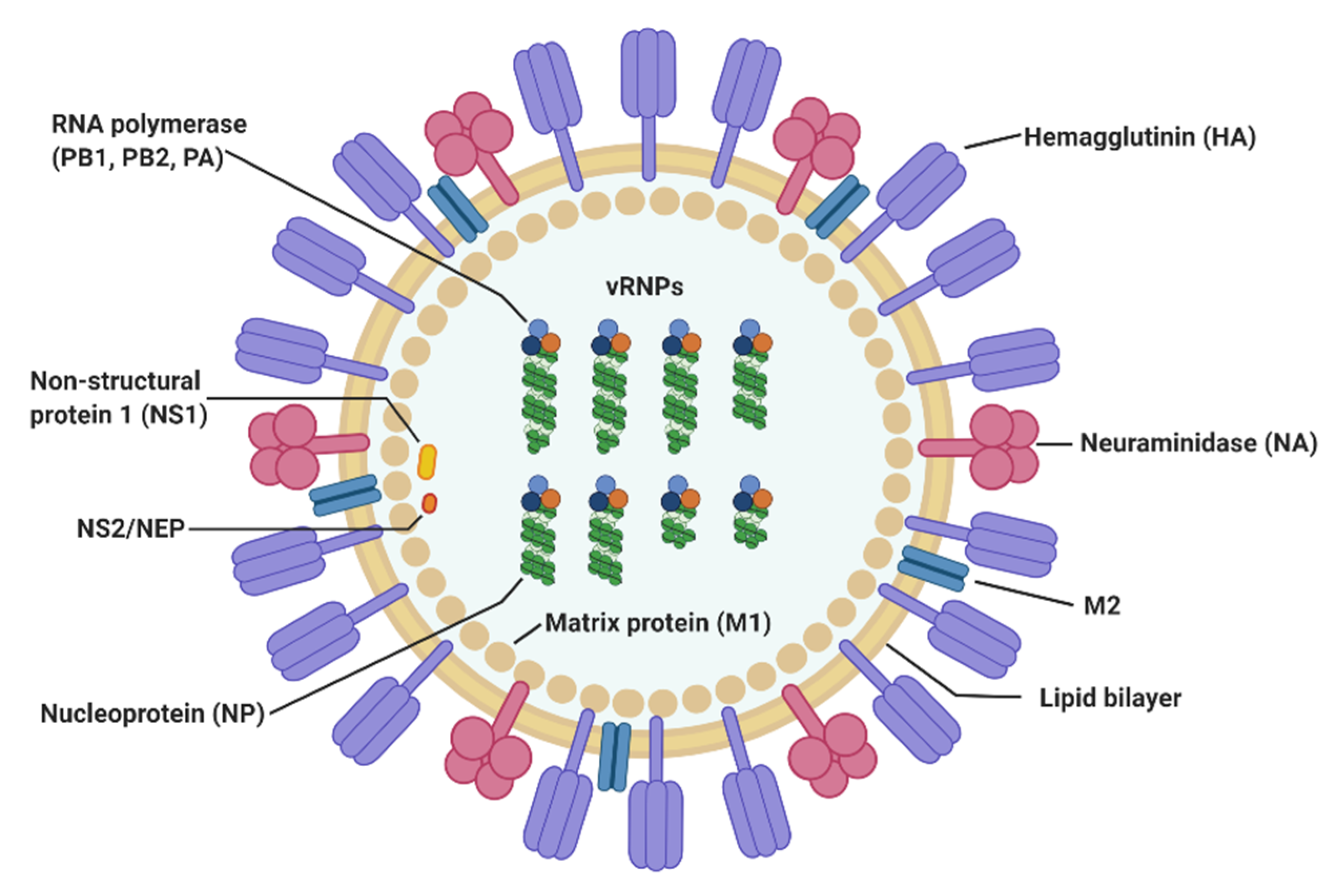
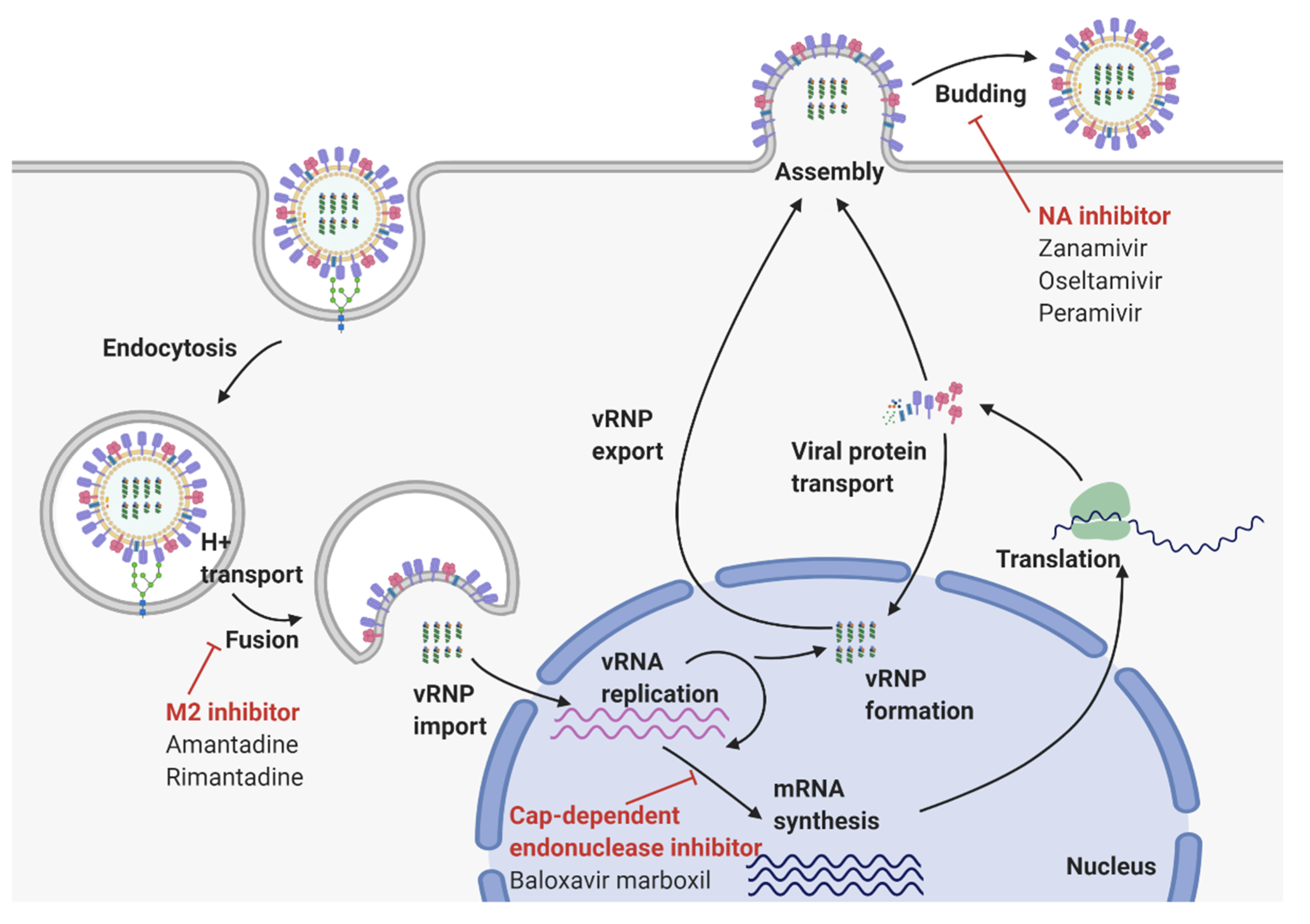
2. Virological Features of Influenza A Virus
3. Innate Sensors Recognizing the Influenza Virus
3.1. TLRs
3.2. RIG-I Signaling Pathway
3.3. Inflammasomes
3.4. cGAS-STING Pathway
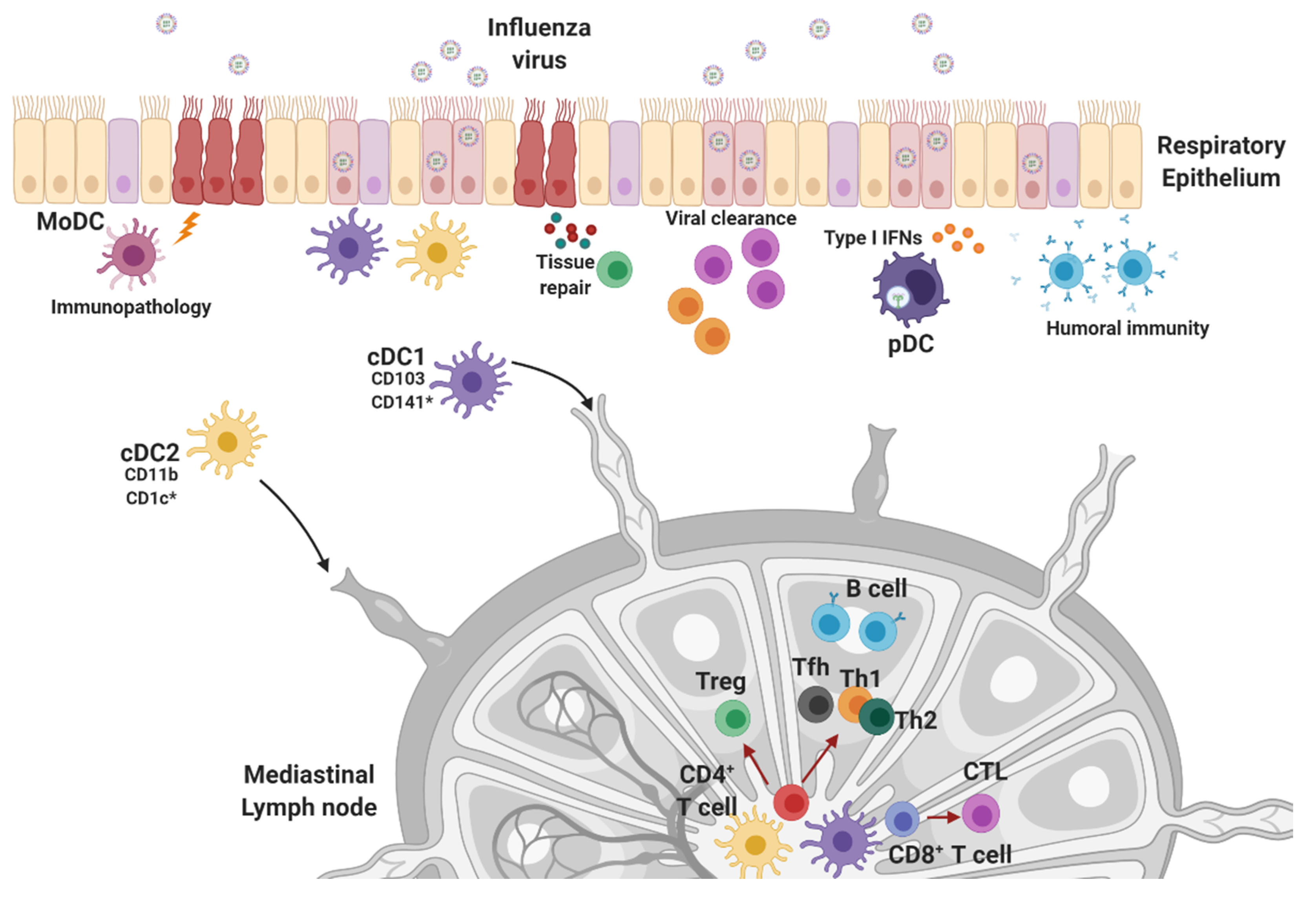
4. Innate Effector Cell Types in the Response to Influenza Infection
4.1. Natural Killer Cells, Neutrophils, and Macrophages
4.2. Dendritic Cells
5. Protective Adaptive Immune Responses against Influenza Infections
5.1. CD4+ Helper T Cell Response
5.2. Cytotoxic CD8+ T Cell Response
5.3. Humoral Immunity
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barr, I.G.; McCauley, J.; Cox, N.; Daniels, R.; Engelhardt, O.G.; Fukuda, K.; Grohmann, G.; Hay, A.; Kelso, A.; Klimov, A.; et al. Epidemiological, antigenic and genetic characteristics of seasonal influenza A(H1N1), A(H3N2) and B influenza viruses: Basis for the WHO recommendation on the composition of influenza vaccines for use in the 2009–2010 northern hemisphere season. Vaccine 2010, 28, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Shindo, N. Influenza virus polymerase inhibitors in clinical development. Curr. Opin. Infect. Dis. 2019, 32, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves first new flu drug in 20 years. Nat. Rev. Drug Discov. 2018, 17, 853. [Google Scholar] [CrossRef] [PubMed]
- Noshi, T.; Kitano, M.; Taniguchi, K.; Yamamoto, A.; Omoto, S.; Baba, K.; Hashimoto, T.; Ishida, K.; Kushima, Y.; Hattori, K.; et al. In vitro characterization of baloxavir acid, a first-in-class cap-dependent endonuclease inhibitor of the influenza virus polymerase PA subunit. Antivir. Res. 2018, 160, 109–117. [Google Scholar] [CrossRef]
- Vijaykrishna, D.; Poon, L.L.; Zhu, H.C.; Ma, S.K.; Li, O.T.; Cheung, C.L.; Smith, G.J.; Peiris, J.S.; Guan, Y. Reassortment of pandemic H1N1/2009 influenza A virus in swine. Science 2010, 328, 1529. [Google Scholar] [CrossRef] [Green Version]
- Dawood, F.S.; Iuliano, A.D.; Reed, C.; Meltzer, M.I.; Shay, D.K.; Cheng, P.Y.; Bandaranayake, D.; Breiman, R.F.; Brooks, W.A.; Buchy, P.; et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: A modelling study. Lancet Infect. Dis. 2012, 12, 687–695. [Google Scholar] [CrossRef] [Green Version]
- Cheung, T.K.; Poon, L.L. Biology of influenza a virus. Ann. N. Y. Acad. Sci. 2007, 1102, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Hayashida, H.; Toh, H.; Kikuno, R.; Miyata, T. Evolution of influenza virus genes. Mol. Biol. Evol. 1985, 2, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Ducatez, M.F.; Pelletier, C.; Meyer, G. Influenza D virus in cattle, France, 2011–2014. Emerg. Infect. Dis. 2015, 21, 368–371. [Google Scholar] [CrossRef]
- Trombetta, C.M.; Marchi, S.; Manini, I.; Kistner, O.; Li, F.; Piu, P.; Manenti, A.; Biuso, F.; Sreenivasan, C.; Druce, J.; et al. Influenza D Virus: Serological Evidence in the Italian Population from 2005 to 2017. Viruses 2019, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Foni, E.; Chiapponi, C.; Baioni, L.; Zanni, I.; Merenda, M.; Rosignoli, C.; Kyriakis, C.S.; Luini, M.V.; Mandola, M.L.; Bolzoni, L.; et al. Influenza D in Italy: Towards a better understanding of an emerging viral infection in swine. Sci. Rep. 2017, 7, 11660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, S.K.; Ma, W.; McDaniel, C.J.; Gray, G.C.; Lednicky, J.A. Serologic evidence of exposure to influenza D virus among persons with occupational contact with cattle. J. Clin. Virol. 2016, 81, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Virk, R.K.; Jayakumar, J.; Mendenhall, I.H.; Moorthy, M.; Lam, P.; Linster, M.; Lim, J.; Lin, C.; Oon, L.L.E.; Lee, H.K.; et al. Divergent evolutionary trajectories of influenza B viruses underlie their contemporaneous epidemic activity. Proc. Natl. Acad. Sci. USA 2020, 117, 619–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumm, R.E.; Heaton, N.S. The Development and Use of Reporter Influenza B Viruses. Viruses 2019, 11, 736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Pizarraya, A.; Perez-Romero, P.; Alvarez, R.; Aydillo, T.A.; Osorio-Gomez, G.; Milara-Ibanez, C.; Sanchez, M.; Pachon, J.; Cordero, E. Unexpected severity of cases of influenza B infection in patients that required hospitalization during the first postpandemic wave. J. Infect. 2012, 65, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Chaves, S.S.; Perez, A.; D’Mello, T.; Kirley, P.D.; Yousey-Hindes, K.; Farley, M.M.; Harris, M.; Sharangpani, R.; Lynfield, R.; et al. Comparing clinical characteristics between hospitalized adults with laboratory-confirmed influenza A and B virus infection. Clin. Infect. Dis. 2014, 59, 252–255. [Google Scholar] [CrossRef] [Green Version]
- Mosnier, A.; Caini, S.; Daviaud, I.; Nauleau, E.; Bui, T.T.; Debost, E.; Bedouret, B.; Agius, G.; van der Werf, S.; Lina, B.; et al. Clinical Characteristics Are Similar across Type A and B Influenza Virus Infections. PLoS ONE 2015, 10, e0136186. [Google Scholar] [CrossRef]
- Ping, J.; Lopes, T.J.; Neumann, G.; Kawaoka, Y. Development of high-yield influenza B virus vaccine viruses. Proc. Natl. Acad. Sci. USA 2016, 113, E8296–E8305. [Google Scholar] [CrossRef] [Green Version]
- Bui, M.; Wills, E.G.; Helenius, A.; Whittaker, G.R. Role of the influenza virus M1 protein in nuclear export of viral ribonucleoproteins. J. Virol. 2000, 74, 1781–1786. [Google Scholar] [CrossRef] [Green Version]
- Palese, P.; Tobita, K.; Ueda, M.; Compans, R.W. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 1974, 61, 397–410. [Google Scholar] [CrossRef]
- Kosik, I.; Yewdell, J.W. Influenza Hemagglutinin and Neuraminidase: Yin(-)Yang Proteins Coevolving to Thwart Immunity. Viruses 2019, 11, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samji, T. Influenza A: Understanding the viral life cycle. Yale J. Biol. Med. 2009, 82, 153–159. [Google Scholar] [PubMed]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, K.; Joseph, T. Scientific barriers to developing vaccines against avian influenza viruses. Nat. Rev. Immunol. 2007, 7, 267–278. [Google Scholar] [CrossRef]
- Nelson, M.I.; Holmes, E.C. The evolution of epidemic influenza. Nat. Rev. Genet. 2007, 8, 196–205. [Google Scholar] [CrossRef]
- Cohen, M.; Zhang, X.Q.; Senaati, H.P.; Chen, H.W.; Varki, N.M.; Schooley, R.T.; Gagneux, P. Influenza A penetrates host mucus by cleaving sialic acids with neuraminidase. Virol. J. 2013, 10, 321. [Google Scholar] [CrossRef] [Green Version]
- Stray, S.J.; Cummings, R.D.; Air, G.M. Influenza virus infection of desialylated cells. Glycobiology 2000, 10, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Thompson, C.I.; Barclay, W.S.; Zambon, M.C.; Pickles, R.J. Infection of human airway epithelium by human and avian strains of influenza a virus. J. Virol. 2006, 80, 8060–8068. [Google Scholar] [CrossRef] [Green Version]
- Reading, P.C.; Miller, J.L.; Anders, E.M. Involvement of the mannose receptor in infection of macrophages by influenza virus. J. Virol. 2000, 74, 5190–5197. [Google Scholar] [CrossRef]
- Upham, J.P.; Pickett, D.; Irimura, T.; Anders, E.M.; Reading, P.C. Macrophage receptors for influenza A virus: Role of the macrophage galactose-type lectin and mannose receptor in viral entry. J. Virol. 2010, 84, 3730–3737. [Google Scholar] [CrossRef] [Green Version]
- Ng, W.C.; Liong, S.; Tate, M.D.; Irimura, T.; Denda-Nagai, K.; Brooks, A.G.; Londrigan, S.L.; Reading, P.C. The macrophage galactose-type lectin can function as an attachment and entry receptor for influenza virus. J. Virol. 2014, 88, 1659–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillaire, M.L.; Nieuwkoop, N.J.; Boon, A.C.; de Mutsert, G.; Vogelzang-van Trierum, S.E.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Binding of DC-SIGN to the hemagglutinin of influenza A viruses supports virus replication in DC-SIGN expressing cells. PLoS ONE 2013, 8, e56164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Londrigan, S.L.; Turville, S.G.; Tate, M.D.; Deng, Y.M.; Brooks, A.G.; Reading, P.C. N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza A viruses into cells expressing DC-SIGN or L-SIGN. J. Virol. 2011, 85, 2990–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.F.; Huang, J.C.; Lee, Y.M.; Liu, S.J.; Chan, Y.J.; Chau, Y.P.; Chong, P.; Chen, Y.M. DC-SIGN mediates avian H5N1 influenza virus infection in cis and in trans. Biochem. Biophys. Res. Commun. 2008, 373, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, L.; Roosendahl, P.; Ng, W.C.; Brooks, A.G.; Reading, P.C.; Londrigan, S.L. Endocytic function is critical for influenza A virus infection via DC-SIGN and L-SIGN. Sci. Rep. 2016, 6, 19428. [Google Scholar] [CrossRef] [PubMed]
- Stencel-Baerenwald, J.E.; Reiss, K.; Reiter, D.M.; Stehle, T.; Dermody, T.S. The sweet spot: Defining virus-sialic acid interactions. Nat. Rev. Microbiol. 2014, 12, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Das, K.; Aramini, J.M.; Ma, L.C.; Krug, R.M.; Arnold, E. Structures of influenza A proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol. 2010, 17, 530–538. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Sieben, C.; Ludwig, K.; Hofer, C.T.; Chiantia, S.; Herrmann, A.; Eghiaian, F.; Schaap, I.A. pH-Controlled two-step uncoating of influenza virus. Biophys. J. 2014, 106, 1447–1456. [Google Scholar] [CrossRef] [Green Version]
- Grambas, S.; Bennett, M.S.; Hay, A.J. Influence of amantadine resistance mutations on the pH regulatory function of the M2 protein of influenza A viruses. Virology 1992, 191, 541–549. [Google Scholar] [CrossRef]
- Wang, C.; Takeuchi, K.; Pinto, L.H.; Lamb, R.A. Ion channel activity of influenza A virus M2 protein: Characterization of the amantadine block. J. Virol. 1993, 67, 5585–5594. [Google Scholar] [CrossRef] [Green Version]
- Dias, A.; Bouvier, D.; Crepin, T.; McCarthy, A.A.; Hart, D.J.; Baudin, F.; Cusack, S.; Ruigrok, R.W. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Guilligay, D.; Tarendeau, F.; Resa-Infante, P.; Coloma, R.; Crepin, T.; Sehr, P.; Lewis, J.; Ruigrok, R.W.; Ortin, J.; Hart, D.J.; et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 2008, 15, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Nayak, D.P. Mutational analysis of the conserved motifs of influenza A virus polymerase basic protein 1. J. Virol. 1994, 68, 1819–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulmanen, I.; Broni, B.A.; Krug, R.M. Role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc. Natl. Acad. Sci. USA 1981, 78, 7355–7359. [Google Scholar] [CrossRef] [Green Version]
- Pflug, A.; Lukarska, M.; Resa-Infante, P.; Reich, S.; Cusack, S. Structural insights into RNA synthesis by the influenza virus transcription-replication machine. Virus Res. 2017, 234, 103–117. [Google Scholar] [CrossRef]
- Fan, H.; Walker, A.P.; Carrique, L.; Keown, J.R.; Serna Martin, I.; Karia, D.; Sharps, J.; Hengrung, N.; Pardon, E.; Steyaert, J.; et al. Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature 2019, 573, 287–290. [Google Scholar] [CrossRef]
- Egorov, A.; Brandt, S.; Sereinig, S.; Romanova, J.; Ferko, B.; Katinger, D.; Grassauer, A.; Alexandrova, G.; Katinger, H.; Muster, T. Transfectant influenza A viruses with long deletions in the NS1 protein grow efficiently in Vero cells. J. Virol. 1998, 72, 6437–6441. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef] [Green Version]
- Kochs, G.; Garcia-Sastre, A.; Martinez-Sobrido, L. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 2007, 81, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Fortes, P.; Beloso, A.; Ortin, J. Influenza virus NS1 protein inhibits pre-mRNA splicing and blocks mRNA nucleocytoplasmic transport. EMBO J. 1994, 13, 704–712. [Google Scholar] [CrossRef]
- Qiu, Y.; Krug, R.M. The influenza virus NS1 protein is a poly(A)-binding protein that inhibits nuclear export of mRNAs containing poly(A). J. Virol. 1994, 68, 2425–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Xie, Y.; Munoz-Moreno, R.; Wang, J.; Zhang, L.; Esparza, M.; Garcia-Sastre, A.; Fontoura, B.M.A.; Ren, Y. Structural basis for influenza virus NS1 protein block of mRNA nuclear export. Nat. Microbiol. 2019, 4, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, M.; Chen, I.Y.; Kawaguchi, A.; Koshiba, T.; Nagata, K.; Takeyama, H.; Hasegawa, H.; Ichinohe, T. The RNA- and TRIM25-Binding Domains of Influenza Virus NS1 Protein Are Essential for Suppression of NLRP3 Inflammasome-Mediated Interleukin-1beta Secretion. J. Virol. 2016, 90, 4105–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruckle, A.; Haasbach, E.; Julkunen, I.; Planz, O.; Ehrhardt, C.; Ludwig, S. The NS1 protein of influenza A virus blocks RIG-I-mediated activation of the noncanonical NF-kappaB pathway and p52/RelB-dependent gene expression in lung epithelial cells. J. Virol. 2012, 86, 10211–10217. [Google Scholar] [CrossRef] [Green Version]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Mibayashi, M.; Martinez-Sobrido, L.; Loo, Y.M.; Cardenas, W.B.; Gale, M., Jr.; Garcia-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Chen, L.M.; Zeng, H.; Gomez, J.A.; Plowden, J.; Fujita, T.; Katz, J.M.; Donis, R.O.; Sambhara, S. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 2007, 36, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Neumann, G.; Hughes, M.T.; Kawaoka, Y. Influenza A virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J. 2000, 19, 6751–6758. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, R.E.; Talon, J.; Palese, P. The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J. 1998, 17, 288–296. [Google Scholar] [CrossRef] [Green Version]
- Gorai, T.; Goto, H.; Noda, T.; Watanabe, T.; Kozuka-Hata, H.; Oyama, M.; Takano, R.; Neumann, G.; Watanabe, S.; Kawaoka, Y. F1Fo-ATPase, F-type proton-translocating ATPase, at the plasma membrane is critical for efficient influenza virus budding. Proc. Natl. Acad. Sci. USA 2012, 109, 4615–4620. [Google Scholar] [CrossRef] [Green Version]
- McAuley, J.L.; Tate, M.D.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.E.; Mansell, A. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.E.; Oh, J.E.; Lee, H.K. Cell-Penetrating Mx1 Enhances Anti-Viral Resistance against Mucosal Influenza Viral Infection. Viruses 2019, 11, 109. [Google Scholar] [CrossRef] [PubMed]
- Wisskirchen, C.; Ludersdorfer, T.H.; Muller, D.A.; Moritz, E.; Pavlovic, J. The cellular RNA helicase UAP56 is required for prevention of double-stranded RNA formation during influenza A virus infection. J. Virol. 2011, 85, 8646–8655. [Google Scholar] [CrossRef] [Green Version]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [Green Version]
- Le Goffic, R.; Pothlichet, J.; Vitour, D.; Fujita, T.; Meurs, E.; Chignard, M.; Si-Tahar, M. Cutting Edge: Influenza A Virus Activates TLR3-Dependent Inflammatory and RIG-I-Dependent Antiviral Responses in Human Lung Epithelial Cells. J. Immunol. 2007, 178, 3368–3372. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Mak, T.W.; Sen, G.; Li, X. Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc. Natl. Acad. Sci. USA 2004, 101, 3533–3538. [Google Scholar] [CrossRef] [Green Version]
- Le Goffic, R.; Balloy, V.; Lagranderie, M.; Alexopoulou, L.; Escriou, N.; Flavell, R.; Chignard, M.; Si-Tahar, M. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006, 2, e53. [Google Scholar] [CrossRef]
- Heer, A.K.; Shamshiev, A.; Donda, A.; Uematsu, S.; Akira, S.; Kopf, M.; Marsland, B.J. TLR signaling fine-tunes anti-influenza B cell responses without regulating effector T cell responses. J. Immunol. 2007, 178, 2182–2191. [Google Scholar] [CrossRef]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Pang, I.K.; Pillai, P.S.; Iwasaki, A. Efficient influenza A virus replication in the respiratory tract requires signals from TLR7 and RIG-I. Proc. Natl. Acad. Sci. USA 2013, 110, 13910–13915. [Google Scholar] [CrossRef] [Green Version]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Stegemann-Koniszewski, S.; Behrens, S.; Boehme, J.D.; Hochnadel, I.; Riese, P.; Guzman, C.A.; Kroger, A.; Schreiber, J.; Gunzer, M.; Bruder, D. Respiratory Influenza A Virus Infection Triggers Local and Systemic Natural Killer Cell Activation via Toll-Like Receptor 7. Front. Immunol. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, C.B.; Moltedo, B.; Alexopoulou, L.; Bonifaz, L.; Flavell, R.A.; Moran, T.M. TLR-independent induction of dendritic cell maturation and adaptive immunity by negative-strand RNA viruses. J. Immunol. 2004, 173, 6882–6889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, S.; Ishii, K.J.; Kumar, H.; Tanimoto, T.; Coban, C.; Uematsu, S.; Kawai, T.; Akira, S. Differential Role of TLR- and RLR-Signaling in the Immune Responses to Influenza A Virus Infection and Vaccination. J. Immunol. 2007, 179, 4711–4720. [Google Scholar] [CrossRef] [PubMed]
- Jeisy-Scott, V.; Kim, J.H.; Davis, W.G.; Cao, W.; Katz, J.M.; Sambhara, S. TLR7 recognition is dispensable for influenza virus A infection but important for the induction of hemagglutinin-specific antibodies in response to the 2009 pandemic split vaccine in mice. J. Virol. 2012, 86, 10988–10998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, S.U.; Kwon, H.J.; Song, J.H.; Byun, Y.H.; Seong, B.L.; Kawai, T.; Akira, S.; Kweon, M.N. MyD88 signaling is indispensable for primary influenza A virus infection but dispensable for secondary infection. J. Virol. 2010, 84, 12713–12722. [Google Scholar] [CrossRef] [Green Version]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS ONE 2012, 7, e43031. [Google Scholar] [CrossRef]
- Khaperskyy, D.A.; Hatchette, T.F.; McCormick, C. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J. 2012, 26, 1629–1639. [Google Scholar] [CrossRef]
- Mok, B.W.; Song, W.; Wang, P.; Tai, H.; Chen, Y.; Zheng, M.; Wen, X.; Lau, S.Y.; Wu, W.L.; Matsumoto, K.; et al. The NS1 protein of influenza A virus interacts with cellular processing bodies and stress granules through RNA-associated protein 55 (RAP55) during virus infection. J. Virol. 2012, 86, 12695–12707. [Google Scholar] [CrossRef] [Green Version]
- Guarda, G.; Zenger, M.; Yazdi, A.S.; Schroder, K.; Ferrero, I.; Menu, P.; Tardivel, A.; Mattmann, C.; Tschopp, J. Differential expression of NLRP3 among hematopoietic cells. J. Immunol. 2011, 186, 2529–2534. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.C.; Kang, H.R.; Yoon, H.; Kang, S.J.; Ting, J.P.; Song, M.J. Influenza A Virus NS1 Protein Inhibits the NLRP3 Inflammasome. PLoS ONE 2015, 10, e0126456. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, P.G.; Dash, P.; Aldridge, J.R., Jr.; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 2009, 30, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Ichinohe, T.; Lee, H.K.; Ogura, Y.; Flavell, R.; Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 2009, 206, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef] [Green Version]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.; Prabakaran, T.; Laustsen, A.; Jorgensen, S.E.; Rahbaek, S.H.; Jensen, S.B.; Nielsen, R.; Leber, J.H.; Decker, T.; Horan, K.A.; et al. Listeria monocytogenes induces IFNbeta expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J. 2014, 33, 1654–1666. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Jacobs, S.R.; West, J.A.; Stopford, C.; Zhang, Z.; Davis, Z.; Barber, G.N.; Glaunsinger, B.A.; Dittmer, D.P.; Damania, B. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 2015, 112, E4306–E4315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriyama, M.; Koshiba, T.; Ichinohe, T. Influenza A virus M2 protein triggers mitochondrial DNA-mediated antiviral immune responses. Nat. Commun. 2019, 10, 4624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veckman, V.; Osterlund, P.; Fagerlund, R.; Melen, K.; Matikainen, S.; Julkunen, I. TNF-alpha and IFN-alpha enhance influenza-A-virus-induced chemokine gene expression in human A549 lung epithelial cells. Virology 2006, 345, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Wareing, M.D.; Lyon, A.; Inglis, C.; Giannoni, F.; Charo, I.; Sarawar, S.R. Chemokine regulation of the inflammatory response to a low-dose influenza infection in CCR2-/- mice. J. Leukoc. Biol. 2007, 81, 793–801. [Google Scholar] [CrossRef]
- Wareing, M.D.; Lyon, A.B.; Lu, B.; Gerard, C.; Sarawar, S.R. Chemokine expression during the development and resolution of a pulmonary leukocyte response to influenza A virus infection in mice. J. Leukoc. Biol. 2004, 76, 886–895. [Google Scholar] [CrossRef]
- Mandelboim, O.; Lieberman, N.; Lev, M.; Paul, L.; Arnon, T.I.; Bushkin, Y.; Davis, D.M.; Strominger, J.L.; Yewdell, J.W.; Porgador, A. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 2001, 409, 1055–1060. [Google Scholar] [CrossRef]
- Arnon, T.I.; Lev, M.; Katz, G.; Chernobrov, Y.; Porgador, A.; Mandelboim, O. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur. J. Immunol. 2001, 31, 2680–2689. [Google Scholar] [CrossRef]
- Mendelson, M.; Tekoah, Y.; Zilka, A.; Gershoni-Yahalom, O.; Gazit, R.; Achdout, H.; Bovin, N.V.; Meningher, T.; Mandelboim, M.; Mandelboim, O.; et al. NKp46 O-glycan sequences that are involved in the interaction with hemagglutinin type 1 of influenza virus. J. Virol. 2010, 84, 3789–3797. [Google Scholar] [CrossRef] [Green Version]
- Jegaskanda, S.; Vanderven, H.A.; Tan, H.X.; Alcantara, S.; Wragg, K.M.; Parsons, M.S.; Chung, A.W.; Juno, J.A.; Kent, S.J. Influenza Virus Infection Enhances Antibody-Mediated NK Cell Functions via Type I Interferon-Dependent Pathways. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.; Scott, J.M.; Kakarla, T.; Duriancik, D.M.; Choi, S.; Cho, C.; Lee, T.; Park, H.; French, A.R.; Beli, E.; et al. Activation mechanisms of natural killer cells during influenza virus infection. PLoS ONE 2012, 7, e51858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, G.E.; Ostridge, K.; Khakoo, S.I.; Wilkinson, T.M.A.; Staples, K.J. Human CD49a(+) Lung Natural Killer Cell Cytotoxicity in Response to Influenza A Virus. Front. Immunol. 2018, 9, 1671. [Google Scholar] [CrossRef] [PubMed]
- Nogusa, S.; Ritz, B.W.; Kassim, S.H.; Jennings, S.R.; Gardner, E.M. Characterization of age-related changes in natural killer cells during primary influenza infection in mice. Mech. Ageing Dev. 2008, 129, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Mori, I.; Hossain, M.J.; Dong, L.; Takeda, K.; Kimura, Y. Interleukin-18 improves the early defence system against influenza virus infection by augmenting natural killer cell-mediated cytotoxicity. J. Gen. Virol. 2004, 85, 423–428. [Google Scholar] [CrossRef]
- Zhou, G.; Juang, S.W.; Kane, K.P. NK cells exacerbate the pathology of influenza virus infection in mice. Eur. J. Immunol. 2013, 43, 929–938. [Google Scholar] [CrossRef]
- Abdul-Careem, M.F.; Mian, M.F.; Yue, G.; Gillgrass, A.; Chenoweth, M.J.; Barra, N.G.; Chew, M.V.; Chan, T.; Al-Garawi, A.A.; Jordana, M.; et al. Critical role of natural killer cells in lung immunopathology during influenza infection in mice. J. Infect. Dis. 2012, 206, 167–177. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Moki, T.; Takizawa, T.; Shiratsuchi, A.; Nakanishi, Y. Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J. Immunol. 2007, 178, 2448–2457. [Google Scholar] [CrossRef] [Green Version]
- Camp, J.V.; Jonsson, C.B. A Role for Neutrophils in Viral Respiratory Disease. Front. Immunol. 2017, 8, 550. [Google Scholar] [CrossRef] [Green Version]
- Tumpey, T.M.; Garcia-Sastre, A.; Taubenberger, J.K.; Palese, P.; Swayne, D.E.; Pantin-Jackwood, M.J.; Schultz-Cherry, S.; Solorzano, A.; Van Rooijen, N.; Katz, J.M.; et al. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: Functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J. Virol. 2005, 79, 14933–14944. [Google Scholar] [CrossRef] [Green Version]
- Tate, M.D.; Brooks, A.G.; Reading, P.C. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir. Res. 2008, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Tate, M.D.; Brooks, A.G.; Reading, P.C.; Mintern, J.D. Neutrophils sustain effective CD8(+) T-cell responses in the respiratory tract following influenza infection. Immunol. Cell Biol. 2012, 90, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Hyun, Y.M.; Lambert-Emo, K.; Capece, T.; Bae, S.; Miller, R.; Topham, D.J.; Kim, M. Neutrophil trails guide influenza-specific CD8(+) T cells in the airways. Science 2015, 349, aaa4352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, M.; Klauschen, F.; Kuchen, S.; Germain, R.N. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 2013, 154, 197–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, B.C.; Whitsett, J.A. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu. Rev. Physiol. 2002, 64, 775–802. [Google Scholar] [CrossRef]
- Kumagai, Y.; Takeuchi, O.; Kato, H.; Kumar, H.; Matsui, K.; Morii, E.; Aozasa, K.; Kawai, T.; Akira, S. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity 2007, 27, 240–252. [Google Scholar] [CrossRef] [Green Version]
- Tate, M.D.; Pickett, D.L.; van Rooijen, N.; Brooks, A.G.; Reading, P.C. Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J. Virol. 2010, 84, 7569–7580. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; Nobs, S.P.; Heer, A.K.; Kurrer, M.; Klinke, G.; van Rooijen, N.; Vogel, J.; Kopf, M. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog. 2014, 10, e1004053. [Google Scholar] [CrossRef] [Green Version]
- Purnama, C.; Ng, S.L.; Tetlak, P.; Setiagani, Y.A.; Kandasamy, M.; Baalasubramanian, S.; Karjalainen, K.; Ruedl, C. Transient ablation of alveolar macrophages leads to massive pathology of influenza infection without affecting cellular adaptive immunity. Eur. J. Immunol. 2014, 44, 2003–2012. [Google Scholar] [CrossRef]
- Cardani, A.; Boulton, A.; Kim, T.S.; Braciale, T.J. Alveolar Macrophages Prevent Lethal Influenza Pneumonia by Inhibiting Infection Of Type-1 Alveolar Epithelial Cells. PLoS Pathog. 2017, 13, e1006140. [Google Scholar] [CrossRef]
- Jung, S.; Unutmaz, D.; Wong, P.; Sano, G.; De los Santos, K.; Sparwasser, T.; Wu, S.; Vuthoori, S.; Ko, K.; Zavala, F.; et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 2002, 17, 211–220. [Google Scholar] [CrossRef] [Green Version]
- GeurtsvanKessel, C.H.; Willart, M.A.; van Rijt, L.S.; Muskens, F.; Kool, M.; Baas, C.; Thielemans, K.; Bennett, C.; Clausen, B.E.; Hoogsteden, H.C.; et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J. Exp. Med. 2008, 205, 1621–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granot, T.; Senda, T.; Carpenter, D.J.; Matsuoka, N.; Weiner, J.; Gordon, C.L.; Miron, M.; Kumar, B.V.; Griesemer, A.; Ho, S.H.; et al. Dendritic Cells Display Subset and Tissue-Specific Maturation Dynamics over Human Life. Immunity 2017, 46, 504–515. [Google Scholar] [CrossRef] [Green Version]
- Plantinga, M.; Guilliams, M.; Vanheerswynghels, M.; Deswarte, K.; Branco-Madeira, F.; Toussaint, W.; Vanhoutte, L.; Neyt, K.; Killeen, N.; Malissen, B.; et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 2013, 38, 322–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.L.; Suzuki, Y.; Nakano, H.; Ramsburg, E.; Gunn, M.D. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J. Immunol. 2008, 180, 2562–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minoda, Y.; Virshup, I.; Leal Rojas, I.; Haigh, O.; Wong, Y.; Miles, J.J.; Wells, C.A.; Radford, K.J. Human CD141(+) Dendritic Cell and CD1c(+) Dendritic Cell Undergo Concordant Early Genetic Programming after Activation in Humanized Mice In Vivo. Front. Immunol. 2017, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Hao, X.; Kim, T.S.; Braciale, T.J. Differential response of respiratory dendritic cell subsets to influenza virus infection. J. Virol. 2008, 82, 4908–4919. [Google Scholar] [CrossRef] [Green Version]
- Helft, J.; Manicassamy, B.; Guermonprez, P.; Hashimoto, D.; Silvin, A.; Agudo, J.; Brown, B.D.; Schmolke, M.; Miller, J.C.; Leboeuf, M.; et al. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J. Clin. Investig. 2012, 122, 4037–4047. [Google Scholar] [CrossRef]
- Waithman, J.; Zanker, D.; Xiao, K.; Oveissi, S.; Wylie, B.; Ng, R.; Togel, L.; Chen, W. Resident CD8(+) and migratory CD103(+) dendritic cells control CD8 T cell immunity during acute influenza infection. PLoS ONE 2013, 8, e66136. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.L.; Teo, Y.J.; Setiagani, Y.A.; Karjalainen, K.; Ruedl, C. Type 1 Conventional CD103(+) Dendritic Cells Control Effector CD8(+) T Cell Migration, Survival, and Memory Responses During Influenza Infection. Front. Immunol. 2018, 9, 3043. [Google Scholar] [CrossRef] [Green Version]
- Pang, I.K.; Ichinohe, T.; Iwasaki, A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8(+) T cell responses to influenza A virus. Nat. Immunol. 2013, 14, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.W.; Prabhu, N.; Betts, R.J.; Ge, M.Q.; Dai, X.; Hutchinson, P.E.; Lew, F.C.; Wong, K.L.; Hanson, B.J.; Macary, P.A.; et al. Lung CD103+ dendritic cells efficiently transport influenza virus to the lymph node and load viral antigen onto MHC class I for presentation to CD8 T cells. J. Immunol. 2011, 187, 6011–6021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mount, A.M.; Smith, C.M.; Kupresanin, F.; Stoermer, K.; Heath, W.R.; Belz, G.T. Multiple dendritic cell populations activate CD4+ T cells after viral stimulation. PLoS ONE 2008, 3, e1691. [Google Scholar] [CrossRef] [PubMed]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.I.; Becker, C.; Metang, P.; Marches, F.; Wang, Y.; Toshiyuki, H.; Banchereau, J.; Merad, M.; Palucka, A.K. Human CD141+ dendritic cells induce CD4+ T cells to produce type 2 cytokines. J. Immunol. 2014, 193, 4335–4343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudziak, D.; Kamphorst, A.O.; Heidkamp, G.F.; Buchholz, V.R.; Trumpfheller, C.; Yamazaki, S.; Cheong, C.; Liu, K.; Lee, H.W.; Park, C.G.; et al. Differential antigen processing by dendritic cell subsets in vivo. Science 2007, 315, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Tato, A.; Leon, B.; Lund, F.E.; Randall, T.D. Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8(+) T cell responses to influenza. Nat. Immunol. 2010, 11, 216–224. [Google Scholar] [CrossRef] [Green Version]
- Ainsua-Enrich, E.; Hatipoglu, I.; Kadel, S.; Turner, S.; Paul, J.; Singh, S.; Bagavant, H.; Kovats, S. IRF4-dependent dendritic cells regulate CD8(+) T-cell differentiation and memory responses in influenza infection. Mucosal Immunol. 2019, 12, 1025–1037. [Google Scholar] [CrossRef]
- Won, H.Y.; Lee, J.Y.; Ryu, D.; Kim, H.T.; Chang, S.Y. The Role of Plasmacytoid Dendritic Cells in Gut Health. Immune Netw. 2019, 19, e6. [Google Scholar] [CrossRef]
- Wolf, A.I.; Buehler, D.; Hensley, S.E.; Cavanagh, L.L.; Wherry, E.J.; Kastner, P.; Chan, S.; Weninger, W. Plasmacytoid dendritic cells are dispensable during primary influenza virus infection. J. Immunol. 2009, 182, 871–879. [Google Scholar] [CrossRef] [Green Version]
- Lui, G.; Manches, O.; Angel, J.; Molens, J.P.; Chaperot, L.; Plumas, J. Plasmacytoid dendritic cells capture and cross-present viral antigens from influenza-virus exposed cells. PLoS ONE 2009, 4, e7111. [Google Scholar] [CrossRef]
- Hemann, E.A.; Sjaastad, L.E.; Langlois, R.A.; Legge, K.L. Plasmacytoid Dendritic Cells Require Direct Infection to Sustain the Pulmonary Influenza A Virus-Specific CD8 T Cell Response. J. Virol. 2015, 90, 2830–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, J.L.; Perez-Giron, J.V.; Ludtke, A.; Gomez-Medina, S.; Ruibal, P.; Idoyaga, J.; Munoz-Fontela, C. Monocyte-derived dendritic cells enhance protection against secondary influenza challenge by controlling the switch in CD8(+) T-cell immunodominance. Eur. J. Immunol. 2017, 47, 345–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Haist, V.; Baumgartner, W.; Schughart, K. Sustained viral load and late death in Rag2-/- mice after influenza A virus infection. Virol. J. 2010, 7, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palladino, G.; Mozdzanowska, K.; Washko, G.; Gerhard, W. Virus-neutralizing antibodies of immunoglobulin G (IgG) but not of IgM or IgA isotypes can cure influenza virus pneumonia in SCID mice. J. Virol. 1995, 69, 2075–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.M.; Lee, S.; Garcia-Hernandez Mde, L.; Swain, S.L. Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. J. Virol. 2012, 86, 6792–6803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.M.; Dilzer, A.M.; Meents, D.L.; Swain, S.L. CD4 T Cell-Mediated Protection from Lethal Influenza: Perforin and Antibody-Mediated Mechanisms Give a One-Two Punch. J. Immunol. 2006, 177, 2888–2898. [Google Scholar] [CrossRef] [Green Version]
- Baumgarth, N.; Kelso, A. In vivo blockade of gamma interferon affects the influenza virus-induced humoral and the local cellular immune response in lung tissue. J. Virol. 1996, 70, 4411–4418. [Google Scholar] [CrossRef] [Green Version]
- Graham, M.B.; Braciale, V.L.; Braciale, T.J. Influenza virus-specific CD4+ T helper type 2 T lymphocytes do not promote recovery from experimental virus infection. J. Exp. Med. 1994, 180, 1273–1282. [Google Scholar] [CrossRef]
- Brown, D.M.; Kamperschroer, C.; Dilzer, A.M.; Roberts, D.M.; Swain, S.L. IL-2 and antigen dose differentially regulate perforin- and FasL-mediated cytolytic activity in antigen specific CD4+ T cells. Cell Immunol. 2009, 257, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Hua, L.; Yao, S.; Pham, D.; Jiang, L.; Wright, J.; Sawant, D.; Dent, A.L.; Braciale, T.J.; Kaplan, M.H.; Sun, J. Cytokine-dependent induction of CD4+ T cells with cytotoxic potential during influenza virus infection. J. Virol. 2013, 87, 11884–11893. [Google Scholar] [CrossRef] [Green Version]
- Betts, R.J.; Prabhu, N.; Ho, A.W.; Lew, F.C.; Hutchinson, P.E.; Rotzschke, O.; Macary, P.A.; Kemeny, D.M. Influenza A virus infection results in a robust, antigen-responsive, and widely disseminated Foxp3+ regulatory T cell response. J. Virol. 2012, 86, 2817–2825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arpaia, N.; Green, J.A.; Moltedo, B.; Arvey, A.; Hemmers, S.; Yuan, S.; Treuting, P.M.; Rudensky, A.Y. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 2015, 162, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Leon, B.; Bradley, J.E.; Lund, F.E.; Randall, T.D.; Ballesteros-Tato, A. FoxP3+ regulatory T cells promote influenza-specific Tfh responses by controlling IL-2 availability. Nat. Commun. 2014, 5, 3495. [Google Scholar] [CrossRef] [PubMed]
- Belz, G.T.; Xie, W.; Altman, J.D.; Doherty, P.C. A previously unrecognized H-2D(b)-restricted peptide prominent in the primary influenza A virus-specific CD8(+) T-cell response is much less apparent following secondary challenge. J. Virol. 2000, 74, 3486–3493. [Google Scholar] [CrossRef] [Green Version]
- Bender, B.S.; Croghan, T.; Zhang, L.; Small, P.A., Jr. Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J. Exp. Med. 1992, 175, 1143–1145. [Google Scholar] [CrossRef] [Green Version]
- Kreijtz, J.H.; Fouchier, R.A.; Rimmelzwaan, G.F. Immune responses to influenza virus infection. Virus Res. 2011, 162, 19–30. [Google Scholar] [CrossRef]
- Brincks, E.L.; Katewa, A.; Kucaba, T.A.; Griffith, T.S.; Legge, K.L. CD8 T cells utilize TRAIL to control influenza virus infection. J. Immunol. 2008, 181, 4918–4925. [Google Scholar] [CrossRef]
- Kim, C.W.; Yoo, H.J.; Park, J.H.; Oh, J.E.; Lee, H.K. Exogenous Interleukin-33 Contributes to Protective Immunity via Cytotoxic T-Cell Priming against Mucosal Influenza Viral Infection. Viruses 2019, 11, 840. [Google Scholar] [CrossRef] [Green Version]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef]
- Schmidt, M.E.; Varga, S.M. The CD8 T Cell Response to Respiratory Virus Infections. Front. Immunol. 2018, 9, 678. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Madan, R.; Karp, C.L.; Braciale, T.J. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat. Med. 2009, 15, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.B.; Braciale, T.J. Resistance to and recovery from lethal influenza virus infection in B lymphocyte-deficient mice. J. Exp. Med. 1997, 186, 2063–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.O.; Rangel-Moreno, J.; Moyron-Quiroz, J.E.; Hartson, L.; Makris, M.; Sprague, F.; Lund, F.E.; Randall, T.D. CD4 T cell-independent antibody response promotes resolution of primary influenza infection and helps to prevent reinfection. J. Immunol. 2005, 175, 5827–5838. [Google Scholar] [CrossRef] [PubMed]
- Waffarn, E.E.; Baumgarth, N. Protective B cell responses to flu--no fluke! J. Immunol. 2011, 186, 3823–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, S.; Kurata, T. Defense mechanisms against influenza virus infection in the respiratory tract mucosa. Jpn. J. Infect. Dis. 2004, 57, 236–247. [Google Scholar] [PubMed]
- Hensley, S.E.; Das, S.R.; Bailey, A.L.; Schmidt, L.M.; Hickman, H.D.; Jayaraman, A.; Viswanathan, K.; Raman, R.; Sasisekharan, R.; Bennink, J.R.; et al. Hemagglutinin receptor binding avidity drives influenza A virus antigenic drift. Science 2009, 326, 734–736. [Google Scholar] [CrossRef] [Green Version]
- Neu, K.E.; Henry Dunand, C.J.; Wilson, P.C. Heads, stalks and everything else: How can antibodies eradicate influenza as a human disease? Curr. Opin. Immunol. 2016, 42, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Angeletti, D.; Gibbs, J.S.; Angel, M.; Kosik, I.; Hickman, H.D.; Frank, G.M.; Das, S.R.; Wheatley, A.K.; Prabhakaran, M.; Leggat, D.J.; et al. Defining B cell immunodominance to viruses. Nat. Immunol. 2017, 18, 456–463. [Google Scholar] [CrossRef]
- van de Sandt, C.E.; Kreijtz, J.H.; Rimmelzwaan, G.F. Evasion of influenza A viruses from innate and adaptive immune responses. Viruses 2012, 4, 1438–1476. [Google Scholar] [CrossRef] [Green Version]
- Treanor, J.J.; Tierney, E.L.; Zebedee, S.L.; Lamb, R.A.; Murphy, B.R. Passively transferred monoclonal antibody to the M2 protein inhibits influenza A virus replication in mice. J. Virol. 1990, 64, 1375–1377. [Google Scholar] [CrossRef] [Green Version]
- Carragher, D.M.; Kaminski, D.A.; Moquin, A.; Hartson, L.; Randall, T.D. A novel role for non-neutralizing antibodies against nucleoprotein in facilitating resistance to influenza virus. J. Immunol. 2008, 181, 4168–4176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jegaskanda, S.; Weinfurter, J.T.; Friedrich, T.C.; Kent, S.J. Antibody-dependent cellular cytotoxicity is associated with control of pandemic H1N1 influenza virus infection of macaques. J. Virol. 2013, 87, 5512–5522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.S.; Baumgarth, N. Dual role for B-1a cells in immunity to influenza virus infection. J. Exp. Med. 2008, 205, 3053–3064. [Google Scholar] [CrossRef] [PubMed]
- Putri, W.; Muscatello, D.J.; Stockwell, M.S.; Newall, A.T. Economic burden of seasonal influenza in the United States. Vaccine 2018, 36, 3960–3966. [Google Scholar] [CrossRef] [PubMed]
- Paget, J.; Spreeuwenberg, P.; Charu, V.; Taylor, R.J.; Iuliano, A.D.; Bresee, J.; Simonsen, L.; Viboud, C.; Global Seasonal Influenza-associated Mortality Collaborator Network; GLaMOR Collaborating Teams. Global mortality associated with seasonal influenza epidemics: New burden estimates and predictors from the GLaMOR Project. J. Glob. Health 2019, 9, 020421. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, C.; Zhang, H.; Liu, G.D.; Xue, C.; Cao, Y. Targeting Hemagglutinin: Approaches for Broad Protection against the Influenza A Virus. Viruses 2019, 11, 405. [Google Scholar] [CrossRef] [Green Version]
- Grodeland, G.; Baranowska-Hustad, M.; Abadejos, J.; Blane, T.R.; Teijaro, J.; Nemazee, D.; Bogen, B. Induction of Cross-Reactive and Protective Antibody Responses After DNA Vaccination With MHCII-Targeted Stem Domain From Influenza Hemagglutinin. Front. Immunol. 2020, 11, 431. [Google Scholar] [CrossRef]
- Hoft, D.F.; Babusis, E.; Worku, S.; Spencer, C.T.; Lottenbach, K.; Truscott, S.M.; Abate, G.; Sakala, I.G.; Edwards, K.M.; Creech, C.B.; et al. Live and inactivated influenza vaccines induce similar humoral responses, but only live vaccines induce diverse T-cell responses in young children. J. Infect. Dis. 2011, 204, 845–853. [Google Scholar] [CrossRef]
- Soema, P.C.; Kompier, R.; Amorij, J.P.; Kersten, G.F. Current and next generation influenza vaccines: Formulation and production strategies. Eur. J. Pharm. Biopharm. 2015, 94, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Huleatt, J.W.; Nakaar, V.; Desai, P.; Huang, Y.; Hewitt, D.; Jacobs, A.; Tang, J.; McDonald, W.; Song, L.; Evans, R.K.; et al. Potent immunogenicity and efficacy of a universal influenza vaccine candidate comprising a recombinant fusion protein linking influenza M2e to the TLR5 ligand flagellin. Vaccine 2008, 26, 201–214. [Google Scholar] [CrossRef]
- Wang, B.Z.; Gill, H.S.; He, C.; Ou, C.; Wang, L.; Wang, Y.C.; Feng, H.; Zhang, H.; Prausnitz, M.R.; Compans, R.W. Microneedle delivery of an M2e-TLR5 ligand fusion protein to skin confers broadly cross-protective influenza immunity. J. Control. Release 2014, 178, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luke, J.M.; Simon, G.G.; Soderholm, J.; Errett, J.S.; August, J.T.; Gale, M., Jr.; Hodgson, C.P.; Williams, J.A. Coexpressed RIG-I agonist enhances humoral immune response to influenza virus DNA vaccine. J. Virol. 2011, 85, 1370–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheu, T.G.; Deyde, V.M.; Okomo-Adhiambo, M.; Garten, R.J.; Xu, X.; Bright, R.A.; Butler, E.N.; Wallis, T.R.; Klimov, A.I.; Gubareva, L.V. Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008. Antimicrob. Agents Chemother. 2008, 52, 3284–3292. [Google Scholar] [CrossRef] [Green Version]
- Samson, M.; Pizzorno, A.; Abed, Y.; Boivin, G. Influenza virus resistance to neuraminidase inhibitors. Antivir. Res. 2013, 98, 174–185. [Google Scholar] [CrossRef]
- Yen, H.L.; McKimm-Breschkin, J.L.; Choy, K.T.; Wong, D.D.; Cheung, P.P.; Zhou, J.; Ng, I.H.; Zhu, H.; Webby, R.J.; Guan, Y.; et al. Resistance to neuraminidase inhibitors conferred by an R292K mutation in a human influenza virus H7N9 isolate can be masked by a mixed R/K viral population. mBio 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Burnham, A.J.; Baranovich, T.; Govorkova, E.A. Neuraminidase inhibitors for influenza B virus infection: Efficacy and resistance. Antivir. Res. 2013, 100, 520–534. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, H.E.; Lee, H.K. Host Protective Immune Responses against Influenza A Virus Infection. Viruses 2020, 12, 504. https://doi.org/10.3390/v12050504
Jung HE, Lee HK. Host Protective Immune Responses against Influenza A Virus Infection. Viruses. 2020; 12(5):504. https://doi.org/10.3390/v12050504
Chicago/Turabian StyleJung, Hi Eun, and Heung Kyu Lee. 2020. "Host Protective Immune Responses against Influenza A Virus Infection" Viruses 12, no. 5: 504. https://doi.org/10.3390/v12050504
APA StyleJung, H. E., & Lee, H. K. (2020). Host Protective Immune Responses against Influenza A Virus Infection. Viruses, 12(5), 504. https://doi.org/10.3390/v12050504