Identification of Combinations of Protein Kinase C Activators and Histone Deacetylase Inhibitors that Potently Reactivate Latent HIV


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Reagents
2.3. Evaluation of HIV-Latency Reversal in Cell Lines
2.4. Evaluation of Cytotoxicity
2.5. Real-Time qRT-PCR (Real-Time Quantitative Reverse Transcription PCR) and HIV-1 RNA Analysis
2.6. Evaluation of HIV Reactivation by Western Blot
2.7. Establishment of HIV Latency in Primary CD4+ T-Cells
2.8. Detection of gp120 Expression
2.9. Drug Interaction Analysis
3. Results
3.1. Evaluation of the Efficacy and Cytotoxicity of the LRAs
3.2. Combinations of PKCas and HDACis Do not Affect the Viability of Uninfected Cells
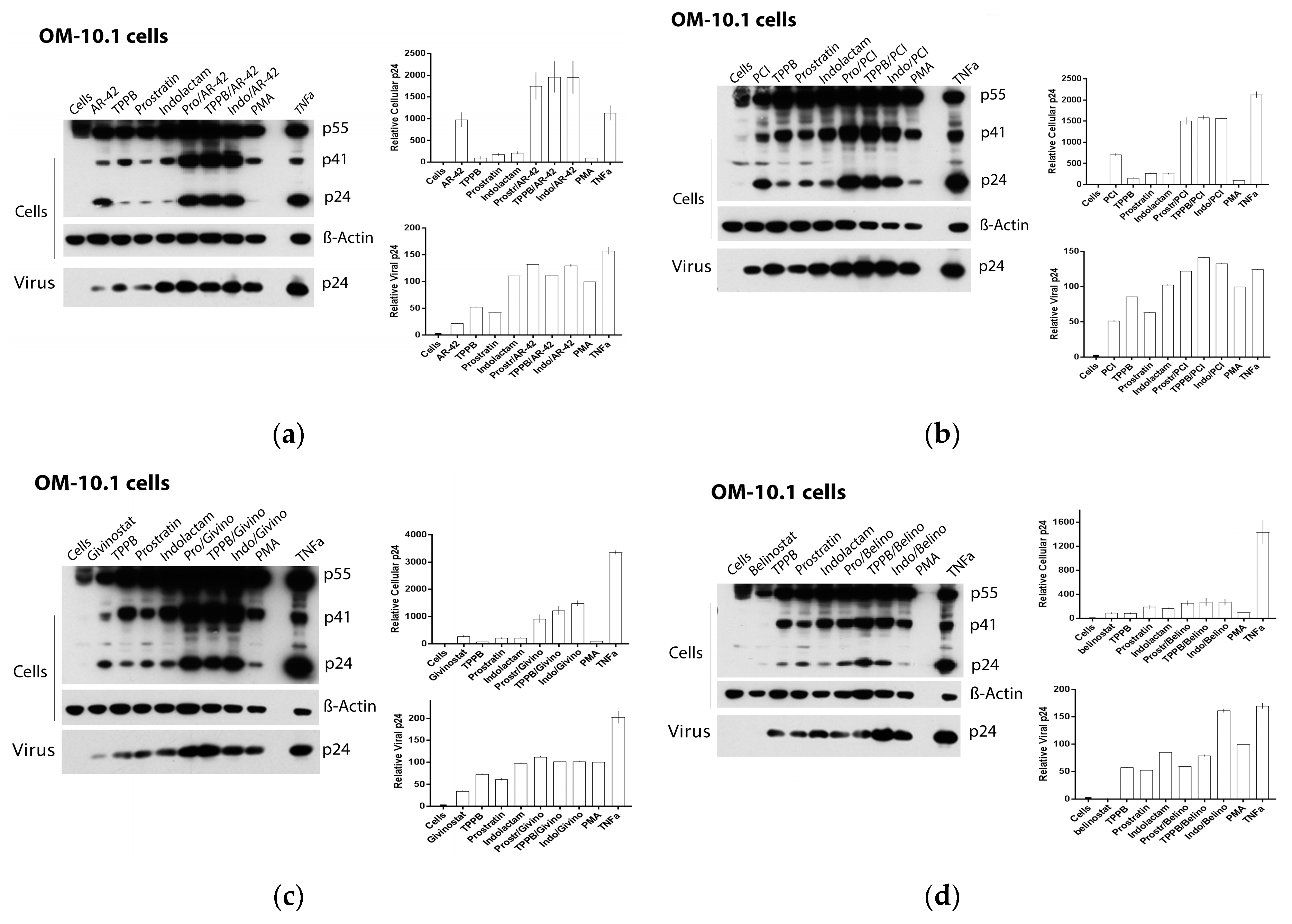
3.3. Combinations of LRAs Reactivate HIV Expression in Latently Infected Cells
3.4. Combinations of PKCas and HDACis Induce High Levels of gp120 Expression
3.5. Indolactam/AR-42 Induces CD4 Downregulation
3.6. Evaluation of HIV Activation in a Primary CD4+ T-Cell Model
3.7. Effect of LRAs Combinations on the Cellular Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bruner, K.M.; Hosmane, N.N.; Siliciano, R.F. Towards an HIV-1 cure: Measuring the latent reservoir. Trends Microbiol. 2015, 23, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Lint, C.; Bouchat, S.; Marcello, A. HIV-1 transcription and latency: An update. Retrovirology 2013, 10, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, M.; Johnston, R. Curing HIV: Moving Forward Faster. AIDS Res. Hum. Retrovir. 2016, 32, 125–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. HIV reservoirs: What, where and how to target them. Nat. Rev. Microbiol. 2016, 14, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Dahabieh, M.S.; Battivelli, E.; Verdin, E. Understanding HIV latency: The road to an HIV cure. Annu. Rev. Med. 2015, 66, 407–421. [Google Scholar] [CrossRef] [Green Version]
- Vanhamel, J.; Bruggemans, A.; Debyser, Z. Establishment of latent HIV-1 reservoirs: What do we really know? J. Virus Erad. 2019, 5, 3–9. [Google Scholar]
- Josefsson, L.; King, M.S.; Makitalo, B.; Brannstrom, J.; Shao, W.; Maldarelli, F.; Kearney, M.F.; Hu, W.S.; Chen, J.; Gaines, H.; et al. Majority of CD4+ T cells from peripheral blood of HIV-1-infected individuals contain only one HIV DNA molecule. Proc. Natl. Acad. Sci. USA 2011, 108, 11199–11204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josefsson, L.; Palmer, S.; Faria, N.R.; Lemey, P.; Casazza, J.; Ambrozak, D.; Kearney, M.; Shao, W.; Kottilil, S.; Sneller, M.; et al. Single cell analysis of lymph node tissue from HIV-1 infected patients reveals that the majority of CD4+ T-cells contain one HIV-1 DNA molecule. PLoS Pathog. 2013, 9, e1003432. [Google Scholar] [CrossRef] [Green Version]
- Taube, R.; Peterlin, M. Lost in transcription: Molecular mechanisms that control HIV latency. Viruses 2013, 5, 902–927. [Google Scholar] [CrossRef] [Green Version]
- Deeks, S.G. HIV: Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef]
- Turner, A.W.; Margolis, D.M. Chromatin Regulation and the Histone Code in HIV Latency. Yale J. Biol. Med. 2017, 90, 229–243. [Google Scholar] [PubMed]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banga, R.; Procopio, F.A.; Cavassini, M.; Perreau, M. In Vitro Reactivation of Replication-Competent and Infectious HIV-1 by Histone Deacetylase Inhibitors. J. Virol. 2016, 90, 1858–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirakawa, K.; Chavez, L.; Hakre, S.; Calvanese, V.; Verdin, E. Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. 2013, 21, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Dandekar, S. Targeting NF-kappaB signaling with protein kinase C agonists as an emerging strategy for combating HIV latency. AIDS Res. Hum. Retrovir. 2015, 31, 4–12. [Google Scholar] [CrossRef]
- McKernan, L.N.; Momjian, D.; Kulkosky, J. Protein Kinase C: One Pathway towards the Eradication of Latent HIV-1 Reservoirs. Adv. Virol. 2012, 2012, 805347. [Google Scholar] [CrossRef] [Green Version]
- Mates, J.M.; de Silva, S.; Lustberg, M.; Van Deusen, K.; Baiocchi, R.A.; Wu, L.; Kwiek, J.J. A Novel Histone Deacetylase Inhibitor, AR-42, Reactivates HIV-1 from Chronically and Latently Infected CD4(+) T-cells. Retrovirology 2015, 7, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, K.; Kobayakawa, T.; Tsuchiya, K.; Hattori, S.I.; Nomura, W.; Gatanaga, H.; Yoshimura, K.; Oka, S.; Endo, Y.; Tamamura, H.; et al. Benzolactam-related compounds promote apoptosis of HIV-infected human cells via protein kinase C-induced HIV latency reversal. J. Biol. Chem. 2019, 294, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Sloan, D.D.; Lam, C.Y.; Irrinki, A.; Liu, L.; Tsai, A.; Pace, C.S.; Kaur, J.; Murry, J.P.; Balakrishnan, M.; Moore, P.A.; et al. Targeting HIV Reservoir in Infected CD4 T Cells by Dual-Affinity Re-targeting Molecules (DARTs) that Bind HIV Envelope and Recruit Cytotoxic T Cells. PLoS Pathog. 2015, 11, e1005233. [Google Scholar] [CrossRef] [Green Version]
- Reuse, S.; Calao, M.; Kabeya, K.; Guiguen, A.; Gatot, J.S.; Quivy, V.; Vanhulle, C.; Lamine, A.; Vaira, D.; Demonte, D.; et al. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: Implications for treatment of latent infection. PLoS ONE 2009, 4, e6093. [Google Scholar] [CrossRef]
- Darcis, G.; Kula, A.; Bouchat, S.; Fujinaga, K.; Corazza, F.; Ait-Ammar, A.; Delacourt, N.; Melard, A.; Kabeya, K.; Vanhulle, C.; et al. An In-Depth Comparison of Latency-Reversing Agent Combinations in Various In Vitro and Ex Vivo HIV-1 Latency Models Identified Bryostatin-1+JQ1 and Ingenol-B+JQ1 to Potently Reactivate Viral Gene Expression. PLoS Pathog. 2015, 11, e1005063. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Lai, W.H.; Zhu, L.; Li, W.; Wei, L.; Lee, K.H.; Xie, L.; Chen, C.H. Elimination of HIV-1 Latently Infected Cells by Gnidimacrin and a Selective HDAC Inhibitor. ACS Med. Chem. Lett. 2018, 9, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Hezareh, M. Prostratin as a new therapeutic agent targeting HIV viral reservoirs. Drug News Perspect. 2005, 18, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Hezareh, M.; Moukil, M.A.; Szanto, I.; Pondarzewski, M.; Mouche, S.; Cherix, N.; Brown, S.J.; Carpentier, J.L.; Foti, M. Mechanisms of HIV receptor and co-receptor down-regulation by prostratin: Role of conventional and novel PKC isoforms. Antivir. Chem. Chemother. 2004, 15, 207–222. [Google Scholar] [CrossRef]
- Miana, G.A.; Riaz, M.; Shahzad-ul-Hussan, S.; Paracha, R.Z.; Paracha, U.Z. Prostratin: An Overview. Mini Rev. Med. Chem. 2015, 15, 1122–1130. [Google Scholar] [CrossRef]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Fenard, D.; Bisgrove, D.; Verdin, E.; Greene, W.C. Prostratin antagonizes HIV latency by activating NF-kappaB. J. Biol. Chem. 2004, 279, 42008–42017. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.Q.; Li, X.; Yang, W.M.; Feng, S.M.; Ma, J.J. Neuroprotective effects of new protein kinase C activator TPPB against Abeta(2)(5)(-)(3)(5) induced neurotoxicity in PC12 cells. Neurochem. Res. 2012, 37, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Matalon, S.; Palmer, B.E.; Nold, M.F.; Furlan, A.; Kassu, A.; Fossati, G.; Mascagni, P.; Dinarello, C.A. The histone deacetylase inhibitor ITF2357 decreases surface CXCR4 and CCR5 expression on CD4(+) T-cells and monocytes and is superior to valproic acid for latent HIV-1 expression in vitro. J Acquir. Immune Defic. Syndr. 2010, 54, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Matalon, S.; Rasmussen, T.A.; Dinarello, C.A. Histone deacetylase inhibitors for purging HIV-1 from the latent reservoir. Mol. Med. 2011, 17, 466–472. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Schmeltz Sogaard, O.; Brinkmann, C.; Wightman, F.; Lewin, S.R.; Melchjorsen, J.; Dinarello, C.; Ostergaard, L.; Tolstrup, M. Comparison of HDAC inhibitors in clinical development: Effect on HIV production in latently infected cells and T-cell activation. Hum. Vaccin. Immunother. 2013, 9, 993–1001. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, E.; Moyal, E.C.; Gregorc, V.; Zucali, P.A.; Menard, J.; Soria, J.C.; Kloos, I.; Hsu, J.; Luan, Y.; Liu, E.; et al. A phase 1 dose-escalation study of the oral histone deacetylase inhibitor abexinostat in combination with standard hypofractionated radiotherapy in advanced solid tumors. Oncotarget 2017, 8, 56199–56209. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.; Wiskocil, R.L.; Stobo, J.D. The role of T3 surface molecules in the activation of human T cells: A two-stimulus requirement for IL 2 production reflects events occurring at a pre-translational level. J. Immunol. 1984, 133, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folks, T.M.; Justement, J.; Kinter, A.; Dinarello, C.A.; Fauci, A.S. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 1987, 238, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Folks, T.M.; Clouse, K.A.; Justement, J.; Rabson, A.; Duh, E.; Kehrl, J.H.; Fauci, A.S. Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T-cell clone. Proc. Natl. Acad. Sci. USA 1989, 86, 2365–2368. [Google Scholar] [CrossRef] [Green Version]
- Butera, S.T.; Roberts, B.D.; Lam, L.; Hodge, T.; Folks, T.M. Human immunodeficiency virus type 1 RNA expression by four chronically infected cell lines indicates multiple mechanisms of latency. J. Virol. 1994, 68, 2726–2730. [Google Scholar] [CrossRef] [Green Version]
- Desire, N.; Dehee, A.; Schneider, V.; Jacomet, C.; Goujon, C.; Girard, P.M.; Rozenbaum, W.; Nicolas, J.C. Quantification of human immunodeficiency virus type 1 proviral load by a TaqMan real-time PCR assay. J. Clin. Microbiol. 2001, 39, 1303–1310. [Google Scholar] [CrossRef] [Green Version]
- Saleh, S.; Solomon, A.; Wightman, F.; Xhilaga, M.; Cameron, P.U.; Lewin, S.R. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: A novel model of HIV-1 latency. Blood 2007, 110, 4161–4164. [Google Scholar] [CrossRef]
- Tripathy, M.K.; McManamy, M.E.; Burch, B.D.; Archin, N.M.; Margolis, D.M. H3K27 Demethylation at the Proviral Promoter Sensitizes Latent HIV to the Effects of Vorinostat in Ex Vivo Cultures of Resting CD4+ T Cells. J. Virol. 2015, 89, 8392–8405. [Google Scholar] [CrossRef] [Green Version]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef]
- Einkauf, K.B.; Lee, G.Q.; Gao, C.; Sharaf, R.; Sun, X.; Hua, S.; Chen, S.M.; Jiang, C.; Lian, X.; Chowdhury, F.Z.; et al. Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J. Clin. Investig. 2019, 129, 988–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashemi, F.B.; Barreto, K.; Bernhard, W.; Hashemi, P.; Lomness, A.; Sadowski, I. HIV Provirus Stably Reproduces Parental Latent and Induced Transcription Phenotypes Regardless of the Chromosomal Integration Site. J. Virol. 2016, 90, 5302–5314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wightman, F.; Lu, H.K.; Solomon, A.E.; Saleh, S.; Harman, A.N.; Cunningham, A.L.; Gray, L.; Churchill, M.; Cameron, P.U.; Dear, A.E.; et al. Entinostat is a histone deacetylase inhibitor selective for class 1 histone deacetylases and activates HIV production from latently infected primary T cells. AIDS 2013, 27, 2853–2862. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Mai, A.; Norelli, S.; El Daker, S.; Valente, S.; Rotili, D.; Altucci, L.; Palamara, A.T.; Garaci, E. “Shock and kill” effects of class I-selective histone deacetylase inhibitors in combination with the glutathione synthesis inhibitor buthionine sulfoximine in cell line models for HIV-1 quiescence. Retrovirology 2009, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Butera, S.T.; Perez, V.L.; Wu, B.Y.; Nabel, G.J.; Folks, T.M. Oscillation of the human immunodeficiency virus surface receptor is regulated by the state of viral activation in a CD4+ cell model of chronic infection. J. Virol. 1991, 65, 4645–4653. [Google Scholar] [CrossRef] [Green Version]
- Butera, S.T.; Roberts, B.D.; Folks, T.M. Regulation of HIV-1 expression by cytokine networks in a CD4+ model of chronic infection. J. Immunol. 1993, 150, 625–634. [Google Scholar]
- Raposo, R.A.; Trudgian, D.C.; Thomas, B.; van Wilgenburg, B.; Cowley, S.A.; James, W. Protein kinase C and NF-kappaB-dependent CD4 downregulation in macrophages induced by T cell-derived soluble factors: Consequences for HIV-1 infection. J. Immunol. 2011, 187, 748–759. [Google Scholar] [CrossRef] [Green Version]
- Rullas, J.; Bermejo, M.; Garcia-Perez, J.; Beltan, M.; Gonzalez, N.; Hezareh, M.; Brown, S.J.; Alcami, J. Prostratin induces HIV activation and downregulates HIV receptors in peripheral blood lymphocytes. Antivir. Ther. 2004, 9, 545–554. [Google Scholar]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef] [Green Version]
- Marquez, N.; Calzado, M.A.; Sanchez-Duffhues, G.; Perez, M.; Minassi, A.; Pagani, A.; Appendino, G.; Diaz, L.; Munoz-Fernandez, M.A.; Munoz, E. Differential effects of phorbol-13-monoesters on human immunodeficiency virus reactivation. Biochem. Pharmacol. 2008, 75, 1370–1380. [Google Scholar] [CrossRef]
- Salio, M.; Valitutti, S.; Lanzavecchia, A. Agonist-induced T cell receptor down-regulation: Molecular requirements and dissociation from T cell activation. Eur. J. Immunol. 1997, 27, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Walker-Sperling, V.E.; Pohlmeyer, C.W.; Tarwater, P.M.; Blankson, J.N. The Effect of Latency Reversal Agents on Primary CD8+ T Cells: Implications for Shock and Kill Strategies for Human Immunodeficiency Virus Eradication. EBioMedicine 2016, 8, 217–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.B.; O’Connor, R.; Mueller, S.; Foley, M.; Szeto, G.L.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M.A.; Trocha, A.; et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog. 2014, 10, e1004287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finzi, D.; Plaeger, S.F.; Dieffenbach, C.W. Defective virus drives human immunodeficiency virus infection, persistence, and pathogenesis. Clin. Vaccine Immunol. 2006, 13, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.L.; Spector, S.A. Autophagy induction by histone deacetylase inhibitors inhibits HIV type 1. J. Biol. Chem. 2015, 290, 5028–5040. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Target | OM-10.1 | U1 | ACH-2 | J-Lat 10.6 | J-Lat A2 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 1 | CC50 1 | EC50 1 | CC50 1 | EC50 1 | CC50 1 | EC50 1 | CC50 1 | EC50 1 | CC50 1 | ||
| Prostratin | PKC | 0.41 ± 0.03 | >8 | 0.3 ± 0.03 | >8 | 0.31 ± 0.07 | >8 | 0.68 ± 0.01 | >8 | 0.87 ± 0.03 | >8 |
| TPPB | PKC | 0.13 ± 0.01 | >8 | 0.21 ± 0.01 | >8 | 0.07 ± 0.01 | >8 | 0.51 ± 0.08 | >8 | 0.49 ± 0.03 | >8 |
| (-)-Indolactam V | PKC | 0.09 ± 0.02 | >8 | 0.12 ± 0.02 | >8 | 0.18 ± 0.03 | >8 | 0.2 ± 0.04 | >8 | 0.32 ± 0.05 | >8 |
| Belinostat | HDAC | 0.88 ± 0.4 | 2.7 ± 0.2 | 0.77 ± 0.01 | >8 | 0.44 ± 0.03 | 1.1 ± 0.2 | 0.94 ± 0.03 | 1.4 ± 0.06 | 0.94 ± 0.05 | 1.4 ± 0.02 |
| PCI-24781 | HDAC | 0.35 ± 0.04 | 0.4 ± 0.02 | 0.46 ± 0.03 | ~8 | 0.37 ± 0.04 | 0.31 ± 0.02 | 0.45 ± 0.02 | 0.4 ± 0.02 | 0.38 ± 0.05 | 0.54 ± 0.03 |
| Givinostat | HDAC | 0.45 ± 0.13 | 0.42 ± 0.01 | 0.63 ± 0.02 | ~4 | 0.32 ± 0.04 | 0.63 ± 0.03 | 0.88 ± 0.04 | 0.93 ± 0.04 | 0.7 ± 0.05 | 1.2 ± 0.02 |
| AR-42 | HDAC | 0.53 ± 0.08 | 0.4 ± 0.01 | 0.26 ± 0.1 | ~8 | 0.24 ± 0.04 | 0.41 ± 0.01 | 0.55 ± 0.02 | 0.45 ± 0.02 | 0.44 ± 0.05 | 0.53 ± 0.05 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curreli, F.; Ahmed, S.; Victor, S.M.B.; Debnath, A.K. Identification of Combinations of Protein Kinase C Activators and Histone Deacetylase Inhibitors that Potently Reactivate Latent HIV. Viruses 2020, 12, 609. https://doi.org/10.3390/v12060609
Curreli F, Ahmed S, Victor SMB, Debnath AK. Identification of Combinations of Protein Kinase C Activators and Histone Deacetylase Inhibitors that Potently Reactivate Latent HIV. Viruses. 2020; 12(6):609. https://doi.org/10.3390/v12060609
Chicago/Turabian StyleCurreli, Francesca, Shahad Ahmed, Sofia M. Benedict Victor, and Asim K. Debnath. 2020. "Identification of Combinations of Protein Kinase C Activators and Histone Deacetylase Inhibitors that Potently Reactivate Latent HIV" Viruses 12, no. 6: 609. https://doi.org/10.3390/v12060609