1. Introduction
The novel coronavirus SARS-CoV-2, the causative agent of Coronavirus disease 2019 (COVID-19), belongs to the betacoronavirus genus, which also includes the highly pathogenic SARS CoV and MERS CoV. SARS-CoV-2, a close relative of bat betacoronaviruses emerged at the end of 2019 in Wuhan, China, and has caused a pandemic [
1]. The pandemic and the public response have greatly impacted public health and caused profound social and economic disruption. As of May 11, 2020, SARS-CoV-2 had caused more than four million infections and more than 250,000 deaths worldwide [
2]. The emergence of this new virus has prompted urgent worldwide efforts to develop diagnostics, vaccines, and antivirals, to define the natural history of human infection; and to better characterize the virus.
The expanded interest in studying SARS-CoV-2 to address the current pandemic requires that many laboratories acquire the capacity to work with the virus. However, despite the rapidly growing body of literature either deposited on preprint servers or in peer-reviewed scientific journals, there remains a lack of information regarding standardized protocols for work with the virus. Among these needs are the means to grow and quantify infectious virus. Additionally, the recommendation that experiments involving the propagation of SARs-CoV-2 be performed at biosafety level 3 (BSL3) necessitates the development of methods to safely inactivate the virus and validate inactivation methods to allow an array of studies to be performed at lower biocontainment levels [
3,
4,
5,
6]. Examples include the isolation of RNA from virus and virus-infected cells to characterize viral genome sequences, monitor viral gene expression and genome replication, and to characterize host responses to infection. The removal of intact, virus-infected cells is critical for studies involving microscopy. Whole inactivated virus and viral proteins are needed for the development of inactivated whole-virus vaccine preparations, and also as a source of antigen for immunoassays.
To help address these needs and to facilitate SARS-CoV-2 research efforts, we describe here methods for the propagation of SARS-CoV-2 in multiple cell lines. We have also determined a more efficient method for quantifying virus by plaque assay and have developed a SARS-CoV-2-specific focus forming assay which can enhance throughput of assays requiring quantification of viral titers. Additionally, we describe validation of methods for the inactivation of SARS-CoV-2 through the use of TRIzol, 10% neutral buffered formalin, beta-propiolactone, and heat. Taken together, the data presented here will serve to provide researchers with a helpful basis of information to aid in their work on SARS-CoV-2.
2. Materials and Methods
2.1. Cells and Virus
Vero E6 (ATCC# CRL-1586), Calu-3 (ATCC# HTB-55), Caco-2 (ATCC# HTB-37), Huh7, A549 (ATCC# CCL-185), and 293T cells were maintained in DMEM (Corning) supplemented with 10% heat inactivated fetal bovine serum (FBS; GIBCO). Cells were kept in a 37 °C, 5% CO2 incubator without antibiotics or antimycotics. SARS-CoV-2, strain USA_WA1/2020, was obtained from the World Reference Collection for Emerging Viruses and Arboviruses at the University of Texas Medical Branch-Galveston.
2.2. Virus Propagation
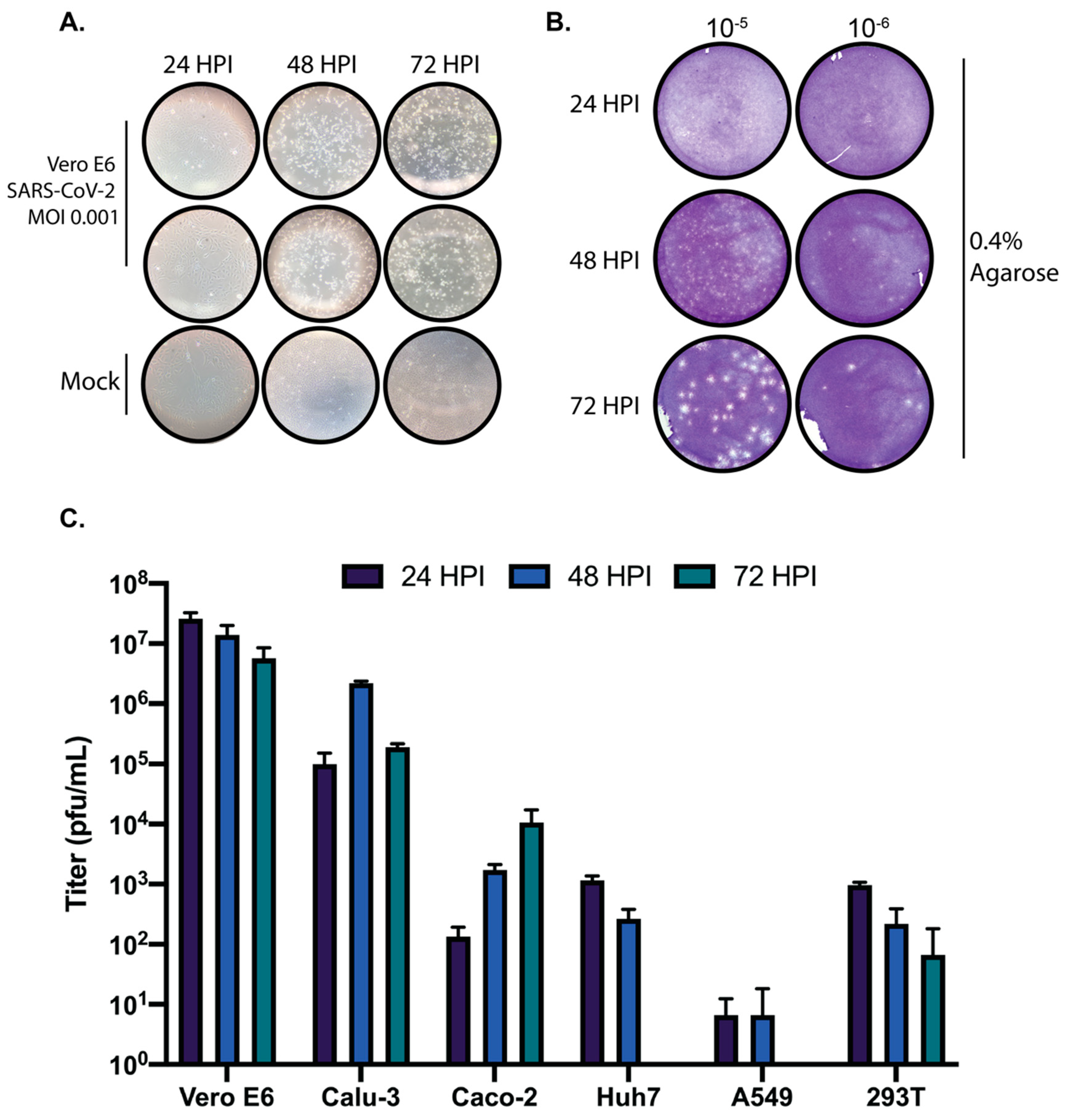
A lyophilized ampule of SARS-CoV-2 was initially resuspended in DMEM supplemented with 2% FBS. VeroE6 cells were inoculated in duplicate with a dilution of 1:100 with an adsorption period of 1 h at 37 °C and shaking every 15 min. Cells were observed for cytopathic effect (CPE) every 24 h. Stock SARS-CoV-2 virus was harvested at 72 h post infection (h.p.i) and supernatants were collected, clarified, aliquoted, and stored at −80 °C. For replication kinetic experiments, cells were seeded into 24-well plates at confluency in DMEM supplemented in 10% FBS. The next day, cells were inoculated with SARS-CoV-2 at a multiplicity of infection (MOI) of 0.01 in DMEM supplemented with 2% FBS. Supernatants were harvested at the indicated timepoints and stored at −80 °C until analysis.
2.3. SARS-CoV-2 Plaque Assay
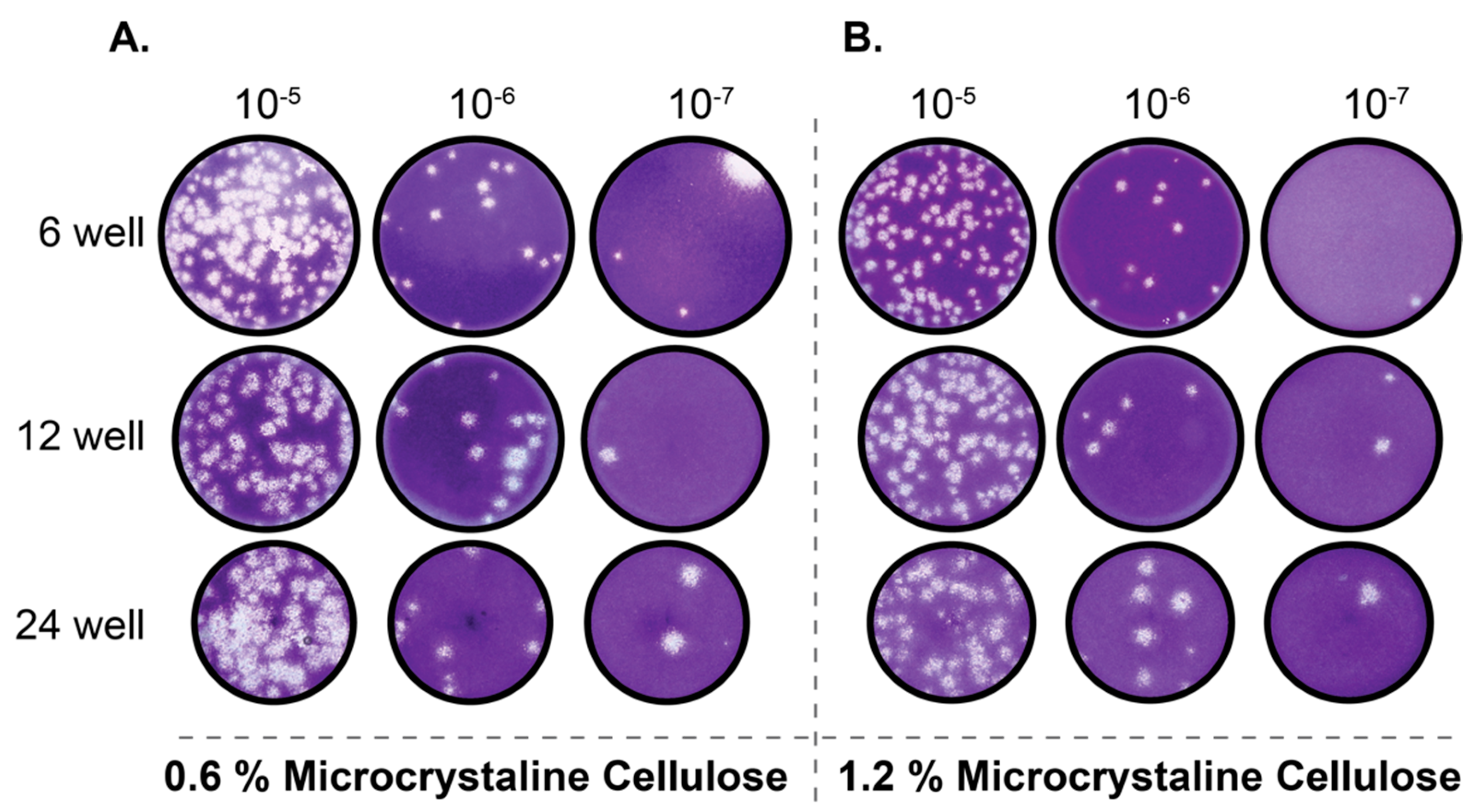
Vero E6 cells were seeded into 6, 12, or 24-well plates 24 h before infection. Ten-fold serial dilutions of SARS-CoV-2 samples were added, adsorbed for 1 h at 37 °C with shaking at 15-min intervals. After the absorption period, 2 mL of 0.3% agarose in DMEM supplemented with 2% FBS or 3 mL of 0.6 or 1.2 percent microcrystalline cellulose (MCC; Sigma 435244, St. Louis, MO, USA) in serum-free DMEM was added. For MCC plaque assays, virus inoculum was not removed. To stain plaque assays performed with an agarose overlay, 10% neutral buffered formalin (NBF) was added on top of the agarose and incubated for one hour at room temperature. The agarose plug was then removed with a pipette tip, and the fixed monolayer was stained with 0.4% crystal violet in 20% methanol. For plaque assays performed with MCC overlay, the MCC was aspirated out, 10% NBF added for one hour at room temp and then removed. Monolayers were then washed with water and stained with 0.4% crystal violet. Plaques were quantified and recorded as PFU/mL.
2.4. Focus Forming Assay
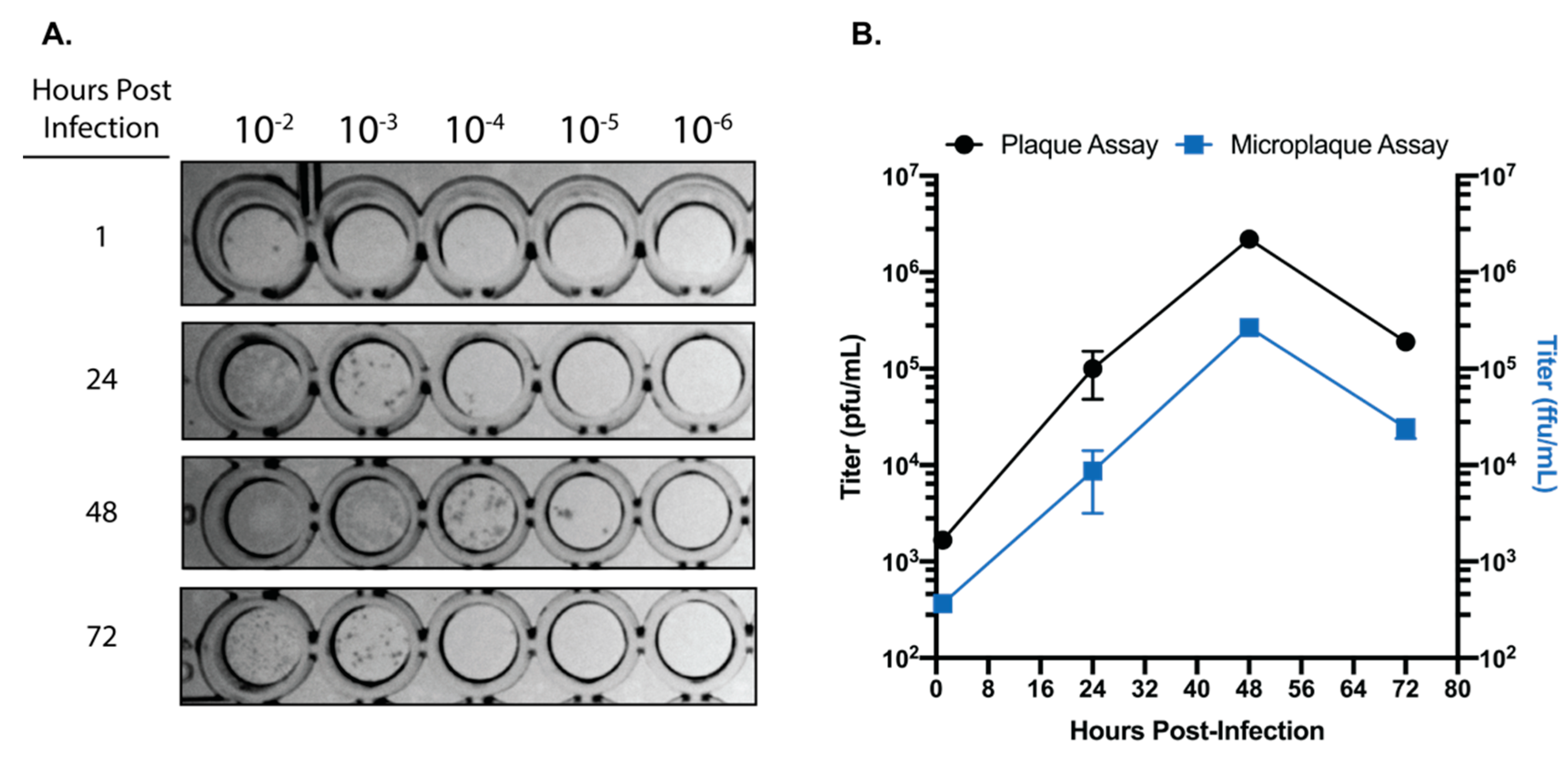
Vero E6 cells were plated into 96-well plates at confluency (75,000 cells/well) in DMEM supplemented with 10% heat-inactivated fetal bovine serum (Gibco). Prior to infection, virus stocks were thawed and serially diluted to obtain dilutions in the range of 10−2 to 10−9. Growth media was removed from the Vero E6 cells and 50 µL of virus dilutions was plated. Virus was adsorbed for 1 h at 37 °C/5% CO2. After adsorption, 50 µL of 2.4% MCC overlay supplemented with DMEM powdered media to a concentration of 1X (Gibco) was added to each well of the 96-well plate to achieve a final MCC overlay concentration of 1.2%. Plates were then incubated at 37 °C/5% CO2 for 24 h. The MCC overlay was gently removed and cells were fixed with 10% NBF for 1 h at room-temperature. After removal of NBF, monolayers were washed with ultrapure water and 100% methanol/0.3% H2O2 was added to permeabilize cells and quench endogenous peroxidase activity. Monolayers were then blocked for 1 h in PBS with 5% non-fat dry milk (NFDM). After blocking, monolayers were incubated with SARS-CoV N primary antibody (Novus Biologicals, Centennial, CO, USA; NB100-56576—1:2000) for 1 h at RT in PBS/5% NFDM. Monolayers were washed with PBS and incubated with an HRP-Conjugated secondary antibody for 1 h at RT in PBS/5% NFDM. Secondary was removed, monolayers were washed with PBS, and then developed using TrueBlue substrate (KPL) for 30 min. Plates were imaged on a Bio-Rad Chemidoc utilizing a phosphorscreen and foci were quantified.
2.5. TRIzol® Treatment
To validate the effectiveness of Trizol® Reagent in the inactivation of SARS-CoV-2, stock virus was separated into the following validation test groups: non-infected control (sterile media), Trizol treated non-infected control (sterile culture media + Trizol), duplicate SARS-CoV-2 positive controls (SARS-CoV-2; 1 × 106 pfu), and triplicate SARS-CoV-2 Trizol treated samples (SARS-CoV-2; 1 × 106 pfu +Trizol). Trizol was added to a final concentration of 10%. All test groups were then incubated at room temperature for 10 min. Samples were then diluted in 25 mL media and plated onto VeroE6 cells in a 15 cm tissue culture dish, incubated at 37 °C and 5% CO2, and monitored daily for CPE. At 48 h post infection, media was removed and the plates fixed with 10% NBF. To determine integrity of the cell monolayer, plates were stained with crystal violet.
2.6. Formalin Treatment
To validate inactivation of SARS-CoV-2 with formalin, 21 wells of a 24-well plate seeded to confluency with VeroE6 cells were infected with SARS-CoV-2 at an MOI of 0.1. Three wells served as mock-infected control. At 24 h post infection, media was removed from wells and treated as follows, using 3 wells per condition: uninfected control, SARS-CoV-2 infected control, SARS-CoV-2 + 2% formaldehyde, SARS-CoV-2 + 1% formaldehyde, SARS-CoV-2 + 0.5% formaldehyde, SARS-CoV-2 + 0.1% formaldehyde, and SARS-CoV-2 + 0.05% formaldehyde. Formaldehyde treatment was carried out at room temperature for 1 h. The cells were then washed with fresh media, scraped off the plate, and overlaid onto uninfected Vero E6 cells. Samples were then incubated at 37 °C, 5% CO2 and monitored daily for CPE. At 72 h post infection, media was removed and the plates fixed with 10% NBF. To determine integrity of the cell monolayer, plates were stained with crystal violet.
2.7. Beta-Propiolactone Treatment
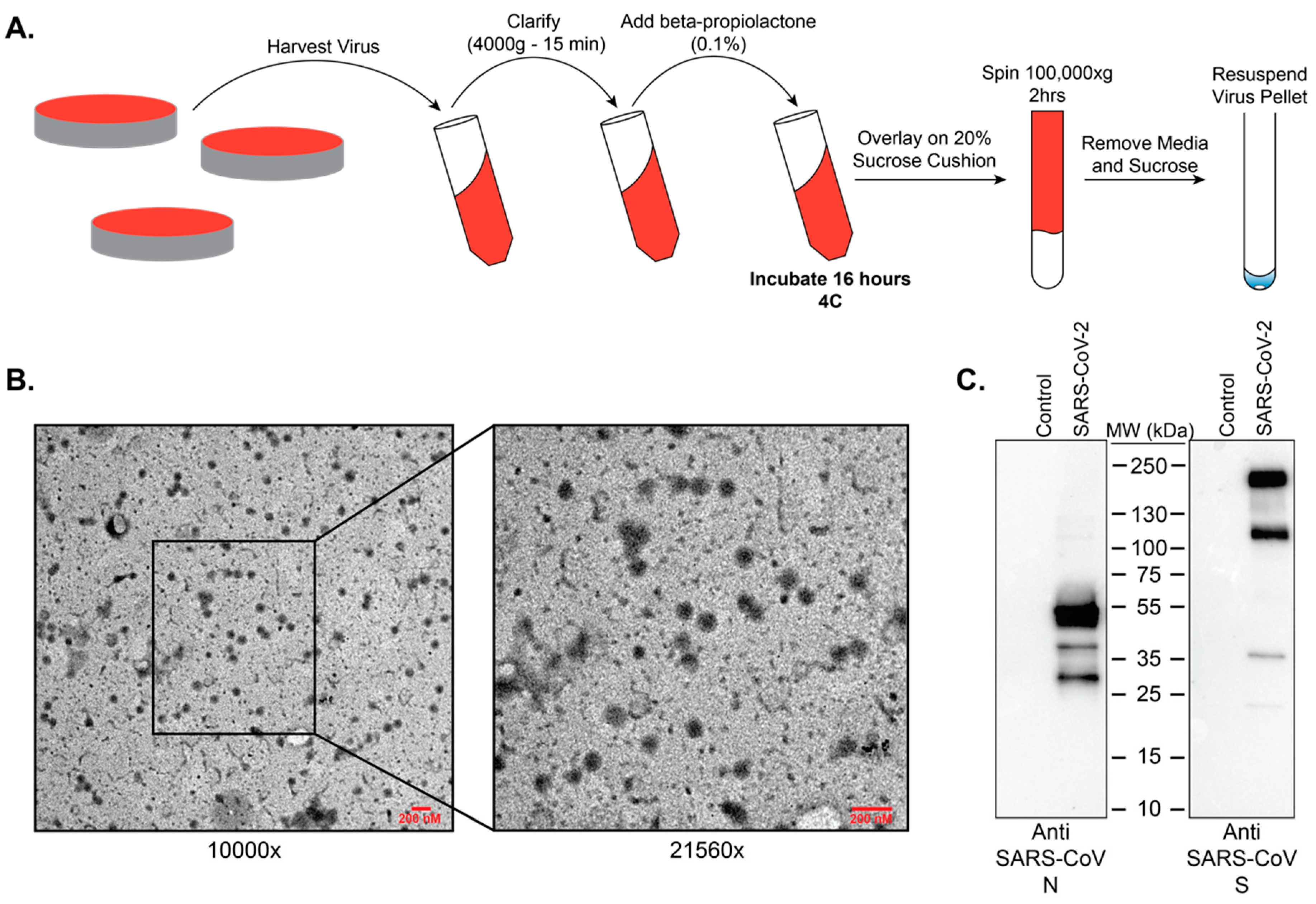
To test treatment of viral particles with beta-propiolactone as means of inactivation, stock SARS-CoV-2 virus was separated into the following validation groups: non-infected control, 4 °C control (1 × 10
6 pfu per replicate), 0.5% beta-propiolactone (1 × 10
6 pfu per replicate), 0.1% beta-propiolactone (1 × 10
6 pfu per replicate), and 0.05% beta-propiolactone (1 × 10
6 pfu per replicate). After incubation at 4 °C for 16 h, samples were transferred to 37 °C for two (2) hours to hydrolyze all residual beta-propiolactone. This step ensures complete hydrolysis of beta-propiolactone to prevent cytotoxicity to mammalian cells [
7]. After hydrolysis, samples were inoculated onto Vero E6 cells, and incubated at 37 °C, 5% CO
2 incubator and monitored daily for CPE. At 72 h post infection, media was removed and plates were fixed with 10% NBF. To determine integrity of the cell monolayer, plates were stained with crystal violet.
2.8. Heat Inactivation
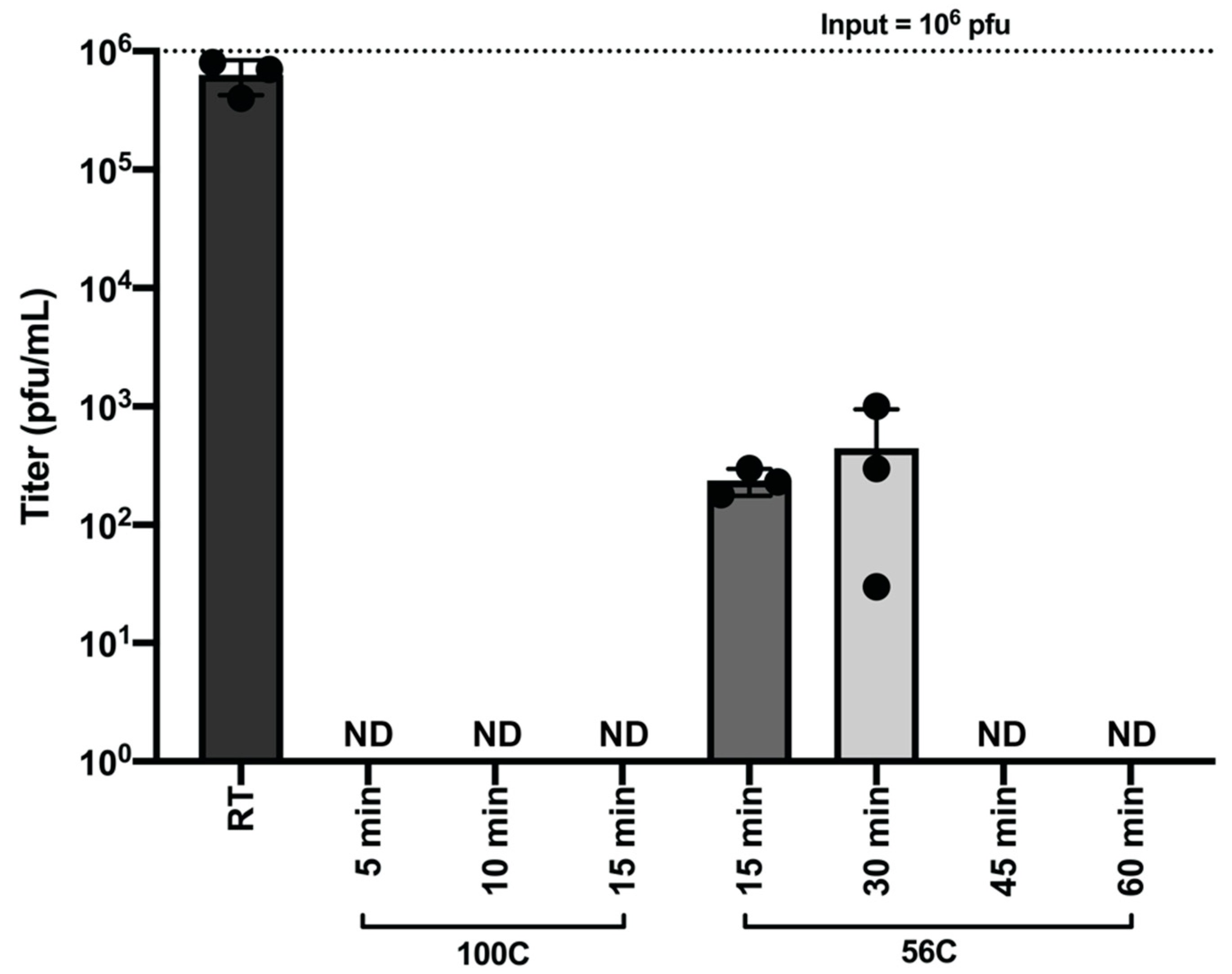
To validate heat treatment as a method to inactivate SARS-CoV-2, SARS-CoV-2 virus was separated into the following validation groups: non-infected control, room temperature control (1 × 106 pfu per replicate), 100 °C for 5 min (1 × 106 pfu per replicate), 100 °C for 10 min (1 × 106 pfu per replicate), and 100 °C for 15 min (1 × 106 pfu per replicate). microcentrifuge polypropylene tubes (1.5 mL) containing the virus (500 µL total volume) were exposed to direct heat in a heat block (Fisher Scientific, Hampton, NH, USA). After heating, all samples were left to cool to room temperature and centrifuged to collect condensation within the tube. Each sample was then inoculated in 5-fold serial dilutions onto VeroE6 cells. Samples were then incubated at 37 °C, 5% CO2. At 72 h post infection, supernatants were harvested, and infectious virus was quantified by plaque assay on Vero E6 cells.
2.9. Electron Microscopy
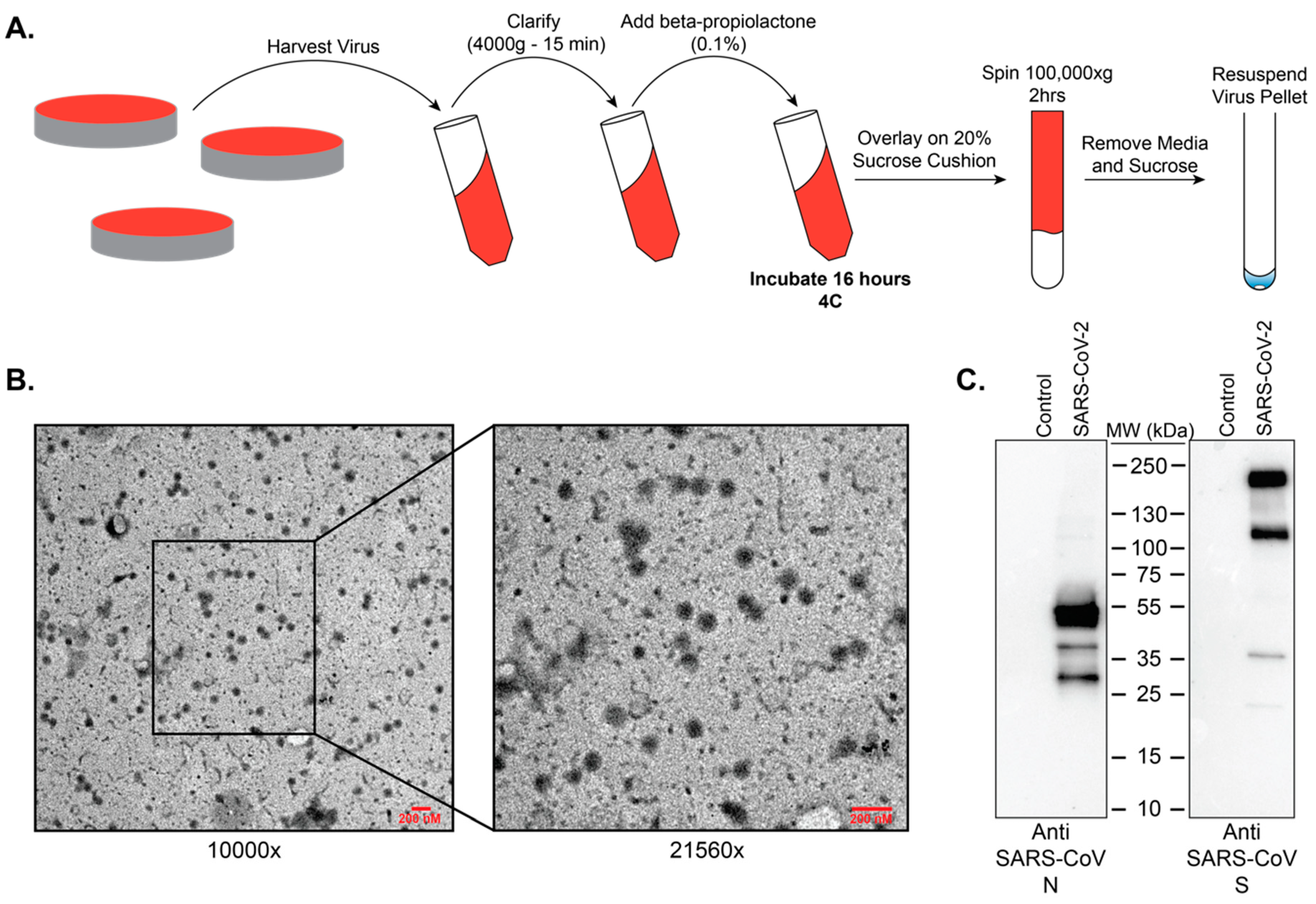
Resuspended purified beta-propiolactone treated SARS-CoV-2 virus preps were adsorbed onto 300 mesh formvar-carbon coated nickel grids for 10 min, washed with 0.2 µM filtered ultrapure water, and negative-stained with UranyLess stain for 15 s. Grids were then washed with 0.2 µM ultrapure water, allowed to dry, and imaged on a LEO 960 TEM at 80 kV.
2.10. Western Blotting
Resuspended purified beta-propiolactone treated SARS-CoV-2 virus preps were separated by SDS-PAGE (Bio-Rad TGX Mini, Bio-Rad, Hercules, CA, USA) and transferred to 0.2 µM PVDF membrane according to the manufacturer’s protocols (Bio-Rad Tansblot Turbo; Bio-Rad, Hercules, CA, USA). After blocking in 5% non-fat dry milk in TBST (10 mM Tris, 150 mM NaCl, 0.5% Tween-20, pH8) for one hour, membranes were incubated overnight at 4 °C with antibodies targeting SARS-CoV N (Novus Biologicals, Centennial, CO, USA; NB100-56576) or SARS-CoV S (Sino Biological, Wayne, PA, USA; 40150-T62). Membranes were washed in TBST and incubated with an HRP-conjugated rabbit secondary antibody (Cell signaling, Danvers, MA, USA; 7074) for 1 Hr at room temperature. Membranes were then washed, developed with ECL, and imaged on a Bio-Rad Chemidoc imaging system.
4. Discussion
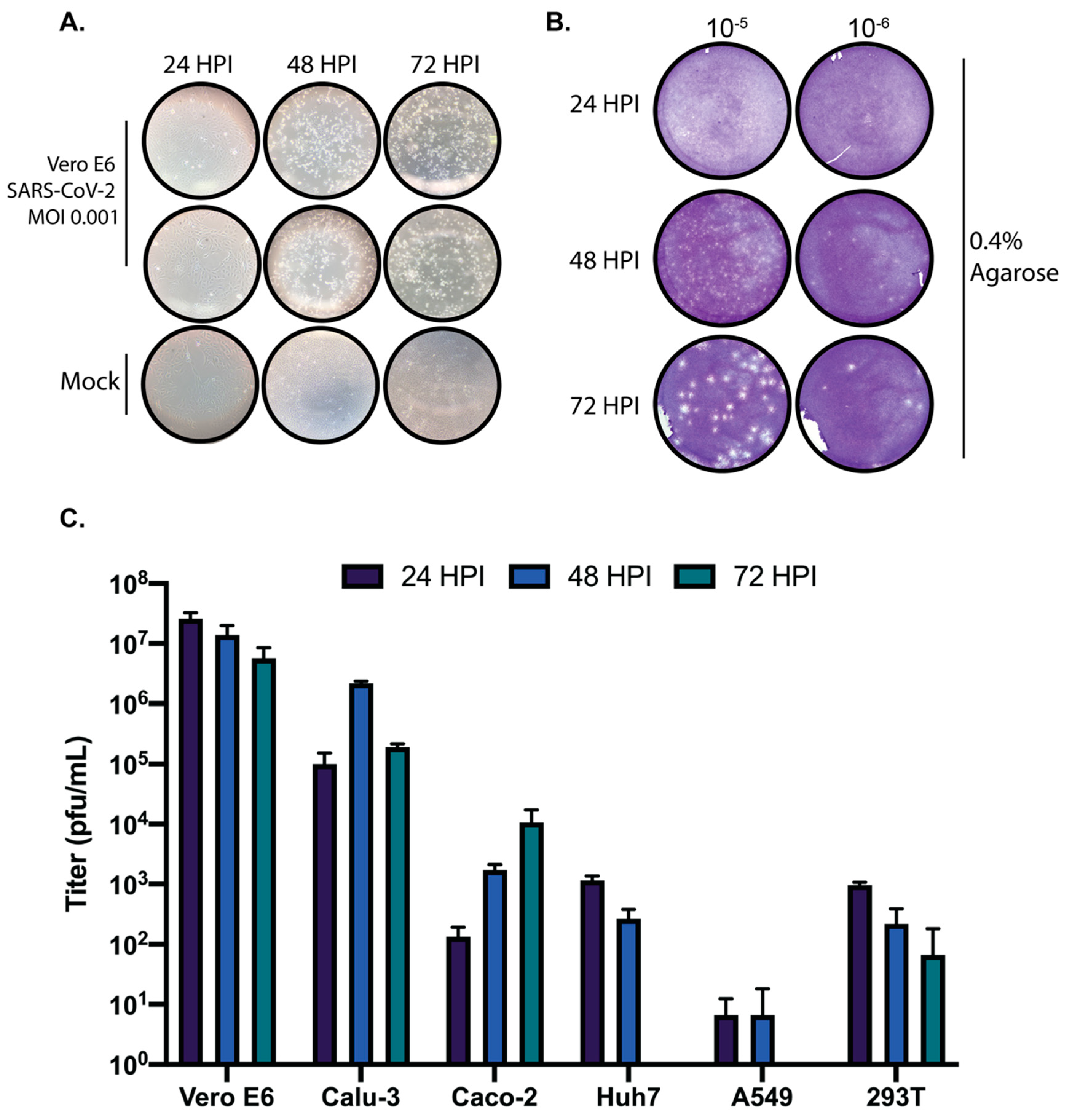
The ability to grow and accurately quantify infectious virus is critical for virological studies. Here, we sought to determine the growth kinetics of SARS-CoV-2 in several commonly used cell lines as well as the most appropriate time post-infection to accurately quantify SARS-CoV-2 by plaque assay. Significant growth was achieved in both Vero E6 and Calu-3 cells at the time points tested. We also observed that the colorectal adenocarcinoma Caco-2 cell line was able to propagate SARS-CoV-2, albeit to lower titers than Vero E6 and Calu-3 cells. Based on the replication kinetics observed in Caco-2 cells, it is likely that if the infection were allowed to progress past 72 h that higher titers would have been achieved. Huh7 and 293T cells exhibited modest growth of SARS-CoV-2, and A549 cells did not support SARS-CoV-2 replication at any of the tested timepoints. Therefore, these cell lines are not ideal hosts for virological studies with SARS-CoV-2 in the absence of modifications such as overexpression of the SARS-CoV-2 entry receptor ACE2 or the cellular protease TMPRSS2 which is necessary for Spike processing [
8,
9].
Plaque assays are among the most commonly used techniques for the accurate quantification of infectious virus. To establish reliable plaque assays for SARS-CoV-2 we compared two separate approaches. One used an agarose overlay and the other used microcrystalline cellulose (MCC). In our hands, both approaches yielded comparable data. However, the MCC overlay approach has significant advantages over the more traditional agarose overlay. MCC overlays do not require heating prior to use which significantly expedites the processing of large numbers of plaque assays and also alleviates concerns of overheating cell monolayers. MCC overlays are also insensitive to virus inoculum remaining in place providing the researcher with the option to remove virus inoculum or not. Additionally, MCC overlays do not require the removal of a “plug” like in the use of agarose overlays as MCC overlays remain liquid throughout their use. They can simply be aspirated, and the monolayer can be further processed. However, due to the fact that MCC overlays remain liquid throughout the course of the assay, it is important to take care that plaque assays utilizing MCC as an overlay are not moved until harvested to ensure reliable data. Our data presented here demonstrate that MCC is an effective and efficient overlay alternative to agarose for SARS-CoV-2 plaque assays.
While plaque assays are perhaps the most commonly used technique for the quantification of infectious virus, immunohistochemical focus forming assays are also commonly used for the quantification of virus [
10,
16,
17,
18]. We have established that SARS-CoV-2 can be reliably quantified by a 96-well plate-based focus forming assay in only 24 h. When compared to traditional plaque assays for SARS-CoV-2 which require 72 h for easily quantifiable plaques, the ability to quantify infectious SARS-CoV-2 within 24 h in a 96-well plate format represents a significant advantage for studies requiring higher-throughput. While it is important to acknowledge that titers acquired with a focus forming assay will generally be one log lower than those acquired by traditional plaque assay, the focus forming assay described here is fully capable of differentiating log-scale changes in virus titer. Taken together, the focus forming assay described here is a rapid and efficient method for quantifying infectious SARS-CoV-2.
Currently the recommendation from the Centers for Disease Control and Prevention (CDC) is for studies involving the propagation of SARS-CoV-2 to be performed within BSL3 laboratories with standard BSL3 practices. While an official risk group determination has not been made for SARS-CoV-2, the related viruses SARS-CoV and MERS are classified as risk-group 3 pathogens. As a BSL3 pathogen, having validated methods to inactivate SARS-CoV-2 is important so as to ensure safety while also allowing removal of samples to lower biosafety levels for analysis. For example, whole inactivated virus can serve as antigen for immunological assays, vaccine preparations, and for the analysis of virus composition. We chose to examine beta-propiolactone as a means to inactivate SARS-CoV-2. This method was chosen because it preserves virus structure and antigenicity, and it has recently been used to generate an inactivated vaccine preparation for SARS-CoV-2 [
15]. Our data show that treatment of SARS-CoV-2 with beta-propiolactone at a concentration of 0.5% for 16 h at 4 °C followed by 2 h at 37 °C will yield intact viral particles that can be utilized safely for downstream purposes.
The isolation of viral RNA and protein from virus or virus infected cells or the isolation RNA from host cells is critical to characterize virus sequence variation and to study the impact of infection on host gene expression. TRIzol is a commonly used reagent for RNA isolation that can also be used to for DNA and protein isolation. Our data show that treatment of SARS-CoV-2 containing samples with TRIzol following the manufacturer’s instructions is an effective method of inactivating SARS-CoV-2.
Formaldehyde is a ubiquitously used reagent in life sciences and has numerous applications such as the fixation of cells for microscopy studies such as indirect immunofluorescence of infected cells and as a disinfectant for scientific equipment. Here, we demonstrate that treatment of SARS-CoV-2 infected cells with formaldehyde at a concentration at or above 0.5% for one hour at room temperature effectively inactivates SARs-CoV-2. Given the wide ranges of uses for formaldehyde, we hope this data will assist in the safe processing of probable or confirmed SARS-CoV-2 containing samples.
Analyses of host responses to viral infection either through the use of Western blotting or the determination of virus specific antibodies in patient sera generally require heating of the sample prior to downstream analysis. To this end, we have determined that heating of SARS-CoV-2 containing samples at 100 °C for longer than 5 min is a safe and effective method for the downstream analysis of samples by Western blot. Likewise, our data demonstrate that heat treatment of laboratory or clinical samples at 56 °C for one hour will serve to effectively inactivate SARS-CoV-2. However, it has been recently reported that heat treatment of clinical samples such as serum can negatively impact sample quality for downstream processes, so researchers utilizing heat treatment as a method of inactivation should take this into account in their experimental design [
19].
Taken together, we hope the methods and data reported here will serve to expedite the much-needed research required to address this unprecedented pandemic.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}