Revisiting the Classification of Percid Perhabdoviruses Using New Full-Length Genomes


 and
and 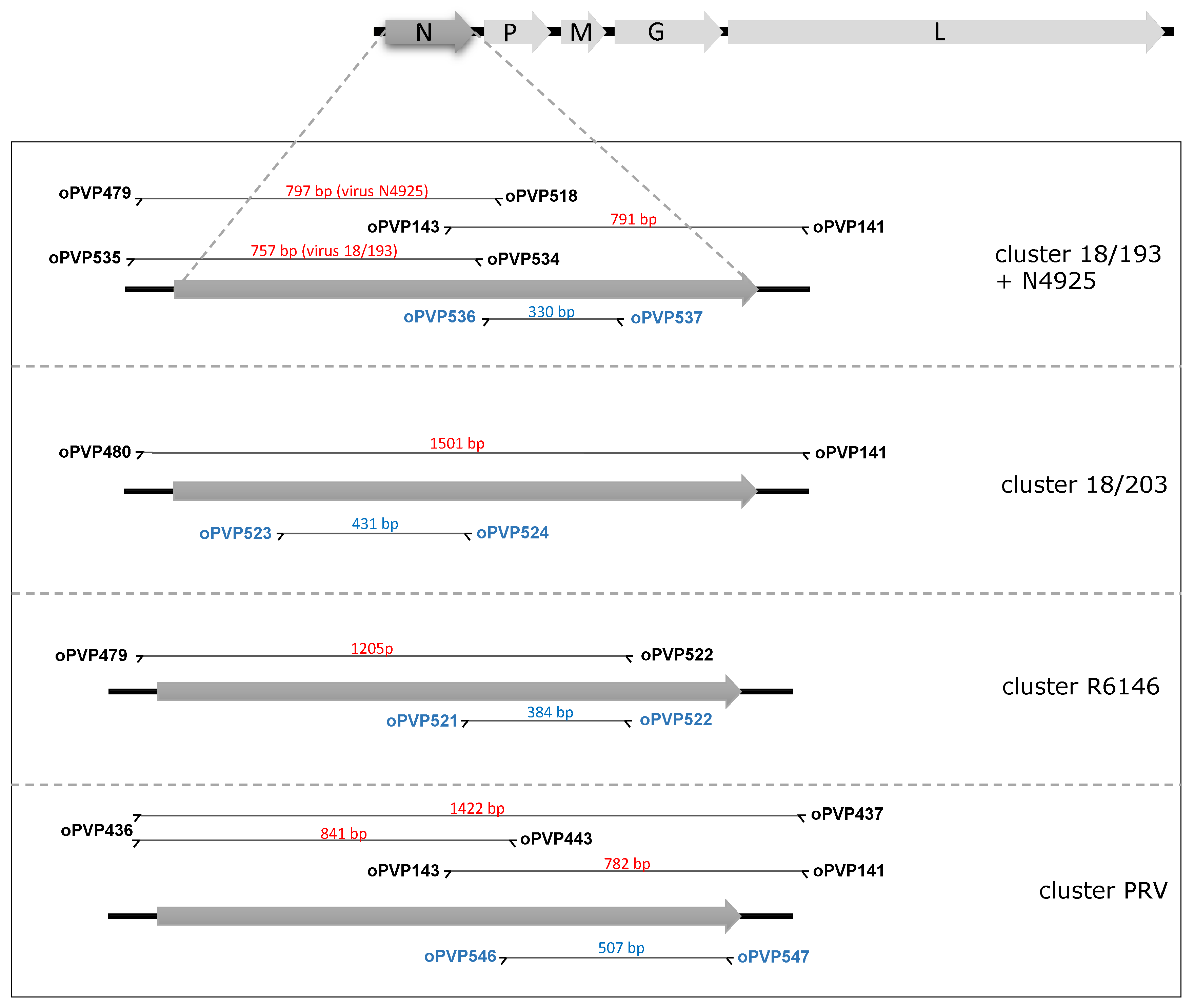{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Isolation
2.2. Nucleic Acid Extraction for RT-PCR Detection
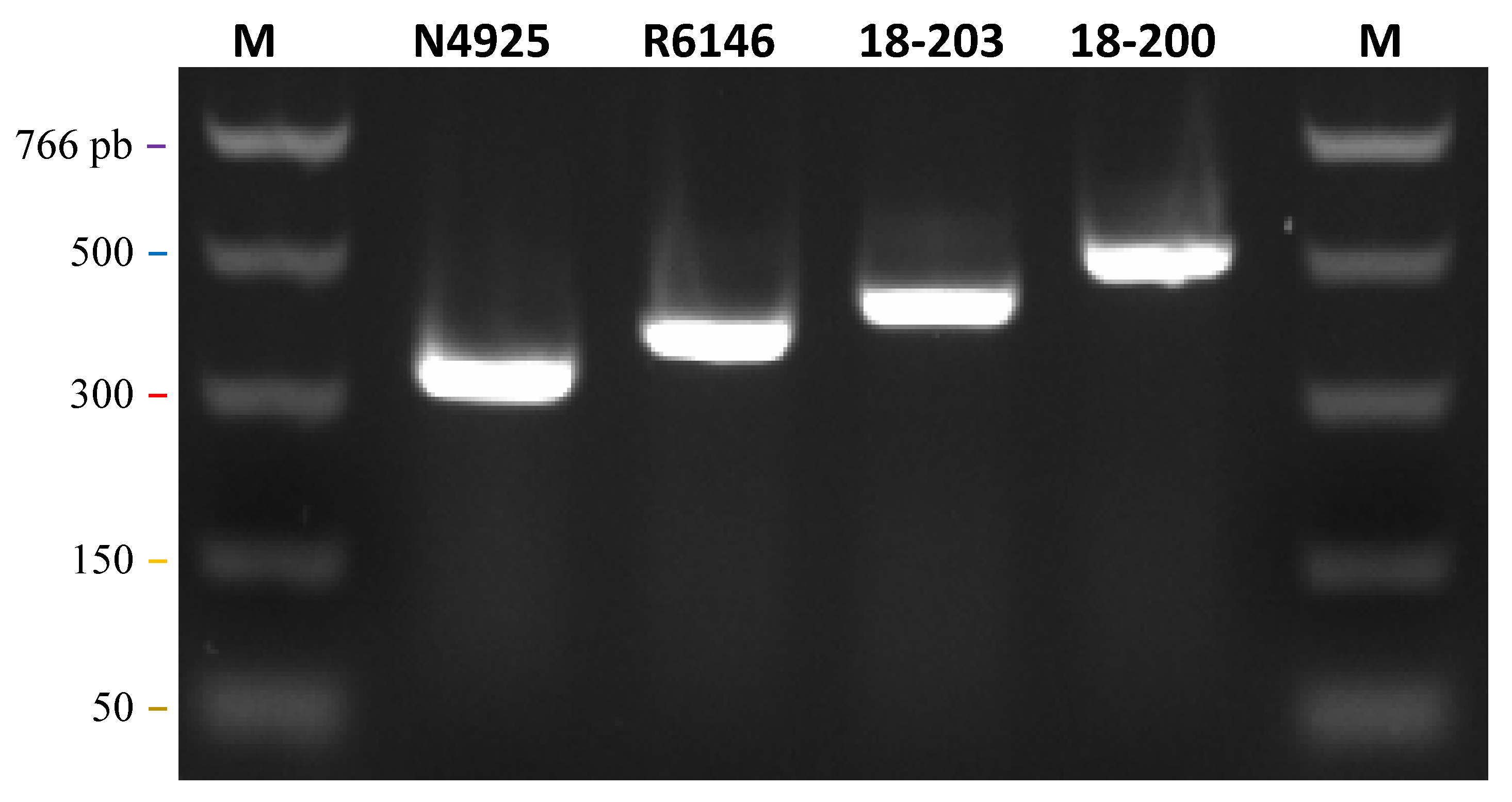
2.3. RT-PCR Amplification for Sequencing
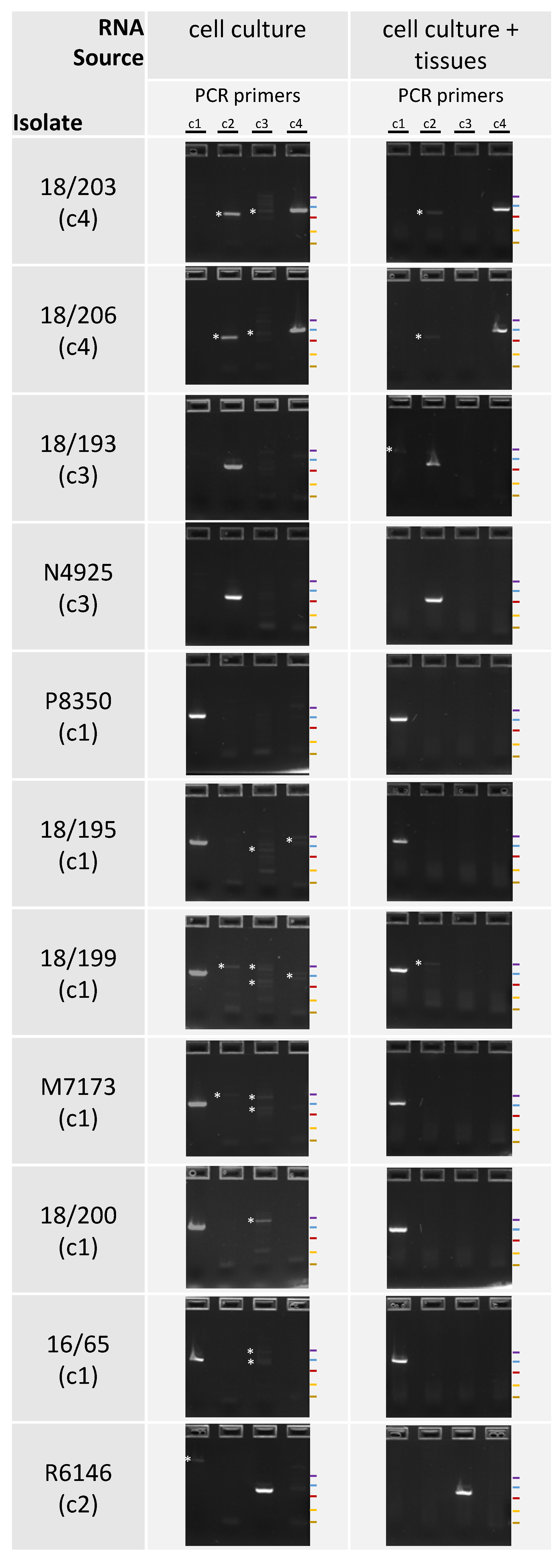
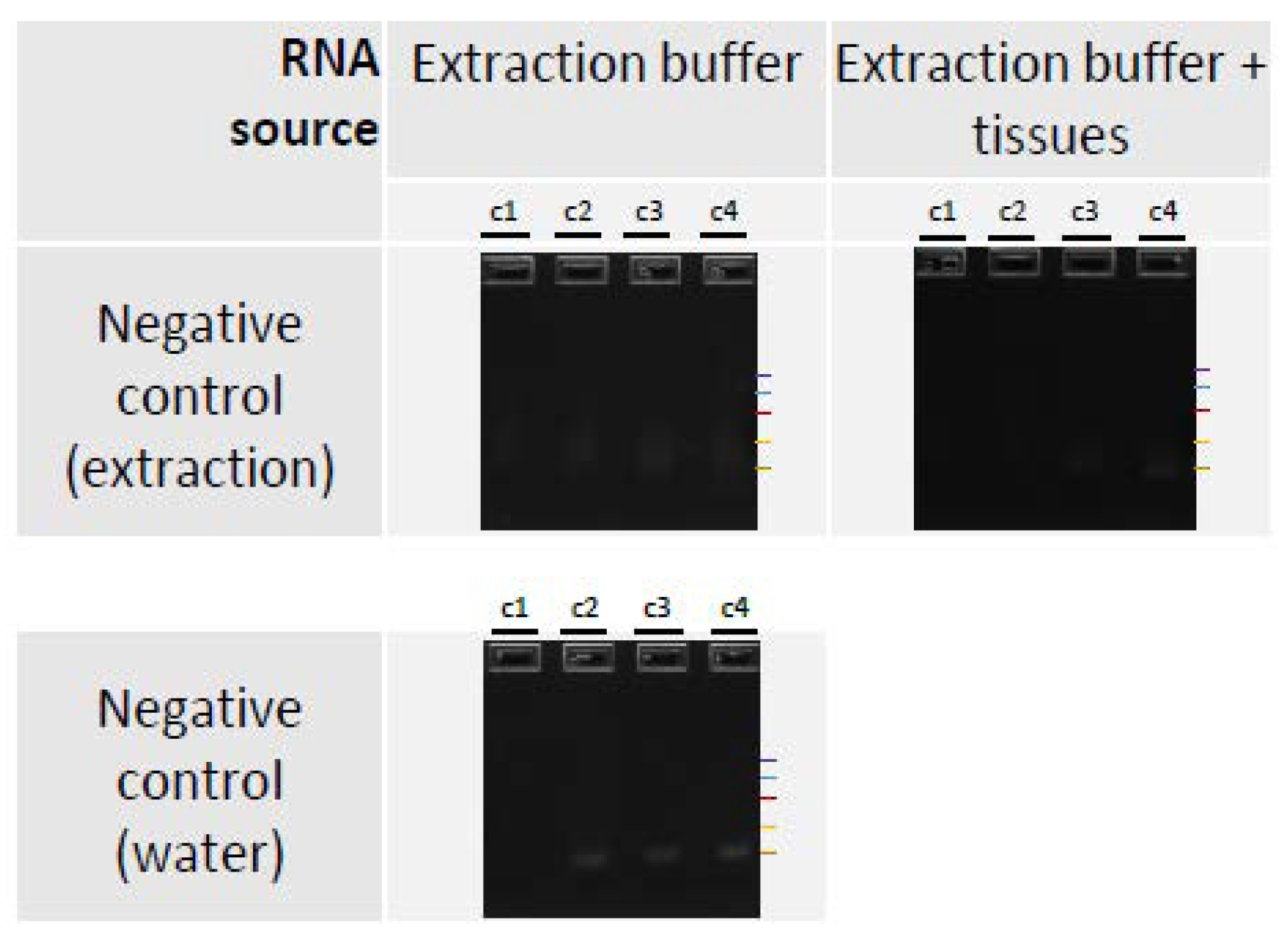
2.4. RT-PCR Detection
2.5. Next-Generation Sequencing
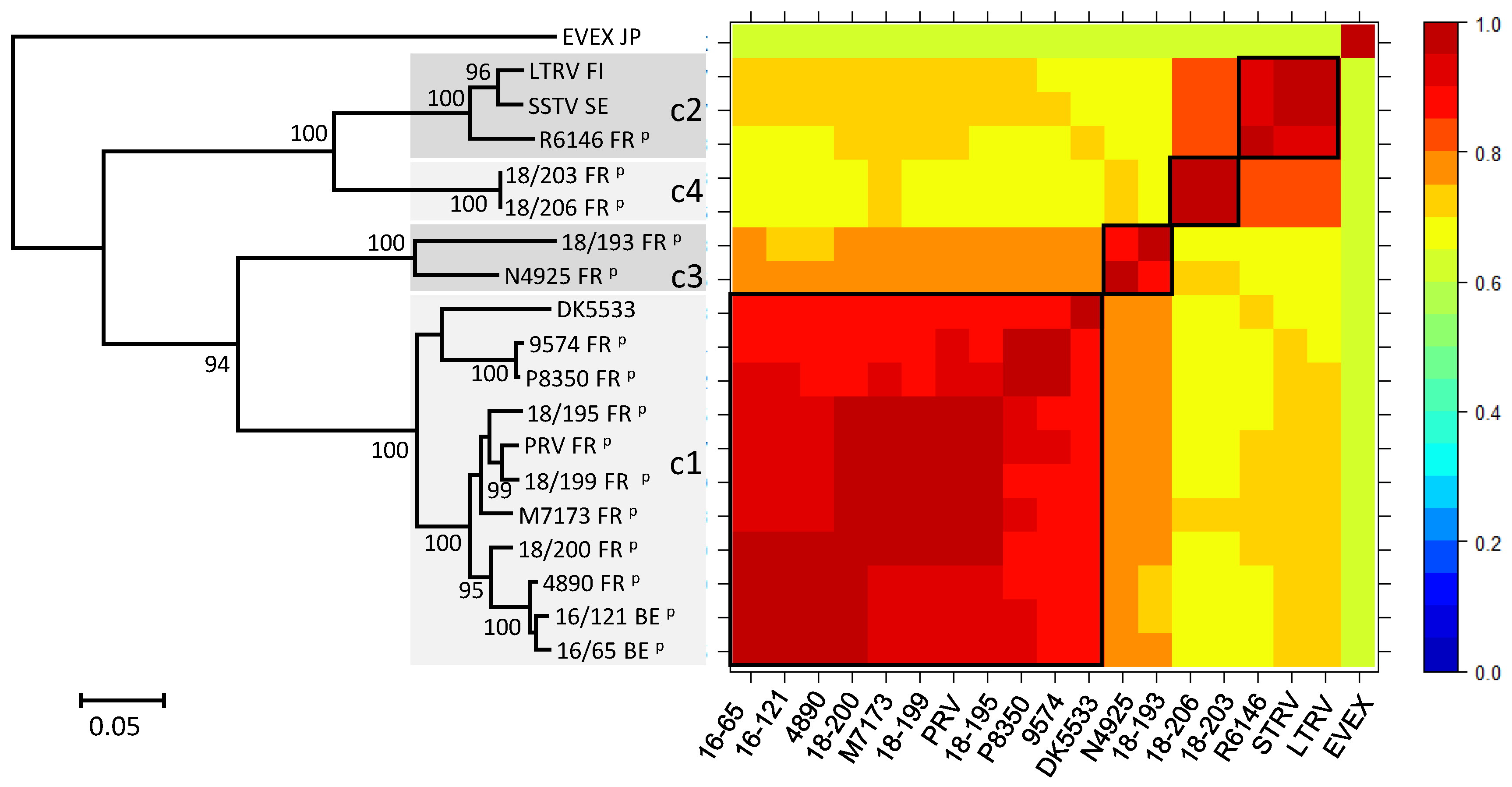
2.6. Phylogenetic Analysis
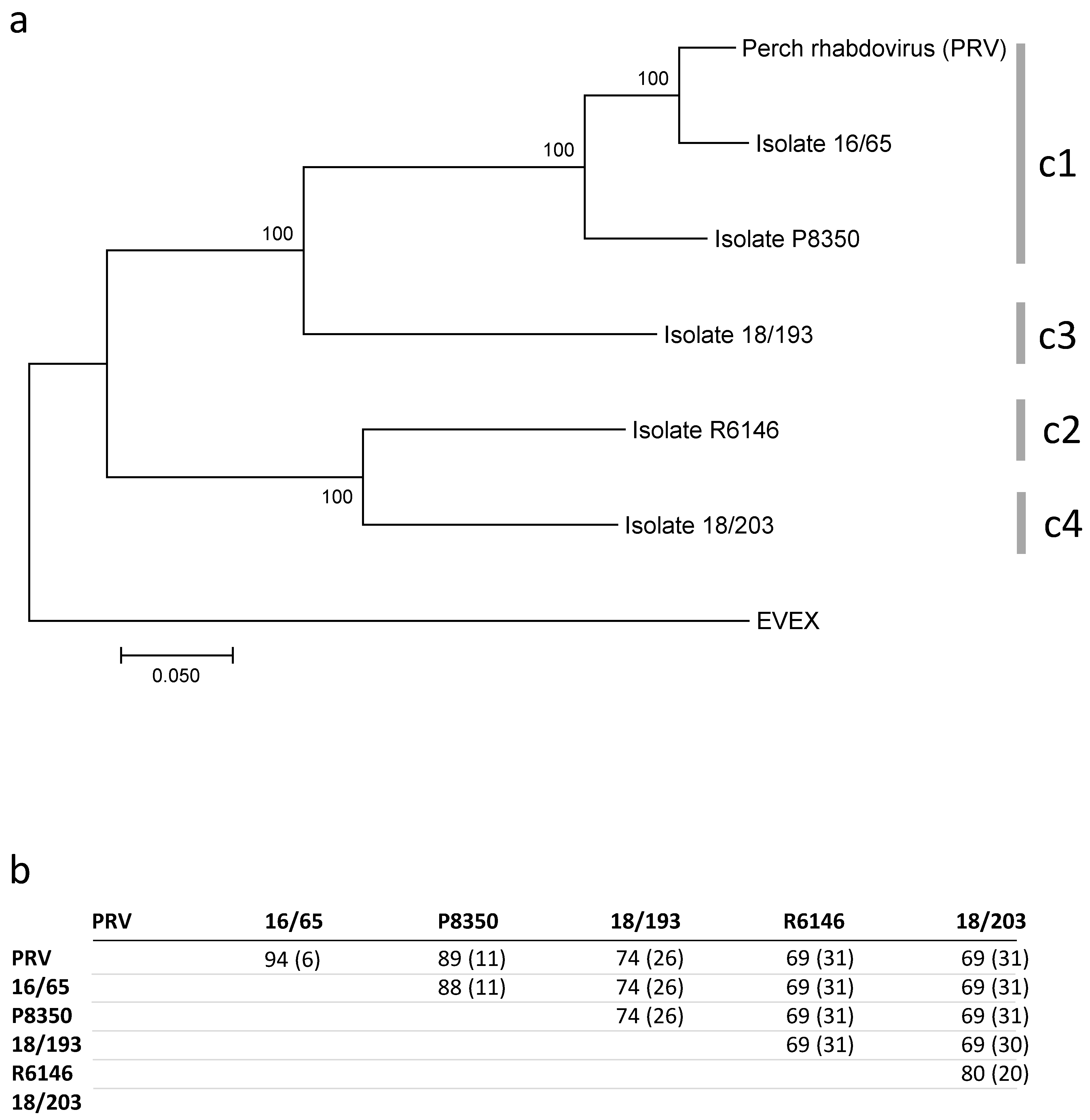
3. Results
3.1. Study of the N Gene
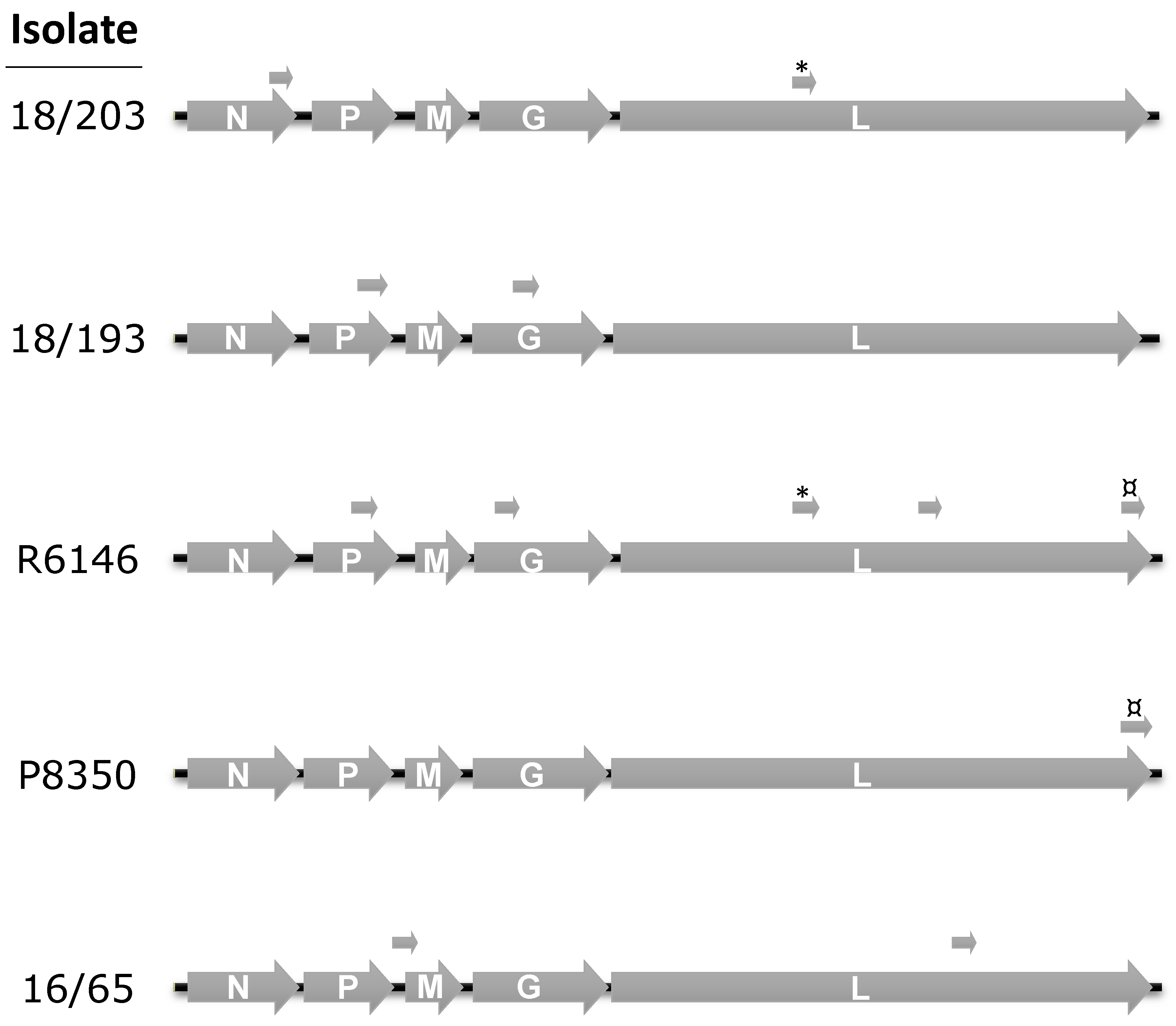
3.2. Complete Genome Sequences
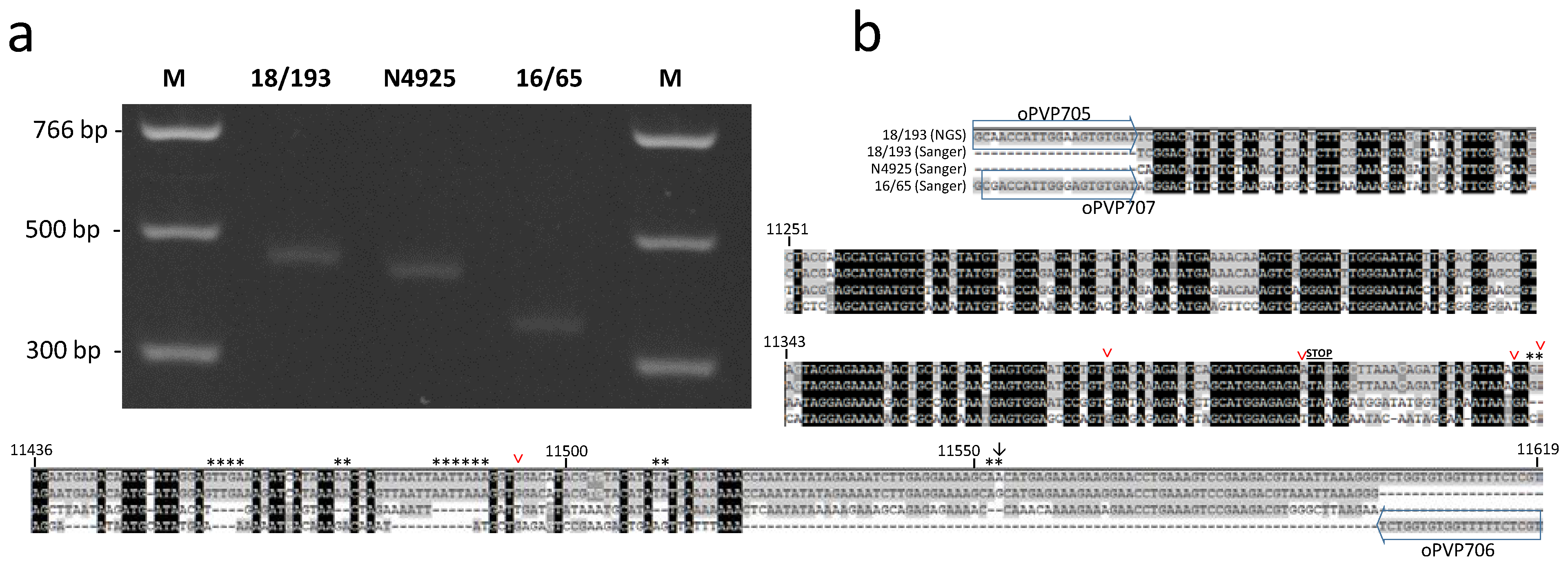
3.3. PCR Detection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Policar, T.; Schaefer, F.J.; Panana, E.; Meyer, S.; Teerlinck, S.; Toner, D.; Żarski, D. Recent progress in European percid fish culture production technology—Tackling bottlenecks. Aquac. Int. 2019, 75, 1151–1174. [Google Scholar] [CrossRef]
- Gadd, T.; Viljamaa-Dirks, S.; Holopainen, R.; Koski, P.; Jakava-Viljanen, M. Characterization of perch rhabdovirus (PRV) in farmed grayling Thymallus thymallus. Dis. Aquat. Organ. 2013, 106, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbi, C.; Cabon, J.; Baud, M.; Bourjaily, M.; de Boisseson, C.; Castric, J.; Bigarré, L. Genetic diversity of perch rhabdoviruses isolates based on the nucleoprotein and glycoprotein genes. Arch. Virol. 2011, 156, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Betts, A.M.; Stone, D.M.; Way, K.; Torhy, C.; Chilmonczyk, S.; Benmansour, A.; de Kinkelin, P. Emerging vesiculo-type virus infections of freshwater fishes in Europe. Dis. Aquat. Organ. 2003, 57, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahli, T.; Bellec, L.; von Siebenthal, B.; Cabon, J.; Schmidt-Posthaus, H.; Morin, T. First isolation of a rhabdovirus from perch Perca fluviatilis in Switzerland. Dis. Aquat. Organ. 2015, 116, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruane, N.M.; Rodger, H.D.; McCarthy, L.J.; Swords, D.; Dodge, M.; Kerr, R.C.; Henshilwood, K.; Stone, D.M. Genetic diversity and associated pathology of rhabdovirus infections in farmed and wild perch Perca fluviatilis in Ireland. Dis. Aquat. Organ. 2014, 112, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannevig, H.; Olesen, N.J.; Jentoft, S.; Kvellestad, A.; Taksdal, T.; Hastein, T. The first isolation of a rhabdovirus from perch (Perca fluviatilis) in Norway. Bull. Eur. Assoc. Fish Pathol. 2001, 21, 186–194. [Google Scholar]
- Bigarré, L.; Plassiart, G.; de Boisseson, C.; Pallandre, L.; Pozet, F.; Ledore, Y.; Fontaine, P.; Lieffrig, F. Molecular investigations of outbreaks of Perch perhabdovirus infections in pike-perch. Dis. Aquat. Organ. 2017, 127, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Nougayrède, P.; de Kinkelin, P.; Chilmonczyk, S.; Vuillaume, A. Isolation of a rhabdovirus from the pike-perch Stizostedion lucioperca (L.1758). Bull. Eur. Assoc. Fish Pathol. 1992, 12, 5–7. [Google Scholar]
- Dorson, M.; Torchy, C.; Chilmonczyk, S.; de Kinkelin, P. A rhabdovirus pathogenic for perch, Perca fluviatilis L.: Isolation and preliminary study. J. Fish Dis. 1984, 7, 241–245. [Google Scholar] [CrossRef]
- Jorgensen, P.E.V.; Olesen, N.J.; Ahne, W.; Wahli, T.; Meier, W. Isolation of a previously undescribed rhabdovirus from pike Esox lucius. Dis. Aquat. Organ. 1993, 16, 171–179. [Google Scholar] [CrossRef]
- Caruso, C.; Gustinelli, A.; Pastorino, P.; Acutis, P.L.; Prato, R.; Masoero, L.; Peletto, S.; Fioravanti, M.L.; Prearo, M. Mortality outbreak by perch rhabdovirus in European perch (Perca fluviatilis) farmed in Italy: Clinical presentation and phylogenetic analysis. J. Fish Dis. 2019, 42, 73–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, D.M.; Kerr, R.C.; Hughes, M.; Radford, A.D.; Darby, A.C. Characterisation of the genomes of four putative vesiculoviruses: Tench rhabdovirus, grass carp rhabdovirus, perch rhabdovirus and eel rhabdovirus European X. Arch. Virol. 2013, 158, 2371–2377. [Google Scholar] [CrossRef] [PubMed]
- Galinier, R.; van Beurden, S.; Amilhat, E.; Castric, J.; Schoehn, G.; Verneau, O.; Fazio, G.; Allienne, J.F.; Engelsma, M.; Sasal, P.; et al. Complete genomic sequence and taxonomic position of eel virus European X (EVEX), a rhabdovirus of European eel. Virus Res. 2012, 166, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Johansson, T.; Nylund, S.; Olesen, N.J.; Bjorklund, H. Molecular characterisation of the nucleocapsid protein gene, glycoprotein gene and gene junctions of rhabdovirus 903/87, a novel fish pathogenic rhabdovirus. Virus Res. 2001, 80, 11–22. [Google Scholar] [CrossRef]
- Johansson, T.; Ostman-Myllyoja, L.; Hellstrom, A.; Martelius, S.; Olesen, N.J.; Bjorklund, H. A novel fish rhabdovirus from sweden is closely related to the Finnish rhabdovirus 903/87. Virus Genes 2002, 25, 127–138. [Google Scholar] [CrossRef]
- Pallandre, L.; Lesne, M.; de Boisseson, C.; Charrier, A.; Daniel, P.; Tragnan, A.; Debeuf, B.; Chesneau, V.; Bigarre, L. Genetic identification of two Acipenser iridovirus-European variants using high-resolution melting analysis. J. Virol. Methods 2019, 265, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Dacheux, L.; Dommergues, L.; Chouanibou, Y.; Domeon, L.; Schuler, C.; Bonas, S.; Luo, D.; Maufrais, C.; Cetre-Sossah, C.; Cardinale, E.; et al. Co-circulation and characterization of novel African arboviruses (genus Ephemerovirus) in cattle, Mayotte island, Indian Ocean, 2017. Transbound. Emerg. Dis. 2019, 66, 2601–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troupin, C.; Dacheux, L.; Tanguy, M.; Sabeta, C.; Blanc, H.; Bouchier, C.; Vignuzzi, M.; Duchene, S.; Holmes, E.C.; Bourhy, H. Large-scale phylogenomic analysis reveals the complex evolutionary history of rabies virus in multiple carnivore hosts. PLoS Pathog. 2016, 12, e1006041. [Google Scholar] [CrossRef] [PubMed]
- Mareuil, F.; Doppel-Azeroual, O.; Ménager, H. A public Galaxy platform at Pasteur used as an execution engine for web services (version 1; not peer reviewed). F1000Research 2017, 6, 1030. [Google Scholar]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Delmas, O.; Holmes, E.C.; Talbi, C.; Larrous, F.; Dacheux, L.; Bouchier, C.; Bourhy, H. Genomic diversity and evolution of the lyssaviruses. PLoS ONE 2008, 3, e2057. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Walker, P.J.; Firth, C.; Widen, S.G.; Blasdell, K.R.; Guzman, H.; Wood, T.G.; Paradkar, P.N.; Holmes, E.C.; Tesh, R.B.; Vasilakis, N. Evolution of genome size and complexity in the rhabdoviridae. PLoS Pathog. 2015, 11, e1004664. [Google Scholar] [CrossRef] [Green Version]
- Peluso, R.W.; Richardson, J.C.; Talon, J.; Lock, M. Identification of a set of proteins (C′ and C) encoded by the bicistronic P gene of the Indiana serotype of vesicular stomatitis virus and analysis of their effect on transcription by the viral RNA polymerase. Virology 1996, 218, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenfeldt, S.; Fontaine, P.; Overton, J.L.; Policar, T.; Toner, D.; Falahaktar, B.; Horvath, A.; Ben Khemis, I.; Hamza, N.; Mhetli, M. Current status of Eurasian percid fishes aquaculture. In Biology and Culture of Percid Fishes; Kestemont, P., Ed.; Springer Science & Business Media: Berlin, Germany, 2015; pp. 817–840. [Google Scholar]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pallandre, L.; Luo, D.; Feuvrier, C.; Lieffrig, F.; Pozet, F.; Dacheux, L.; Bigarré, L. Revisiting the Classification of Percid Perhabdoviruses Using New Full-Length Genomes. Viruses 2020, 12, 649. https://doi.org/10.3390/v12060649
Pallandre L, Luo D, Feuvrier C, Lieffrig F, Pozet F, Dacheux L, Bigarré L. Revisiting the Classification of Percid Perhabdoviruses Using New Full-Length Genomes. Viruses. 2020; 12(6):649. https://doi.org/10.3390/v12060649
Chicago/Turabian StylePallandre, Laurane, Dongsheng Luo, Claudette Feuvrier, François Lieffrig, Françoise Pozet, Laurent Dacheux, and Laurent Bigarré. 2020. "Revisiting the Classification of Percid Perhabdoviruses Using New Full-Length Genomes" Viruses 12, no. 6: 649. https://doi.org/10.3390/v12060649
APA StylePallandre, L., Luo, D., Feuvrier, C., Lieffrig, F., Pozet, F., Dacheux, L., & Bigarré, L. (2020). Revisiting the Classification of Percid Perhabdoviruses Using New Full-Length Genomes. Viruses, 12(6), 649. https://doi.org/10.3390/v12060649