The Interplay between HIV-1 Gag Binding to the Plasma Membrane and Env Incorporation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Gag Assembly on the Plasma Membrane
3. Structure and Function of MA
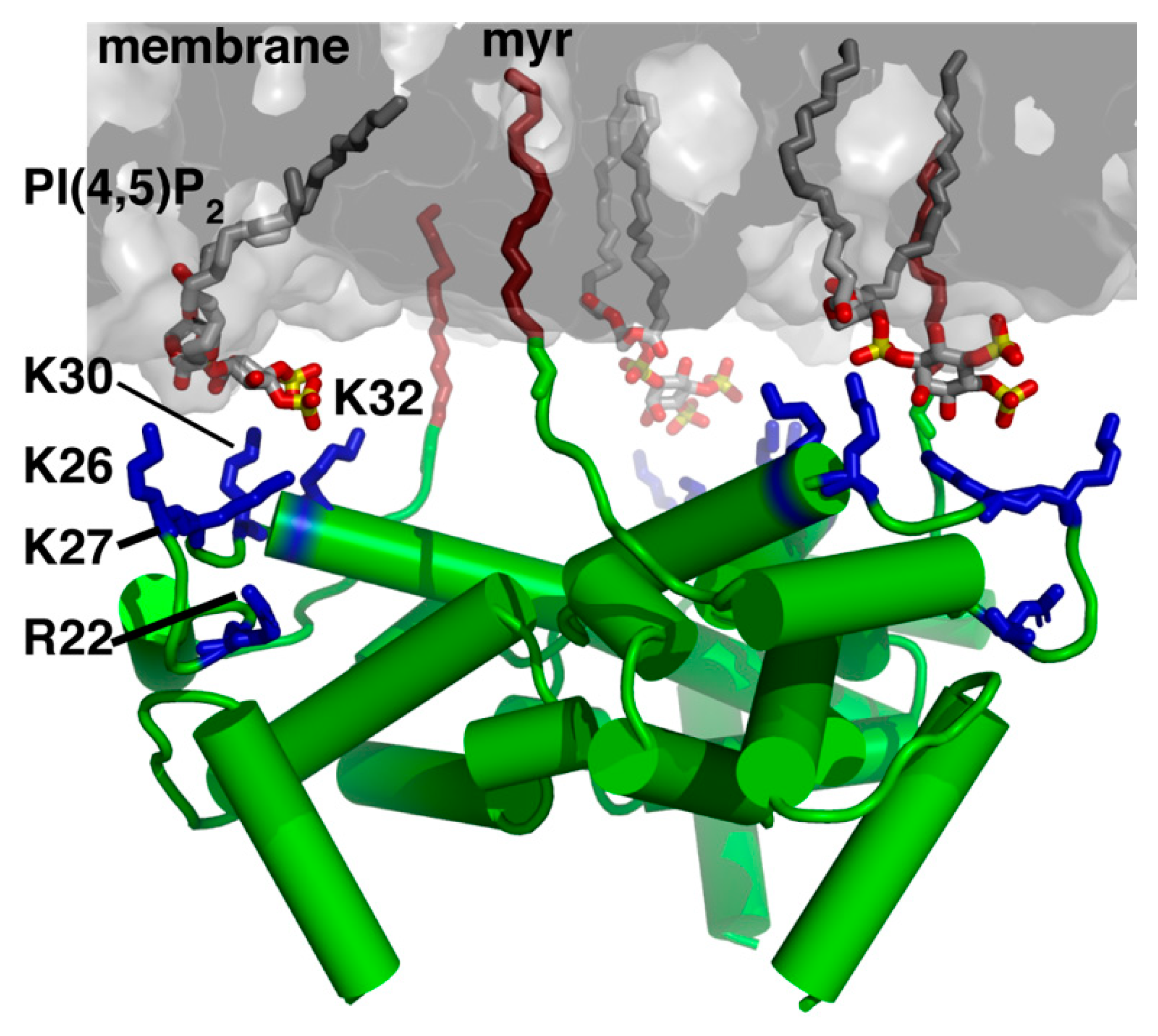
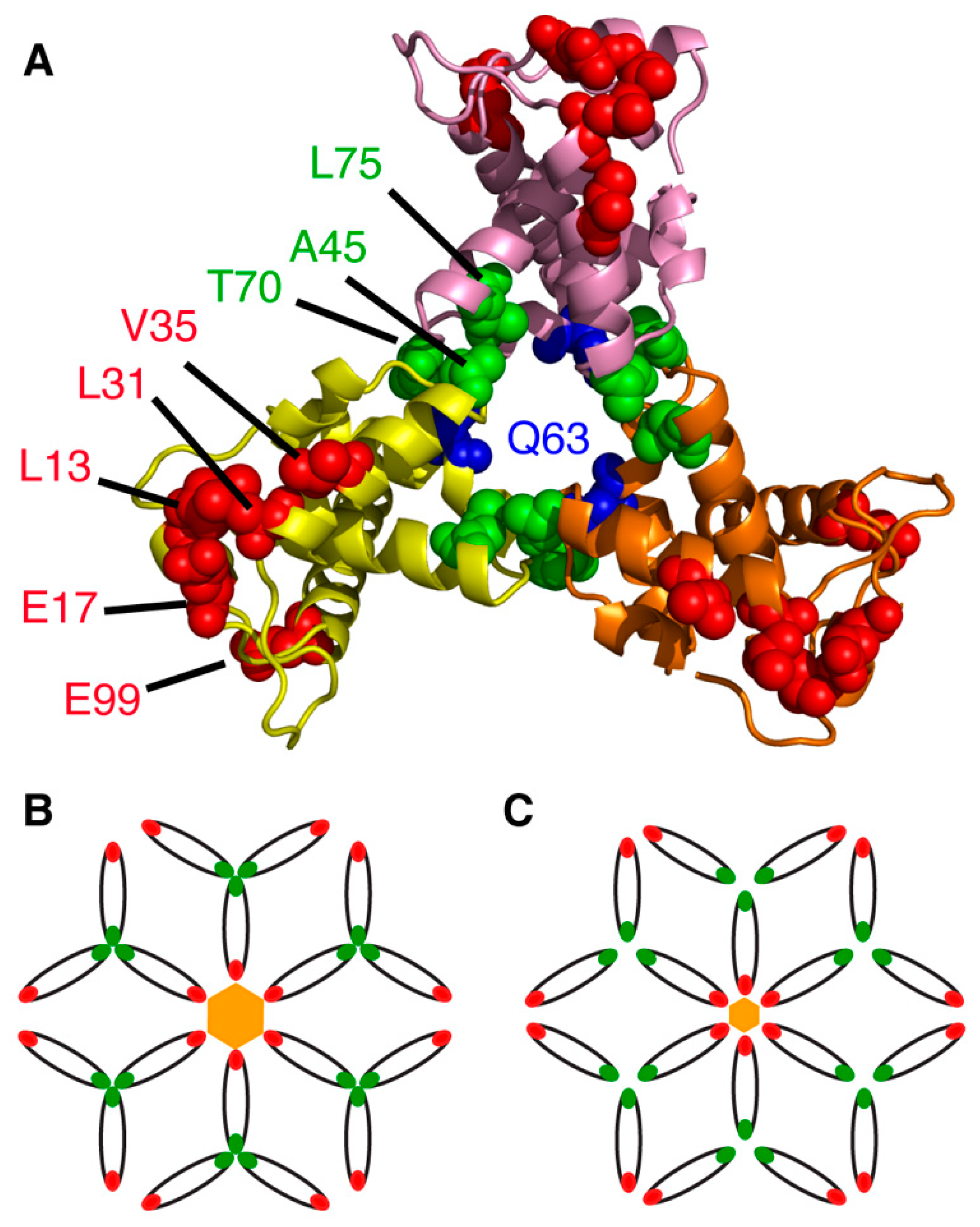
4. Structural Organization of MA on the Membrane
5. Structure and Function of the Envelope Glycoprotein
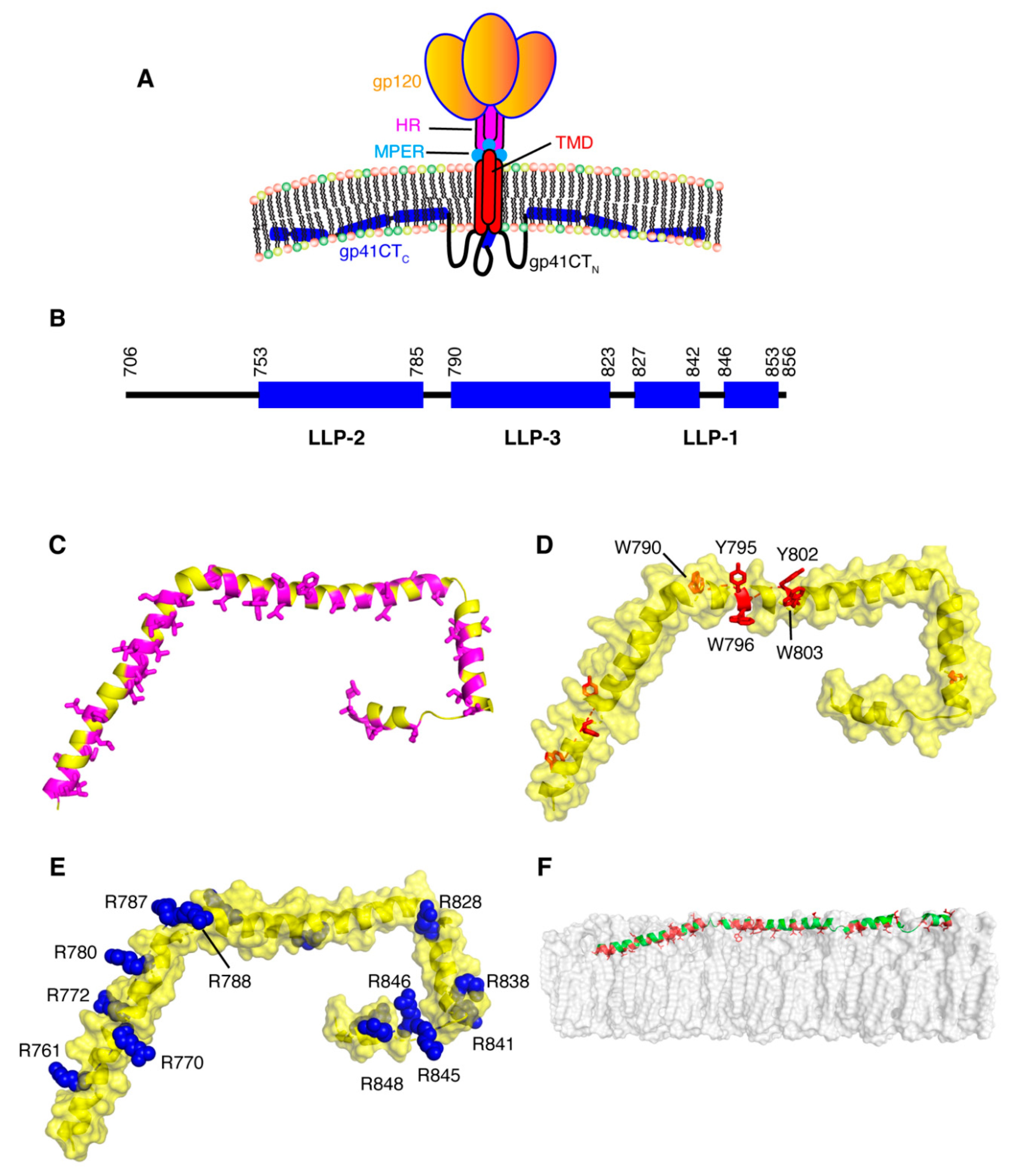
6. Structure and Topology of gp41CT
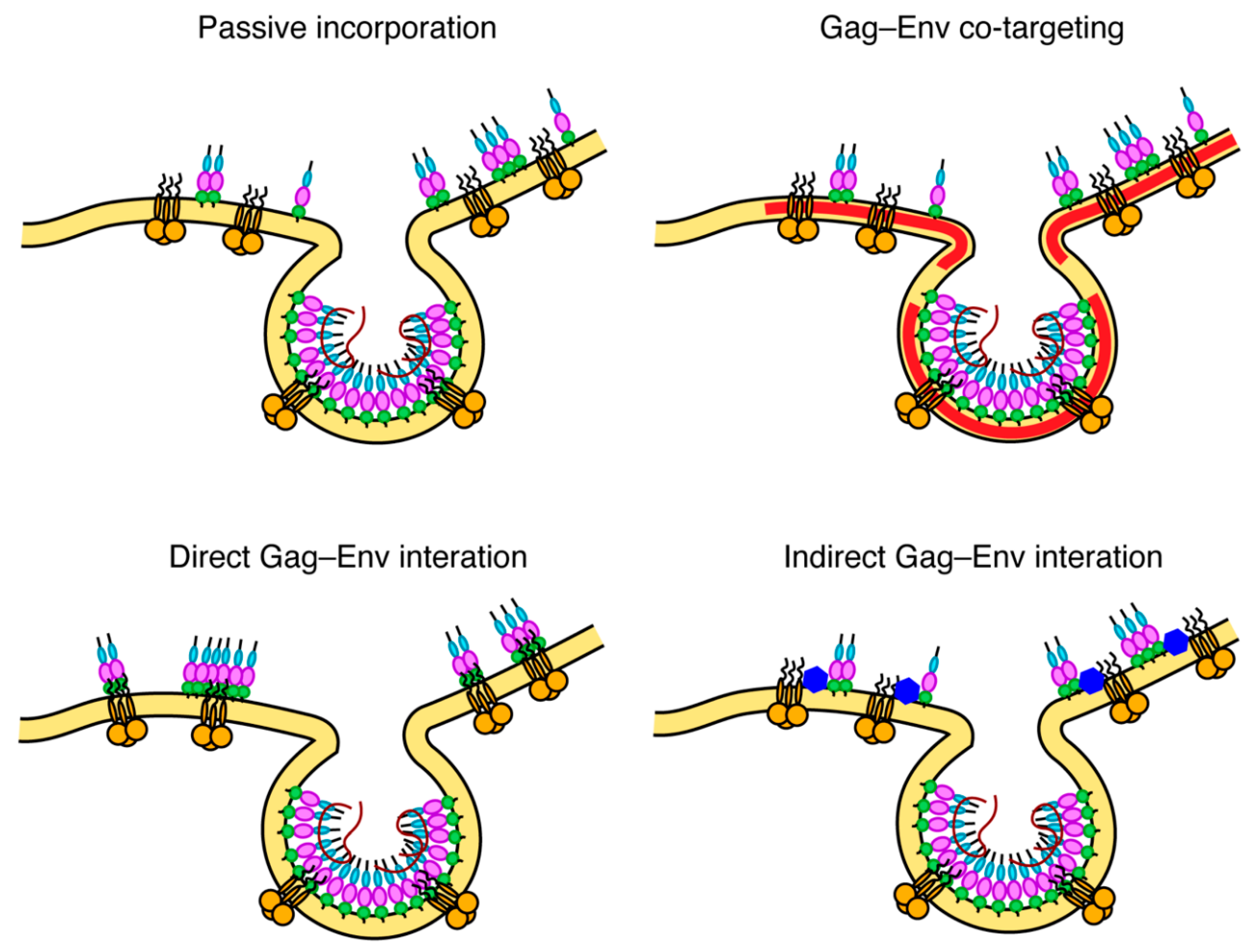
7. Mechanisms of Env Incorporation
8. Gag–Env–Membrane Complex as a Therapeutic Target
Author Contributions
Funding
Conflicts of Interest
References
- Rodger, A.J.; Lodwick, R.; Schechter, M.; Deeks, S.; Amin, J.; Gilson, R.; Paredes, R.; Bakowska, E.; Engsig, F.N.; Phillips, A. Mortality in well controlled HIV in the continuous antiretroviral therapy arms of the SMART and ESPRIT trials compared with the general population. AIDS 2013, 27, 973–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.S.; Chen, Y.Q.; McCauley, M.; Gamble, T.; Hosseinipour, M.C.; Kumarasamy, N.; Hakim, J.G.; Kumwenda, J.; Grinsztejn, B.; Pilotto, J.H. Antiretroviral therapy for the prevention of HIV-1 transmission. N. Engl. J. Med. 2016, 375, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [PubMed]
- Duarte, H.A.; Panpradist, N.; Beck, I.A.; Lutz, B.; Lai, J.; Kanthula, R.M.; Kantor, R.; Tripathi, A.; Saravanan, S.; MacLeod, I.J.; et al. Current Status of Point-of-Care Testing for Human Immunodeficiency Virus Drug Resistance. J. Infect. Dis. 2017, 216, S824–S828. [Google Scholar] [CrossRef] [Green Version]
- Menendez-Arias, L. Molecular basis of human immunodeficiency virus type 1 drug resistance: Overview and recent developments. Antivir. Res. 2013, 98, 93–120. [Google Scholar] [CrossRef]
- Shukla, E.; Chauhan, R. Host-HIV-1 Interactome: A Quest for Novel Therapeutic Intervention. Cells 2019, 8, 1155. [Google Scholar] [CrossRef] [Green Version]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Houck-Loomis, B.; Durney, M.A.; Salguero, C.; Shankar, N.; Nagle, J.M.; Goff, S.P.; D’Souza, V.M. An equilibrium-dependent retroviral mRNA switch regulates translational recoding. Nature 2011, 480, 561–564. [Google Scholar] [CrossRef]
- Jacks, T.; Power, M.D.; Masiarz, F.R.; Luciw, P.A.; Barr, P.J.; Varmus, H.E. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 1988, 331, 280–283. [Google Scholar] [CrossRef]
- Mucksch, F.; Laketa, V.; Muller, B.; Schultz, C.; Krausslich, H.G. Synchronized HIV assembly by tunable PIP2 changes reveals PIP2 requirement for stable Gag anchoring. eLife 2017, 6, e25287. [Google Scholar] [CrossRef]
- Hendrix, J.; Baumgartel, V.; Schrimpf, W.; Ivanchenko, S.; Digman, M.A.; Gratton, E.; Krausslich, H.G.; Muller, B.; Lamb, D.C. Live-cell observation of cytosolic HIV-1 assembly onset reveals RNA-interacting Gag oligomers. J. Cell Biol. 2015, 210, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Gousset, K.; Ablan, S.D.; Coren, L.V.; Ono, A.; Soheilian, F.; Nagashima, K.; Ott, D.E.; Freed, E.O. Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog. 2008, 4, e1000015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouvenet, N.; Neil, S.J.D.; Bess, C.; Johnson, M.C.; Virgen, C.A.; Simon, S.M.; Bieniasz, P.D. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006, 4, e435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsch, S.; Keppler, O.T.; Habermann, A.; Allespach, I.; Krijnse-Locker, J.; Kräusslich, H.-G. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 2007, 3, e36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chukkapalli, V.; Hogue, I.B.; Boyko, V.; Hu, W.-S.; Ono, A. Interaction between HIV-1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient Gag-membrane binding. J. Virol. 2008, 82, 2405–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 14889–14894. [Google Scholar] [CrossRef] [Green Version]
- Olety, B.; Ono, A. Roles played by acidic lipids in HIV-1 Gag membrane binding. Virus Res. 2014, 193, 108–115. [Google Scholar] [CrossRef] [Green Version]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef]
- D’Souza, V.; Summers, M.F. How retroviruses select their genomes. Nat. Rev. Microbiol. 2005, 3, 643–655. [Google Scholar] [CrossRef]
- Rein, A. RNA Packaging in HIV. Trends Microbiol. 2019, 27, 715–723. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Yeager, M.; Sundquist, W.I. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 2008, 18, 203–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundquist, W.I.; Kräusslich, H.-G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef]
- Chukkapalli, V.; Inlora, J.; Todd, G.C.; Ono, A. Evidence in support of RNA-mediated inhibition of phosphatidylserine-dependent HIV-1 Gag membrane binding in cells. J. Virol. 2013, 87, 7155–7159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chukkapalli, V.; Ono, A. Molecular Determinants that Regulate Plasma Membrane Association of HIV-1 Gag. J. Mol. Biol. 2011, 410, 512–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purohit, P.; Dupont, S.; Stevenson, M.; Green, M.R. Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA 2001, 7, 576–584. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Dou, J.; Ding, L.; Spearman, P. Myristoylation is required for human immunodeficiency virus type 1 Gag-Gag multimerization in mammalian cells. J. Virol. 2007, 81, 12899–12910. [Google Scholar] [CrossRef] [Green Version]
- Dalton, A.K.; Ako-Adjei, D.; Murray, P.S.; Murray, D.; Vogt, M.V. Electrostatic Interactions Drive Membrane Association of the Human Immunodeficiency Virus Type 1 Gag MA Domain. J. Virol. 2007, 81, 6434–6445. [Google Scholar] [CrossRef] [Green Version]
- Dick, R.A.; Goh, S.L.; Feigenson, G.W.; Vogt, V.M. HIV-1 Gag protein can sense the cholesterol and acyl chain environment in model membranes. Proc. Natl. Acad. Sci. USA 2012, 109, 18761–18767. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.R.; Schooler, J.B.; Ding, H.J.; Kieffer, C.; Fillmore, C.; Sundquist, W.I.; Jensen, G.J. Electron crytomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 2007, 26, 2218–2226. [Google Scholar] [CrossRef] [Green Version]
- Briggs, J.A.; Riches, J.D.; Glass, B.; Bartonova, V.; Zanetti, G.; Krausslich, H.G. Structure and assembly of immature HIV. Proc. Natl. Acad. Sci. USA 2009, 106, 11090–11095. [Google Scholar] [CrossRef] [Green Version]
- Ganser-Pornillos, B.K.; Yeager, M.; Pornillos, O. Assembly and architecture of HIV. Adv. Exp. Med. Biol. 2012, 726, 441–465. [Google Scholar]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Lippincott-Schwartz, J.; Freed, E.O.; van Engelenburg, S.B. A Consensus View of ESCRT-Mediated Human Immunodeficiency Virus Type 1 Abscission. Annu. Rev. Virol. 2017, 4, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Ganser-Pornillos, B.K. Maturation of retroviruses. Curr. Opin. Virol. 2019, 36, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Massiah, M.A.; Starich, M.R.; Paschall, C.; Summers, M.F.; Christensen, A.M.; Sundquist, W.I. Three dimensional structure of the human immunodeficiency virus type 1 matrix protein. J. Mol. Biol. 1994, 244, 198–223. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.; Barlow, P.; Boyd, J.; Barton, G.; Russell, R.; Mills, H.; Cunningham, M.; Meyers, N.; Burns, N.; Clark, N.; et al. Structural similarity between the p17 matrix protein of HIV-1 and interferon-g. Nature (London) 1994, 370, 666–668. [Google Scholar] [CrossRef]
- Matthews, S.; Barlow, P.; Clark, N.; Kingsman, S.; Kingsman, A.; Campbell, I. Refined solution structure of p17, the HIV matrix protein. Biochem. Soc. Trans. 1995, 23, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.P.; Worthylake, D.; Bancroft, D.P.; Christensen, A.M.; Sundquist, W.I. Crystal Structures of the Trimeric HIV-1 Matrix Protein: Implications for Membrane Association. Proc. Natl. Acad. Sci. USA 1996, 93, 3099–3104. [Google Scholar] [CrossRef] [Green Version]
- Massiah, M.A.; Worthylake, D.; Christensen, A.M.; Sundquist, W.I.; Hill, C.P.; Summers, M.F. Comparison of the NMR and X-ray structures of the HIV-1 matrix protein: Evidence for conformational changes during viral assembly. Protein Sci. 1996, 5, 2391–2398. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Ndassa, Y.; Summers, M.F. Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Nat. Struct. Biol. 2002, 9, 537–543. [Google Scholar] [CrossRef]
- Tang, C.; Loeliger, E.; Luncsford, P.; Kinde, I.; Beckett, D.; Summers, M.F. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl. Acad. Sci. USA 2004, 101, 517–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, J.S.; Miller, J.; Tai, J.; Kim, A.; Ghanam, R.H.; Summers, M.F. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. USA 2006, 103, 11364–11369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, M.; Ratner, L. Myristoylation-Dependent Replication and Assembly of Human Immunodeficiency Virus 1. Proc. Natl. Acad. Sci. USA 1990, 87, 523–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Resh, M.D. Differential membrane binding of the human immunodeficiency virus type 1 matrix protein. J. Virol. 1996, 70, 8540–8548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spearman, P.; Horton, R.; Ratner, L.; Kuli-Zade, I. Membrane binding of human immunodeficiency virus type 1 matrix protein in vivo supports a conformational myristyl switch mechanism. J. Virol. 1997, 71, 6582–6592. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.H.; Hildreth, J.E. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J. Virol. 2000, 74, 3264–3272. [Google Scholar] [CrossRef] [Green Version]
- Provitera, P.; El-Maghrabi, R.; Scarlata, S. The effect of HIV-1 Gag myristoylation on membrane binding. J. Mol. Biol. 2006, 119, 23–32. [Google Scholar] [CrossRef]
- Hermida-Matsumoto, L.; Resh, M.D. Human immunodeficiency virus type 1 protease triggers a myristoyl switch that modulates membrane binding fo Pr55gag and p17MA. J. Virol. 1999, 73, 1902–1908. [Google Scholar] [CrossRef] [Green Version]
- Paillart, J.-C.; Gottlinger, H.G. Opposing effects of human immunodeficiency virus type 1 matrix mutations support a myristyl switch model of Gag membrane targeting. J. Virol. 1999, 73, 2604–2612. [Google Scholar] [CrossRef] [Green Version]
- Ono, A.; Freed, E.O. Binding of Human Immunodeficiency Virus Type 1 gag to membrane: Role of the matrix amino terminus. J. Virol. 1999, 73, 4136–4144. [Google Scholar] [CrossRef] [Green Version]
- Fledderman, E.L.; Fujii, K.; Ghanam, R.H.; Waki, K.; Prevelige, P.E.; Freed, E.O.; Saad, J.S. Myristate exposure in the human immunodeficiency virus type 1 matrix protein is modulated by pH. Biochemistry 2010, 49, 9551–9562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanam, R.H.; Fernandez, T.F.; Fledderman, E.L.; Saad, J.S. Binding of calmodulin to the HIV-1 matrix protein triggers myristate exposure. J. Biol. Chem. 2010, 285, 41911–41920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermida-Matsumoto, L.; Resh, M.D. Localization of Human Immunodeficiency virus Type 1 Gag and env at the Plasma Membrane by Confocal Imagine. J. Virol. 2000, 74, 8670–8679. [Google Scholar] [CrossRef] [Green Version]
- Ono, A.; Demirov, D.; Freed, E.O. Relationship between human immunodeficiency virus Type-1 Gag multimerization and membrane binding. J. Virol. 2000, 74, 5142–5150. [Google Scholar] [CrossRef]
- Ono, A.; Freed, E.O. Cell-Type-Dependent Tageting of Human Immunodeficiency Virus Type 1 Assembly to the Plasma Membrane and the Multivesicular body. J. Virol. 2004, 78, 1552–1563. [Google Scholar] [CrossRef] [Green Version]
- Chukkapalli, V.; Oh, S.J.; Ono, A. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc. Natl. Acad. Sci. USA 2010, 107, 1600–1605. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.R.; Fischer, M.J.; Anderson, K.E.; Holdich, J.; Koteci, A.; Balla, T.; Irvine, R.F. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 2012, 337, 727–730. [Google Scholar] [CrossRef] [Green Version]
- Kolay, S.; Basu, U.; Raghu, P. Control of diverse subcellular processes by a single multi-functional lipid phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2]. Biochem. J. 2016, 473, 1681–1692. [Google Scholar] [CrossRef]
- Behnia, R.; Munro, S. Organelle identity and the signposts for membrane traffic. Nature 2005, 438, 597–604. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, S.; Murray, D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature 2005, 438, 605–611. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, S.; Wang, J.; Gambhir, A.; Murray, D. PIP2 and Proteins: Interactions, Organization, and Information Flow. Annu. Rev. Biophys. Biomol. Struct. 2002, 31, 151–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonsen, A.; Wurmser, A.E.; Emr, S.D.; Stenmark, H. The role of phosphoinositides in membrane transport. Curr. Opin. Cell Biol. 2001, 13, 485–492. [Google Scholar] [CrossRef]
- Wenk, M.R.; De Camilli, P. Protein-lipid interactions and phosphoinositide metabolism in membrane traffic: Insights from vesicle recycling in nerve terminals. Proc. Natl. Acad. Sci. USA 2004, 101, 8262–8269. [Google Scholar] [CrossRef] [Green Version]
- Agamasu, C.; Ghanam, R.H.; Xu, F.; Sun, Y.; Chen, Y.; Saad, J.S. The Interplay between Calmodulin and Membrane Interactions with the Pleckstrin Homology Domain of Akt. J. Biol. Chem. 2017, 292, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; He, L.; Li, Y.; Xiao, F.; Hu, G. Modeling of PH Domains and Phosphoinositides Interactions and Beyond. Adv. Exp. Med. Biol. 2019, 1111, 19–32. [Google Scholar]
- Hurley, J.H.; Meyer, T. Subcellular targeting by membrane lipids. Curr. Opin. Cell Biol. 2001, 13, 146–152. [Google Scholar] [CrossRef]
- Yamamoto, E.; Kalli, A.C.; Yasuoka, K.; Sansom, M.S. Interactions of Pleckstrin Homology Domains with Membranes: Adding Back the Bilayer via High-Throughput Molecular Dynamics. Structure 2016, 24, 1421–1431. [Google Scholar] [CrossRef] [Green Version]
- Stansell, E.; Apkarian, R.; Haubova, S.; Diehl, W.E.; Tytler, E.M.; Hunter, E. Basic residues in the Mason-Pfizer monkey virus gag matrix domain regulate intracellular trafficking and capsid-membrane interactions. J. Virol. 2007, 81, 8977–8988. [Google Scholar] [CrossRef] [Green Version]
- Hamard-Peron, E.; Juillard, F.; Saad, J.S.; Roy, C.; Roingeard, P.; Summers, M.F.; Darlix, J.L.; Picart, C.; Muriaux, D. Targeting of murine leukemia virus gag to the plasma membrane is mediated by PI(4,5)P2/PS and a polybasic region in the matrix. J. Virol. 2010, 84, 503–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prchal, J.; Srb, P.; Hunter, E.; Ruml, T.; Hrabal, R. The Structure of Myristoylated Mason-Pfizer Monkey Virus Matrix Protein and the Role of Phosphatidylinositol-(4,5)-Bisphosphate in Its Membrane Binding. J. Mol. Biol. 2012, 423, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, J.S.; Ablan, S.D.; Ghanam, R.H.; Kim, A.; Andrews, K.; Nagashima, K.; Soheilian, F.; Freed, E.O.; Summers, M.F. Structure of the myristylated HIV-2 MA protein and the role of phosphatidylinositol-(4,5)-bisphosphate in membrane targeting. J. Mol. Biol. 2008, 382, 434–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, L.A.; Cox, C.; Baptiste, J.; Summers, H.; Button, R.; Bahlow, K.; Spurrier, V.; Kyser, J.; Luttge, B.G.; Kuo, L.; et al. NMR structure of the myristylated feline immunodeficiency virus matrix protein. Viruses 2015, 7, 2210–2229. [Google Scholar] [CrossRef]
- Watanabe, S.M.; Medina, G.N.; Eastep, G.N.; Ghanam, R.H.; Vlach, J.; Saad, J.S.; Carter, C.A. The matrix domain of the Gag protein from avian sarcoma virus contains a PI(4,5)P2-binding site that targets Gag to the cell periphery. J. Biol. Chem. 2018, 293, 18841–18853. [Google Scholar] [CrossRef] [Green Version]
- Ono, A.; Freed, E.O. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA 2001, 98, 13925–13930. [Google Scholar] [CrossRef] [Green Version]
- Hogue, I.B.; Grover, J.R.; Soheilian, F.; Nagashima, K.; Ono, A. Gag induces the coalescence of clustered lipid rafts and tetraspanin-enriched microdomains at HIV-1 assembly sites on the plasma membrane. J. Virol. 2011, 85, 9749–9766. [Google Scholar] [CrossRef] [Green Version]
- Brügger, B.; Glass, B.; Haberkant, P.; Leibrecht, I.; Wieland, F.T.; Krausslich, H.-G. The HIV lipidome: A raft with an unusual composition. Proc. Natl. Acad. Sci. USA 2006, 103, 2641–2646. [Google Scholar] [CrossRef] [Green Version]
- Charlier, L.; Louet, M.; Chaloin, L.; Fuchs, P.; Martinez, J.; Muriaux, D.; Favard, C.; Floquet, N. Coarse-Grained Simulations of the HIV-1 Matrix Protein Anchoring: Revisiting Its Assembly on Membrane Domains. Biophys. J. 2014, 106, 577–585. [Google Scholar] [CrossRef] [Green Version]
- Yandrapalli, N.; Lubart, Q.; Tanwar, H.S.; Picart, C.; Mak, J.; Muriaux, D.; Favard, C. Self assembly of HIV-1 Gag protein on lipid membranes generates PI(4,5)P2/Cholesterol nanoclusters. Sci. Rep. 2016, 6, 39332. [Google Scholar] [CrossRef] [Green Version]
- Favard, C.; Chojnacki, J.; Merida, P.; Yandrapalli, N.; Mak, J.; Eggeling, C.; Muriaux, D. HIV-1 Gag specifically restricts PI(4,5)P2 and cholesterol mobility in living cells creating a nanodomain platform for virus assembly. Sci. Adv. 2019, 5, eaaw8651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfadhli, A.; Still, A.; Barklis, E. Analysis of Human Immunodeficiency Virus Type 1 Matrix Binding to Membranes and Nucleic Acids. J. Virol. 2009, 83, 12196–12203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, L.S.; Fong, S.; Scarlata, S.; Zybarth, G.; Carter, C. Partitioning of HIV-1 Gag and Gag-related proteins to membranes. Biochemistry 1996, 35, 3933–3943. [Google Scholar] [CrossRef]
- Scarlata, S.; Ehrlich, L.S.; Carter, C.A. Membrane-Induced Alterations in HIV-1 Gag and Matrix Protein-Protein Interactions. J. Mol. Biol. 1998, 277, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Mariani, C.; Desdouits, M.; Favard, C.; Benaroch, P.; Muriaux, D.M. Role of Gag and lipids during HIV-1 assembly in CD4(+) T cells and macrophages. Front. Microbiol. 2014, 5, 312. [Google Scholar] [CrossRef] [Green Version]
- Kutluay, S.B.; Zang, T.; Blanco-Melo, D.; Powell, C.; Jannain, D.; Errando, M.; Bieniasz, P.D. Global Changes in the RNA Binding Specificity of HIV-1 Gag Regulate Virion Genesis. Cell 2014, 159, 1096–1109. [Google Scholar] [CrossRef] [Green Version]
- Inlora, J.; Collins, D.R.; Trubin, M.E.; Chung, J.Y.; Ono, A. Membrane binding and subcellular localization of retroviral Gag proteins are differentially regulated by MA interactions with phosphatidylinositol-(4,5)-bisphosphate and RNA. mBio 2014, 5, e02202. [Google Scholar] [CrossRef] [Green Version]
- Saad, J.S.; Loeliger, E.; Luncsford, P.; Liriano, M.; Tai, J.; Kim, A.; Miller, J.; Joshi, A.; Freed, E.O.; Summers, M.F. Point mutations in the HIV-1 matrix protein turn off the myristyl switch. J. Mol. Biol. 2007, 366, 574–585. [Google Scholar] [CrossRef] [Green Version]
- Vlach, J.; Saad, J.S. Trio engagement via plasma membrane phospholipids and the myristoyl moiety governs HIV-1 matrix binding to bilayers. Proc. Natl. Acad. Sci. USA 2013, 110, 3525–3530. [Google Scholar] [CrossRef] [Green Version]
- Vlach, J.; Eastep, G.N.; Ghanam, R.H.; Watanabe, S.M.; Carter, C.A.; Saad, J.S. Structural basis for targeting avian sarcoma virus Gag polyprotein to the plasma membrane for virus assembly. J. Biol. Chem. 2018, 293, 18828–18840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercredi, P.Y.; Bucca, N.; Loeliger, B.; Gaines, C.R.; Mehta, M.; Bhargava, P.; Tedbury, P.R.; Charlier, L.; Floquet, N.; Muriaux, D.; et al. Structural and Molecular Determinants of Membrane Binding by the HIV-1 Matrix Protein. J. Mol. Biol. 2016, 428, 1637–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anraku, K.; Fukuda, R.; Takamune, N.; Misumi, S.; Okamoto, Y.; Otsuka, M.; Fujita, M. Highly sensitive analysis of the interaction between HIV-1 Gag and phosphoinositide derivatives based on surface plasmon resonance. Biochemistry 2010, 49, 5109–5116. [Google Scholar] [CrossRef] [PubMed]
- Shkriabai, N.; Datta, S.A.; Zhao, Z.; Hess, S.; Rein, A.; Kvaratskhelia, M. Interactions of HIV-1 Gag with assembly cofactors. Biochemistry 2006, 45, 4077–4083. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.; Chen, K.; Ehrlich, L.S.; Jin, J.; Chen, M.H.; Medina, G.N.; Symons, M.; Montelaro, R.; Donaldson, J.; Tjandra, N.; et al. Phosphoinositides direct equine infectious anemia virus gag trafficking and release. Traffic 2011, 12, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Murphy, R.E.; Samal, A.B.; Vlach, J.; Mas, V.; Prevelige, P.E.; Saad, J.S. Structural and biophysical characterizations of HIV-1 matrix trimer binding to lipid nanodiscs shed light on virus assembly. J. Biol. Chem. 2019, 294, 18600–18612. [Google Scholar] [CrossRef] [PubMed]
- Borch, J.; Hamann, T. The nanodisc: A novel tool for membrane protein studies. Biol. Chem. 2009, 390, 805–814. [Google Scholar] [CrossRef]
- Bayburt, T.H.; Sligar, S.G. Membrane protein assembly into Nanodiscs. FEBS Lett. 2010, 584, 1721–1727. [Google Scholar] [CrossRef] [Green Version]
- Hagn, F.; Etzkorn, M.; Raschle, T.; Wagner, G. Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J. Am. Chem. Soc. 2013, 135, 1919–1925. [Google Scholar] [CrossRef] [Green Version]
- Kobashigawa, Y.; Harada, K.; Yoshida, N.; Ogura, K.; Inagaki, F. Phosphoinositide-incorporated lipid-protein nanodiscs: A tool for studying protein-lipid interactions. Anal. Biochem. 2011, 410, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, T.K.; Grinkova, Y.V.; Bayburt, T.H.; Denisov, I.G.; Zolnerciks, J.K.; Atkins, W.M.; Sligar, S.G. Chapter 11-Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 2009, 464, 211–231. [Google Scholar] [PubMed] [Green Version]
- Yokogawa, M.; Kobashigawa, Y.; Yoshida, N.; Ogura, K.; Harada, K.; Inagaki, F. NMR Analyses of the Interaction between the FYVE Domain of Early Endosome Antigen 1 (EEA1) and Phosphoinositide Embedded in a Lipid Bilayer. J. Biol. Chem. 2012, 287, 34936–34945. [Google Scholar] [CrossRef] [Green Version]
- Ganser-Pornillos, B.K.; Cheng, A.; Yeager, M. Structure of full-length HIV-1 CA: A model for the mature capsid lattice. Cell 2007, 131, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharat, T.A.; Davey, N.E.; Ulbrich, P.; Riches, J.D.; de Marco, A.; Rumlova, M.; Sachse, C.; Ruml, T.; Briggs, J.A. Structure of the immature retroviral capsid at 8 Å resolution by cryo-electron microscopy. Nature 2012, 487, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C.; et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Bharat, T.A.; Castillo Menendez, L.R.; Hagen, W.J.; Lux, V.; Igonet, S.; Schorb, M.; Schur, F.K.; Krausslich, H.G.; Briggs, J.A. Cryo-electron microscopy of tubular arrays of HIV-1 Gag resolves structures essential for immature virus assembly. Proc. Natl. Acad. Sci. USA 2014, 111, 8233–8238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schur, F.K.; Hagen, W.J.; Rumlova, M.; Ruml, T.; Muller, B.; Krausslich, H.G.; Briggs, J.A. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 A resolution. Nature 2015, 517, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Alfadhli, A.; Barklis, R.L.; Barklis, E. HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4,5)-bisphosphate. Virology 2009, 387, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Tedbury, P.R.; Novikova, M.; Ablan, S.D.; Freed, E.O. Biochemical evidence of a role for matrix trimerization in HIV-1 envelope glycoprotein incorporation. Proc. Natl. Acad. Sci. USA 2016, 113, E182–E190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouamr, F.; Scarlata, S.; Carter, C.A. Role of myristylation in HIV-1 Gag assembly. Biochemistry 2003, 42, 6408–6417. [Google Scholar] [CrossRef]
- Alfadhli, A.; Huseby, D.; Kapit, E.; Colman, D.; Barklis, E. Human Immunodeficiency Virus Type 1 Matrix Protein Assembles on Membranes as a Hexamer. J. Virol. 2007, 81, 1472–1478. [Google Scholar] [CrossRef] [Green Version]
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 2011, 410, 582–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otteken, A.; Earl, P.L.; Moss, B. Folding, assembly, and intracellular trafficking of the human immunodeficiency virus type 1 envelope glycoprotein analyzed with monoclonal antibodies recognizing maturational intermediates. J. Virol. 1996, 70, 3407–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Kirschman, J.; Qi, M.; Ding, L.; Hammonds, J.; Dienger-Stambaugh, K.; Wang, J.J.; Lapierre, L.A.; Goldenring, J.R.; Spearman, P. HIV-1 Envelope Glycoprotein Trafficking through the Endosomal Recycling Compartment Is Required for Particle Incorporation. J. Virol. 2018, 92, e01893-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, M.; Williams, J.A.; Chu, H.; Chen, X.; Wang, J.J.; Ding, L.; Akhirome, E.; Wen, X.; Lapierre, L.A.; Goldenring, J.R.; et al. Rab11-FIP1C and Rab14 direct plasma membrane sorting and particle incorporation of the HIV-1 envelope glycoprotein complex. PLoS Pathog. 2013, 9, e1003278. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Chu, H.; Chen, X.; Choi, J.; Wen, X.; Hammonds, J.; Ding, L.; Hunter, E.; Spearman, P. A tyrosine-based motif in the HIV-1 envelope glycoprotein tail mediates cell-type- and Rab11-FIP1C-dependent incorporation into virions. Proc. Natl. Acad. Sci. USA 2015, 112, 7575–7580. [Google Scholar] [CrossRef] [Green Version]
- Pancera, M.; Majeed, S.; Ban, Y.E.; Chen, L.; Huang, C.C.; Kong, L.; Kwon, Y.D.; Stuckey, J.; Zhou, T.; Robinson, J.E.; et al. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc. Natl. Acad. Sci. USA 2010, 107, 1166–1171. [Google Scholar] [CrossRef] [Green Version]
- Merk, A.; Subramaniam, S. HIV-1 envelope glycoprotein structure. Curr. Opin. Struct. Biol. 2013, 23, 268–276. [Google Scholar] [CrossRef] [Green Version]
- Montero, M.; van Houten, N.E.; Wang, X.; Scott, J.K. The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: Dominant site of antibody neutralization and target for vaccine design. Microbiol. Mol. Biol. Rev. 2008, 72, 54–84. [Google Scholar] [CrossRef] [Green Version]
- Alam, S.M.; Morelli, M.; Dennison, S.M.; Liao, H.X.; Zhang, R.; Xia, S.M.; Rits-Volloch, S.; Sun, L.; Harrison, S.C.; Haynes, B.F.; et al. Role of HIV membrane in neutralization by two broadly neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2009, 106, 20234–20239. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Fenwick, C.; Caillat, C.; Silacci, C.; Guseva, S.; Dehez, F.; Chipot, C.; Barbieri, S.; Minola, A.; Jarrossay, D.; et al. Structural Basis for Broad HIV-1 Neutralization by the MPER-Specific Human Broadly Neutralizing Antibody LN01. Cell Host. Microbe 2019, 26, 623–637.e628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Q.; Shaik, M.M.; Cai, Y.; Ghantous, F.; Piai, A.; Peng, H.; Rits-Volloch, S.; Liu, Z.; Harrison, S.C.; Seaman, M.S.; et al. Structure of the membrane proximal external region of HIV-1 envelope glycoprotein. Proc. Natl. Acad. Sci. USA 2018, 115, E8892–E8899. [Google Scholar] [CrossRef] [Green Version]
- Blumenthal, R.; Durell, S.; Viard, M. HIV entry and envelope glycoprotein-mediated fusion. J. Biol. Chem. 2012, 287, 40841–40849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dev, J.; Park, D.; Fu, Q.; Chen, J.; Ha, H.J.; Ghantous, F.; Herrmann, T.; Chang, W.; Liu, Z.; Frey, G.; et al. Structural basis for membrane anchoring of HIV-1 envelope spike. Science 2016, 353, 172–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichart, T.M.; Baksh, M.M.; Rhee, J.K.; Fiedler, J.D.; Sligar, S.G.; Finn, M.; Zwick, M.B.; Dawson, P.E. Trimerization of the HIV transmembrane domain in lipid bilayers modulates broadly neutralizing antibody binding. Angew. Chem. Int. Edit. 2016, 55, 2688–2692. [Google Scholar] [CrossRef]
- Dai, Z.; Tao, Y.; Liu, N.; Brenowitz, M.D.; Girvin, M.E.; Lai, J.R. Conditional trimerization and lytic activity of HIV-1 gp41 variants containing the membrane-associated segments. Biochemistry 2015, 54, 1589–1599. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Ozorowski, G.; Ward, A.B. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 2016, 351, 1043–1048. [Google Scholar] [CrossRef] [Green Version]
- Chiliveri, S.C.; Louis, J.M.; Ghirlando, R.; Baber, J.L.; Bax, A. Tilted, Uninterrupted, Monomeric HIV-1 gp41 Transmembrane Helix from Residual Dipolar Couplings. J. Am. Chem. Soc. 2018, 140, 34–37. [Google Scholar] [CrossRef]
- Postler, T.S.; Desrosiers, R.C. The tale of the long tail: The cytoplasmic domain of HIV-1 gp41. J. Virol. 2013, 87, 2–15. [Google Scholar] [CrossRef] [Green Version]
- Kirchhoff, F.; Kestler, H.; Desrosiers, R.C. Upstream U3 sequences in simian immunodeficiency virus are selectively deleted in vivo in the absence of an intact nef gene. J. Virol. 1994, 68, 2031–2037. [Google Scholar] [CrossRef] [Green Version]
- Kirchoff, F.; Greenough, T.C.; Brettler, D.B.; Sullivan, J.L.; Desrosiers, R.C. Absence of intact nef sequences in a long-term servivor with nonprogressive HIV-1 infection. N. Engl. J. Med. 1995, 332, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, S.K.; Srinivas, R.V.; Anantharamaiah, G.M.; Compans, R.W.; Segrest, J.P. Cytosolic domain of the human immunodeficiency virus envelope glycoproteins binds to calmodulin and inhibits calmodulin-regulated proteins. J. Biol. Chem. 1993, 268, 22895–22899. [Google Scholar] [PubMed]
- Radding, W.; Pan, Z.Q.; Hunter, E.; Johnston, P.; Williams, J.P.; McDonald, J.M. Expression of HIV-1 Envelope Glycoprotein Alters Cellular Calmodulin. Biochem. Biophys. Res. Commun. 1996, 218, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Wyss, S.; Berlioz-Torrent, C.; Boge, M.; Blot, G.; Höning, S.; Benarous, R.; Thali, M. The highly conserved C-terminal dileucine motif in the cytosolic domain of the human immunodeficiency virus type 1 envelope glycoprotein is critical for its association with the AP-1 clathrin adapter. J. Virol. 2001, 75, 2982–2992. [Google Scholar] [CrossRef] [Green Version]
- Ohno, H.; Aguilar, R.C.; Fournier, M.C.; Hennecke, S.; Cosson, P.; Bonifacino, J.S. Interaction of endocytic signals from the HIV-1 envelope glycoprotein complex with members of the adaptor medium chain family. Virology 1997, 238, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Boge, M.; Wyss, S.; Bonifacino, J.S.; Thali, M. A membrane-proximal tyrosine-based signal mediates internalization of the HIV-1 envelope glycoprotein via interaction with the AP-2 clathrin adaptor. J. Biol. Chem. 1998, 273, 15773–15778. [Google Scholar] [CrossRef] [Green Version]
- Berlioz-Torrent, C.; Shacklett, B.L.; Erdtmann, L.; Delamarre, L.; Bouchaert, I.; Sonigo, P.; Dokhelar, M.C.; Benarous, R. Interactions of the cytoplasmic domains of human and simian retroviral transmembrane proteins with components of the clathrin adaptor complexes modulate intracellular and cell surface expression of envelope glycoproteins. J. Virol. 1999, 73, 1350–1361. [Google Scholar] [CrossRef] [Green Version]
- Tedbury, P.R.; Ablan, S.D.; Freed, E.O. Global Rescue of Defects in HIV-1 Envelope Glycoprotein Incorporation: Implications for Matrix Structure. PLoS Pathog. 2013, 9, e1003739. [Google Scholar] [CrossRef]
- Tedbury, P.R.; Freed, E.O. The role of matrix in HIV-1 envelope glycoprotein incorporation. Trends Microbiol. 2014, 22, 372–378. [Google Scholar] [CrossRef] [Green Version]
- Tedbury, P.R.; Freed, E.O. The cytoplasmic tail of retroviral envelope glycoproteins. Prog. Mol. Biol. Transl. Sci. 2015, 129, 253–284. [Google Scholar]
- Costin, J.M.; Rausch, J.M.; Garry, R.F.; Wimley, W.C. Viroporin potential of the lentivirus lytic peptide (LLP) domains of the HIV-1 gp41 protein. Virol. J. 2007, 4, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steckbeck, J.D.; Sun, C.; Sturgeon, T.J.; Montelaro, R.C. Topology of the C-terminal tail of HIV-1 gp41: Differential exposure of the Kennedy epitope on cell and viral membranes. PLoS ONE 2010, 5, e15261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boscia, A.L.; Akabori, K.; Benamram, Z.; Michel, J.A.; Jablin, M.S.; Steckbeck, J.D.; Montelaro, R.C.; Nagle, J.F.; Tristram-Nagle, S. Membrane structure correlates to function of LLP2 on the cytoplasmic tail of HIV-1 gp41 protein. Biophys. J. 2013, 105, 657–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steckbeck, J.D.; Kuhlmann, A.S.; Montelaro, R.C. C-terminal tail of human immunodeficiency virus gp41: Functionally rich and structurally enigmatic. J. Gen. Virol. 2013, 94, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Steckbeck, J.D.; Sun, C.; Sturgeon, T.J.; Montelaro, R.C. Detailed topology mapping reveals substantial exposure of the "cytoplasmic" C-terminal tail (CTT) sequences in HIV-1 Env proteins at the cell surface. PLoS ONE 2013, 8, e65220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.A.; Garry, R.F.; Jaynes, J.M.; Montelaro, R.C. A structural correlation between lentivirus transmembrane proteins and natural cytolytic peptides. AIDS Res. Hum. Retroviruses 1991, 7, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, S.K.; Srinivas, R.V.; Anantharamaiah, G.M.; Segrest, J.P.; Compans, R.W. Membrane interactions of synthetic peptides corresponding to amphipathic helical segments of the human immunodeficiency virus type-1 envelope glycoprotein. J. Biol. Chem. 1992, 267, 7121–7127. [Google Scholar]
- Steckbeck, J.D.; Craigo, J.K.; Barnes, C.O.; Montelaro, R.C. Highly conserved structural properties of the C-terminal tail of HIV-1 gp41 protein despite substantial sequence variation among diverse clades: Implications for functions in viral replication. J. Biol. Chem. 2011, 286, 27156–27166. [Google Scholar] [CrossRef] [Green Version]
- Viard, M.; Ablan, S.D.; Zhou, M.; Veenstra, T.D.; Freed, E.O.; Raviv, Y.; Blumenthal, R. Photoinduced reactivity of the HIV-1 envelope glycoprotein with a membrane-embedded probe reveals insertion of portions of the HIV-1 Gp41 cytoplasmic tail into the viral membrane. Biochemistry 2008, 47, 1977–1983. [Google Scholar] [CrossRef] [Green Version]
- Bültmann, A.; Muranyi, W.; Seed, B.; Haas, J. Identification of two sequences in the cytoplasmic tail of the human immunodeficiency virus type 1 envelope glycoprotein that inhibit cell surface expression. J. Virol. 2001, 75, 5263–5276. [Google Scholar] [CrossRef] [Green Version]
- Kalia, V.; Sarkar, S.; Gupta, P.; Montelaro, R.C. Rational site-directed mutations of the LLP-1 and LLP-2 lentivirus lytic peptide domains in the intracytoplasmic tail of human immunodeficiency virus type 1 gp41 indicate common functions in cell-cell fusion but distinct roles in virion envelope incorporation. J. Virol. 2003, 77, 3634–3646. [Google Scholar] [PubMed] [Green Version]
- Lee, S.F.; Ko, C.Y.; Wang, C.T.; Chen, S.S. Effect of point mutations in the N terminus of the lentivirus lytic peptide-1 sequence of human immunodeficiency virus type 1 transmembrane protein gp41 on Env stability. J. Biol. Chem. 2002, 277, 15363–15375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piller, S.C.; Dubay, J.W.; Derdeyn, C.A.; Hunter, E. Mutational analysis of conserved domains within the cytoplasmic tail of gp41 from human immunodeficiency virus type 1: Effects on glycoprotein incorporation and infectivity. J. Virol. 2000, 74, 11717–11723. [Google Scholar] [CrossRef] [Green Version]
- Murphy, R.E.; Samal, A.B.; Vlach, J.; Saad, J.S. Solution Structure and Membrane Interaction of the Cytoplasmic Tail of HIV-1 gp41 Protein. Structure 2017, 25, 1708–1718.e5. [Google Scholar] [CrossRef] [Green Version]
- Cleveland, S.M.; McLain, L.; Cheung, L.; Jones, T.D.; Hollier, M.; Dimmock, N.J. A region of the C-terminal tail of the gp41 envelope glycoprotein of human immunodeficiency virus type 1 contains a neutralizing epitope: Evidence for its exposure on the surface of the virion. J. Gen. Virol. 2003, 84, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Hollier, M.J.; Dimmock, N.J. The C-terminal tail of the gp41 transmembrane envelope glycoprotein of HIV-1 clades A, B, C, and D may exist in two conformations: An analysis of sequence, structure, and function. Virology 2005, 337, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhlmann, A.S.; Steckbeck, J.D.; Sturgeon, T.J.; Craigo, J.K.; Montelaro, R.C. Unique functional properties of conserved arginine residues in the lentivirus lytic peptide domains of the C-terminal tail of HIV-1 gp41. J. Biol. Chem. 2014, 289, 7630–7640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akari, H.; Fukumori, T.; Adachi, A. Cell-dependent requirement of human immunodeficiency virus type 1 gp41 cytoplasmic tail for Env incorporation into virions. J. Virol. 2000, 74, 4891–4893. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Freed, E.O. The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc. Natl. Acad. Sci. USA 2000, 97, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Hales, C.M.; Griner, R.; Hobdy-Henderson, K.C.; Dorn, M.C.; Hardy, D.; Kumar, R.; Navarre, J.; Chan, E.K.; Lapierre, L.A.; Goldenring, J.R. Identification and characterization of a family of Rab11-interacting proteins. J. Biol. Chem. 2001, 276, 39067–39075. [Google Scholar] [CrossRef] [Green Version]
- Roy, N.H.; Chan, J.; Lambele, M.; Thali, M. Clustering and mobility of HIV-1 Env at viral assembly sites predict its propensity to induce cell-cell fusion. J. Virol. 2013, 87, 7516–7525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muranyi, W.; Malkusch, S.; Müller, B.; Heilemann, M.; Kräusslich, H.G. Super-Resolution Microscopy Reveals Specific Recruitment of HIV-1 Envelope Proteins to Viral Assembly Sites Dependent on the Envelope C-Terminal Tail. PLoS Pathog. 2013, 9, e1103198. [Google Scholar] [CrossRef] [PubMed]
- Buttler, C.A.; Pezeshkian, N.; Fernandez, M.V.; Aaron, J.; Norman, S.; Freed, E.O.; van Engelenburg, S.B. Single molecule fate of HIV-1 envelope reveals late-stage viral lattice incorporation. Nat. Commun. 2018, 9, 1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, R.J.; Compans, R.W. Expression of the human immunodeficiency virus envelope glycoprotein is restricted to basolateral surfaces of polarized epithelial cells. J. Virol. 1989, 63, 978–982. [Google Scholar] [CrossRef] [Green Version]
- Owens, R.J.; Dubay, J.W.; Hunter, E.; Compans, R.W. Human Immunodeficiency Virus Envelope Protein Determines the Site of Virus Release in Polarized Epithelial Cells. Proc. Natl. Acad. Sci. USA 1991, 88, 3987–3991. [Google Scholar] [CrossRef] [Green Version]
- Lodge, R.; Gottlinger, H.; Gabuzda, D.; Cohen, E.A.; Lemay, G. The intracytoplasmic domain of gp41 mediates polarized budding of human immunodeficiency virus type 1 in MDCK cells. J. Virol. 1994, 68, 4857–4861. [Google Scholar] [CrossRef] [Green Version]
- Kiernan, R.E.; Freed, E.O. Cleavage of the murine leukemia virus transmembrane env protein by human immunodeficiency virus type 1 protease: Transdominant inhibition by matrix mutations. J. Virol. 1998, 72, 9621–9627. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.E.; Olinger, G.Y.; Janaka, S.K.; Johnson, M.C. Sequence Determinants in Gammaretroviral Env Cytoplasmic Tails Dictate Virus-Specific Pseudotyping Compatibility. J. Virol. 2019, 93, e02172-18. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Yuan, X.; Matsuda, Z.; Lee, T.-H.; Essex, M. The Matrix Protein of Human Immunodeficiency Virus Type I is Required for Incorporation of Viral Envelope Protein into Mature Virions. J. Virol. 1992, 66, 4966–4971. [Google Scholar] [CrossRef] [Green Version]
- Freed, O.E.; Martin, A.M. Virion Incorporation of Envelope Glycoproteins with Long but Not Short Cytoplasmic Tails Is Blocked by Specific, Single Amino Acid Substitutions in the Human Immunodeficiency Virus Type 1 Matrix. J. Virol. 1995, 69, 1984–1989. [Google Scholar] [CrossRef] [Green Version]
- Dorfman, T.; Mammano, F.; Haseltine, W.A.; Göttlinger, H.G. Role of the Matrix Protein in the Virion Association of the Human Immunodeficiency Virus Type 1 Envelope Glycoprotein. J. Virol. 1994, 68, 1689–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed, E.O.; Martin, A.M. Domains of the Human Immonodeficiency Virus Type 1 Matrix and gp41 Cytoplasmic Tail Required for Envelope Incorporation into Virions. J. Virol. 1996, 70, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosson, P. Direct interaction between the envelope and matrix proteins of HIV-1. EMBO J. 1996, 15, 5783–5788. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Huang, M.; Freed, E.O. Characterization of human immunodeficiency virus type 1 matrix revertants: Effects on virus assembly, Gag processing, and Env incorporation into virions. J. Virol. 1997, 71, 4409–4418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Ablan, S.; Freed, E.O.; Tanaka, Y. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J. Virol. 2004, 78, 1026–1031. [Google Scholar] [CrossRef] [Green Version]
- Wyma, D.J.; Jiang, J.; Shi, J.; Zhou, J.; Lineberger, J.E.; Miller, M.D.; Aiken, C. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: A novel role of the gp41 cytoplasmic tail. J. Virol. 2004, 78, 3429–3435. [Google Scholar] [CrossRef] [Green Version]
- Pezeshkian, N.; Groves, N.S.; van Engelenburg, S.B. Single-molecule imaging of HIV-1 envelope glycoprotein dynamics and Gag lattice association exposes determinants responsible for virus incorporation. Proc. Natl. Acad. Sci. USA 2019, 116, 25269–25277. [Google Scholar] [CrossRef] [Green Version]
- Alfadhli, A.; Staubus, A.O.; Tedbury, P.R.; Novikova, M.; Freed, E.O.; Barklis, E. Analysis of HIV-1 Matrix-Envelope Cytoplasmic Tail Interactions. J. Virol. 2019, 93, e01079-19. [Google Scholar] [CrossRef]
- Tedbury, P.R.; Novikova, M.; Alfadhli, A.; Hikichi, Y.; Kagiampakis, I.; KewalRamani, V.N.; Barklis, E.; Freed, E.O. HIV-1 Matrix Trimerization-Impaired Mutants Are Rescued by Matrix Substitutions That Enhance Envelope Glycoprotein Incorporation. J. Virol. 2019, 94, e01526-19. [Google Scholar] [CrossRef]
- Tedbury, P.R.; Mercredi, P.Y.; Gaines, C.R.; Summers, M.F.; Freed, E.O. Elucidating the mechanism by which compensatory mutations rescue an HIV-1 matrix mutant defective for gag membrane targeting and envelope glycoprotein incorporation. J. Mol. Biol. 2015, 427, 1413–1427. [Google Scholar] [CrossRef] [Green Version]
- Alfadhli, A.; Mack, A.; Ritchie, C.; Cylinder, I.; Harper, L.; Tedbury, P.R.; Freed, E.O.; Barklis, E. Trimer Enhancement Mutation Effects on HIV-1 Matrix Protein Binding Activities. J. Virol. 2016, 90, 5657–5664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waheed, A.A.; Freed, E.O. HIV Type 1 Gag as a Target for Antiviral Therapy. AIDS Res. Hum. Retroviruses 2012, 28, 54–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, A.; Cocklin, S. Recent Advances in HIV-1 Gag Inhibitor Design and Development. Molecules 2020, 25, 1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaPlante, S.R.; Forgione, P.; Boucher, C.; Coulombe, R.; Gillard, J.; Hucke, O.; Jakalian, A.; Joly, M.A.; Kukolj, G.; Lemke, C.; et al. Enantiomeric atropisomers inhibit HCV polymerase and/or HIV matrix: Characterizing hindered bond rotations and target selectivity. J. Med. Chem. 2014, 57, 1944–1951. [Google Scholar] [CrossRef] [PubMed]
- Zentner, I.; Sierra, L.J.; Fraser, A.K.; Maciunas, L.; Mankowski, M.K.; Vinnik, A.; Fedichev, P.; Ptak, R.G.; Martín-García, J.; Cocklin, S. Identification of a Small-Molecule Inhibitor of HIV-1 Assembly that Targets the Phosphatidylinositol (4,5)-bisphosphate Binding Site of the HIV-1 Matrix Protein. ChemMedChem 2013, 8, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Zentner, I.; Sierra, L.J.; Maciunas, L.; Vinnik, A.; Fedichev, P.; Mankowski, M.K.; Ptak, R.G.; Martín-García, J.; Cocklin, S. Discovery of a small-molecule antiviral targeting the HIV-1 matrix protein. Bioorg. Med. Chem. Lett. 2013, 23, 1132–1135. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murphy, R.E.; Saad, J.S. The Interplay between HIV-1 Gag Binding to the Plasma Membrane and Env Incorporation. Viruses 2020, 12, 548. https://doi.org/10.3390/v12050548
Murphy RE, Saad JS. The Interplay between HIV-1 Gag Binding to the Plasma Membrane and Env Incorporation. Viruses. 2020; 12(5):548. https://doi.org/10.3390/v12050548
Chicago/Turabian StyleMurphy, R. Elliot, and Jamil S. Saad. 2020. "The Interplay between HIV-1 Gag Binding to the Plasma Membrane and Env Incorporation" Viruses 12, no. 5: 548. https://doi.org/10.3390/v12050548
APA StyleMurphy, R. E., & Saad, J. S. (2020). The Interplay between HIV-1 Gag Binding to the Plasma Membrane and Env Incorporation. Viruses, 12(5), 548. https://doi.org/10.3390/v12050548