The Contribution of Neutrophils to the Pathogenesis of RSV Bronchiolitis

Abstract
:1. Introduction
2. Neutrophils Deploy a Diverse Anti-Microbial Arsenal against Invading Pathogens
3. Neutrophils Influence Innate and Adaptive Immunity
4. The Pathophysiology of RSV Bronchiolitis
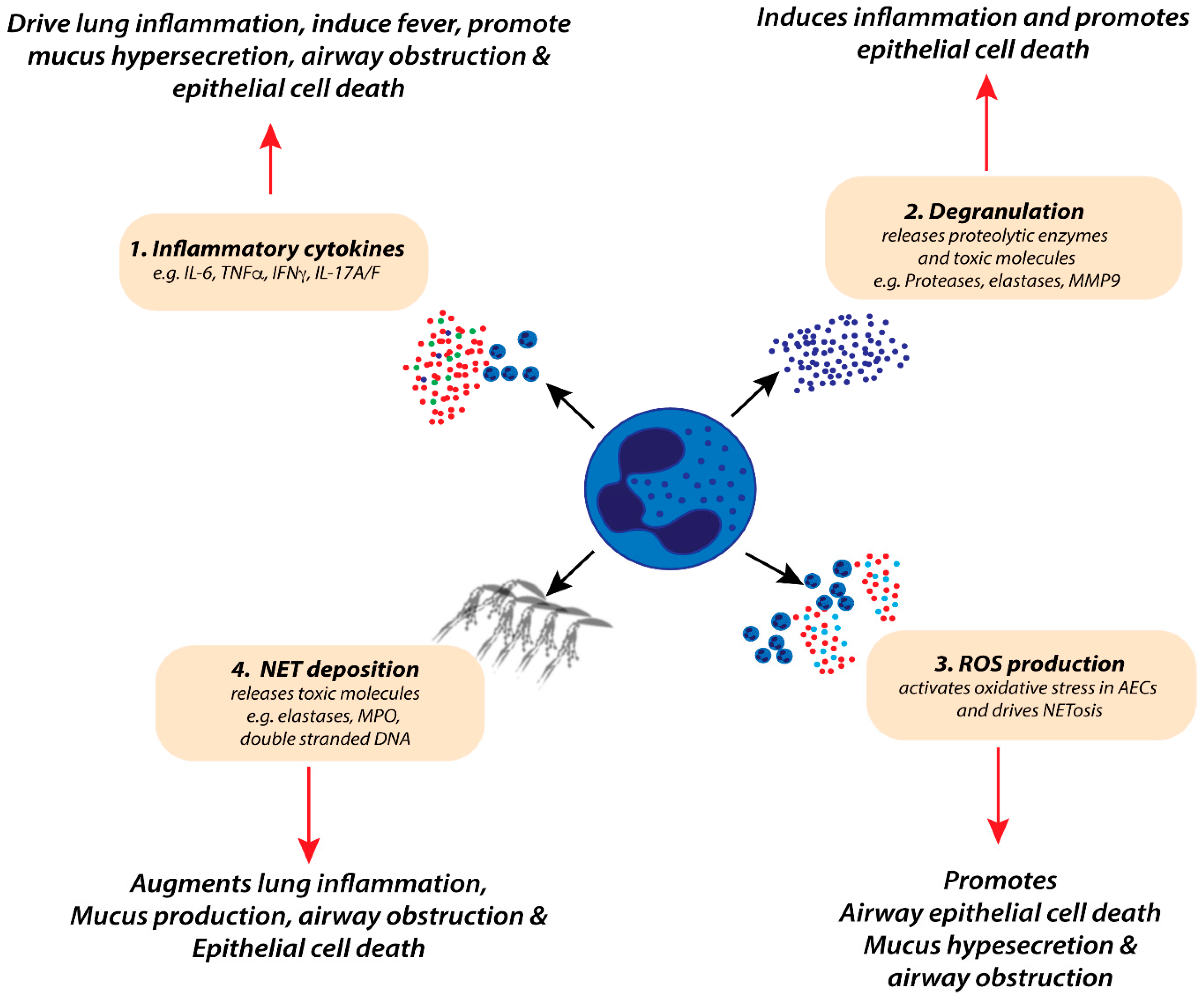
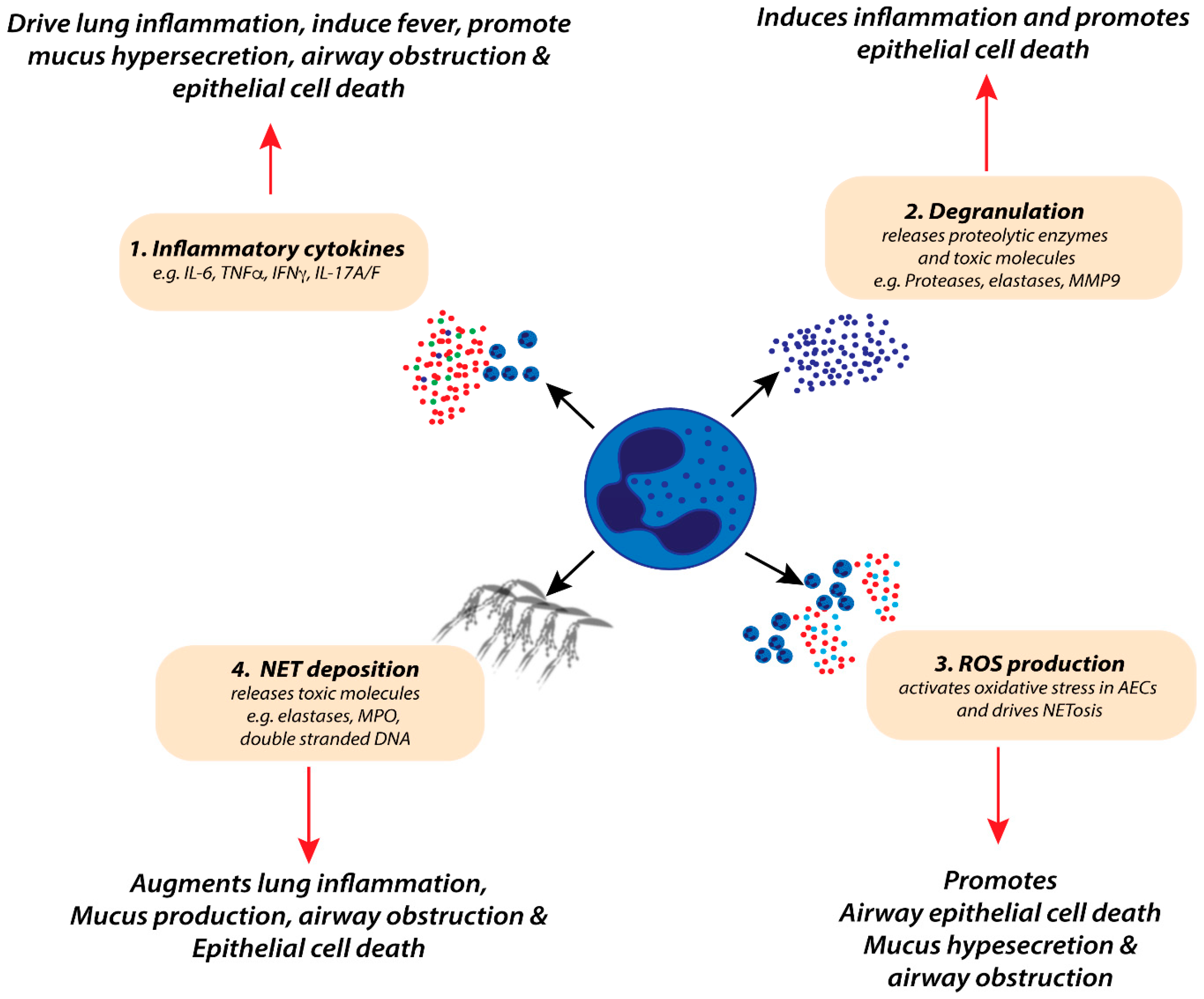
4.1. Neutrophil Inflammatory Mediators and Lung Pathology during RSV-Induced Bronchiolitis
4.2. Neutrophil Diversity in RSV-Induced Bronchiolitis
4.3. Are Neutrophils Beneficial in Host Immunity against RSV Infection?
4.4. Are Neutrophils Deleterious during Severe RSV Bronchiolitis?
4.5. Therapeutic Regulation of Neutrophil-Induced Pathology in RSV Bronchiolitis
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Florin, T.A.; Plint, A.C.; Zorc, J.J. Viral bronchiolitis. Lancet 2017, 389, 211–224. [Google Scholar] [CrossRef]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.B. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 2001, 344, 1917–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, J.L. Respiratory syncytial virus vaccine development. Expert Rev. Vaccines 2011, 10, 1415–1433. [Google Scholar] [CrossRef] [Green Version]
- Graham, B.S.; Modjarrad, K.; McLellan, J.S. Novel antigens for RSV vaccines. Curr. Opin. Immunol. 2015, 35, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jartti, T.; Gern, J.E. Role of viral infections in the development and exacerbation of asthma in children. J. Allergy Clin. Immunol. 2017, 140, 895–906. [Google Scholar] [CrossRef] [Green Version]
- Halfhide, C.P.; Flanagan, B.F.; Brearey, S.P.; Hunt, J.A.; Fonceca, A.M.; McNamara, P.S.; Howarth, D.; Edwards, S.; Smyth, R.L. Respiratory syncytial virus binds and undergoes transcription in neutrophils from the blood and airways of infants with severe bronchiolitis. J. Infect. Dis. 2011, 204, 451–458. [Google Scholar] [CrossRef]
- McNamara, P.S.; Ritson, P.; Selby, A.; Hart, C.A.; Smyth, R.L. Bronchoalveolar lavage cellularity in infants with severe respiratory syncytial virus bronchiolitis. Arch. Dis. Child 2003, 88, 922–926. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.E.; Gonzales, R.A.; Olson, S.J.; Wright, P.F.; Graham, B.S. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod. Pathol. 2007, 20, 108–119. [Google Scholar] [CrossRef]
- Dancey, J.T.; Deubelbeiss, K.A.; Harker, L.A.; Finch, C.A. Neutrophil kinetics in man. J. Clin. Investig. 1976, 58, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Ng, L.G.; Ostuni, R.; Hidalgo, A. Heterogeneity of neutrophils. Nat. Rev. Immunol. 2019, 19, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Kruger, P.; Saffarzadeh, M.; Weber, A.N.; Rieber, N.; Radsak, M.; von Bernuth, H.; Benarafa, C.; Roos, D.; Skokowa, J.; Hartl, D. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015, 11, e1004651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil diversity in health and disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Harrison, R.E.; Grinstein, S. Phagocytosis by neutrophils. Microbes Infect 2003, 5, 1299–1306. [Google Scholar] [CrossRef]
- Allen, L.H.; Criss, A.K. Cell intrinsic functions of neutrophils and their manipulation by pathogens. Curr. Opin. Immunol. 2019, 60, 124–129. [Google Scholar] [CrossRef]
- Cassatella, M.A.; Ostberg, N.K.; Tamassia, N.; Soehnlein, O. Biological roles of neutrophil-derived granule proteins and cytokines. Trends Immunol. 2019, 40, 648–664. [Google Scholar] [CrossRef]
- Mollinedo, F. Neutrophil degranulation, plasticity, and cancer metastasis. Trends Immunol. 2019, 40, 228–242. [Google Scholar] [CrossRef]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer. 2016, 16, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Soehnlein, O.; Wantha, S.; Simsekyilmaz, S.; Doring, Y.; Megens, R.T.; Mause, S.F.; Drechsler, M.; Smeets, R.; Weinandy, S.; Schreiber, F.; et al. Neutrophil-derived cathelicidin protects from neointimal hyperplasia. Sci. Transl. Med. 2011, 3, 103ra98. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.R.; Robb, C.T.; Perretti, M.; Rossi, A.G. The role of neutrophils in inflammation resolution. Semin. Immunol. 2016, 28, 137–145. [Google Scholar] [CrossRef]
- Belaaouaj, A.; McCarthy, R.; Baumann, M.; Gao, Z.; Ley, T.J.; Abraham, S.N.; Shapiro, S.D. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat. Med. 1998, 4, 615–618. [Google Scholar] [CrossRef]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Butin-Israeli, V.; Bui, T.M.; Wiesolek, H.L.; Mascarenhas, L.; Lee, J.J.; Mehl, L.C.; Knutson, K.R.; Adam, S.A.; Goldman, R.D.; Beyder, A.; et al. Neutrophil-induced genomic instability impedes resolution of inflammation and wound healing. J. Clin. Investig. 2019, 129, 712–726. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Dejonckheere, E.; Libert, C. A therapeutic role for matrix metalloproteinase inhibitors in lung diseases? Eur. Respir. J. 2011, 38, 1200–1214. [Google Scholar] [CrossRef]
- Durr, U.H.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1408–1425. [Google Scholar] [CrossRef] [Green Version]
- Hasty, K.A.; Pourmotabbed, T.F.; Goldberg, G.I.; Thompson, J.P.; Spinella, D.G.; Stevens, R.M.; Mainardi, C.L. Human neutrophil collagenase. A distinct gene product with homology to other matrix metalloproteinases. J. Biol. Chem. 1990, 265, 11421–11424. [Google Scholar]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Ley, K.; Hoffman, H.M.; Kubes, P.; Cassatella, M.A.; Zychlinsky, A.; Hedrick, C.C.; Catz, S.D. Neutrophils: New insights and open questions. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Sollberger, G.; Tilley, D.O.; Zychlinsky, A. Neutrophil extracellular traps: The biology of chromatin externalization. Dev. Cell. 2018, 44, 542–553. [Google Scholar] [CrossRef] [Green Version]
- D’Cruz, A.A.; Speir, M.; Bliss-Moreau, M.; Dietrich, S.; Wang, S.; Chen, A.A.; Gavillet, M.; Al-Obeidi, A.; Lawlor, K.E.; Vince, J.E.; et al. The pseudokinase MLKL activates PAD4-dependent NET formation in necroptotic neutrophils. Sci. Signal 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.M.; Carbo, C.; Curtis, B.R.; Martinod, K.; Mazo, I.B.; Schatzberg, D.; Cifuni, S.M.; Fuchs, T.A.; von Andrian, U.H.; Hartwig, J.H.; et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood 2012, 119, 6335–6343. [Google Scholar] [CrossRef] [Green Version]
- Toussaint, M.; Jackson, D.J.; Swieboda, D.; Guedan, A.; Tsourouktsoglou, T.D.; Ching, Y.M.; Radermecker, C.; Makrinioti, H.; Aniscenko, J.; Bartlett, N.W.; et al. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat. Med. 2017, 23, 681–691. [Google Scholar] [CrossRef] [Green Version]
- Knight, J.S.; Zhao, W.; Luo, W.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Hodgin, J.B.; Eitzman, D.T.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J. Clin. Investig. 2013, 123, 2981–2993. [Google Scholar] [CrossRef]
- Smolen, J.S.; Beaulieu, A.; Rubbert-Roth, A.; Ramos-Remus, C.; Rovensky, J.; Alecock, E.; Woodworth, T.; Alten, R.; Investigators, O. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): A double-blind, placebo-controlled, randomised trial. Lancet 2008, 371, 987–997. [Google Scholar] [CrossRef]
- Marcos, V.; Zhou, Z.; Yildirim, A.O.; Bohla, A.; Hector, A.; Vitkov, L.; Wiedenbauer, E.M.; Krautgartner, W.D.; Stoiber, W.; Belohradsky, B.H.; et al. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat. Med. 2010, 16, 1018–1023. [Google Scholar] [CrossRef]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, F.; Means, T.K.; Luster, A.D. Toll-like receptors stimulate human neutrophil function. Blood 2003, 102, 2660–2669. [Google Scholar] [CrossRef] [PubMed]
- Tamassia, N.; Le Moigne, V.; Rossato, M.; Donini, M.; McCartney, S.; Calzetti, F.; Colonna, M.; Bazzoni, F.; Cassatella, M.A. Activation of an immunoregulatory and antiviral gene expression program in poly(I:C)-transfected human neutrophils. J. Immunol. 2008, 181, 6563–6573. [Google Scholar] [CrossRef] [Green Version]
- Clarke, T.B.; Davis, K.M.; Lysenko, E.S.; Zhou, A.Y.; Yu, Y.; Weiser, J.N. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat. Med. 2010, 16, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.H.; Hwang, S.M.; Chung, I.Y. S100A8, S100A9 and S100A12 activate airway epithelial cells to produce MUC5AC via extracellular signal-regulated kinase and nuclear factor-kappaB pathways. Immunology 2015, 144, 79–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardif, M.R.; Chapeton-Montes, J.A.; Posvandzic, A.; Page, N.; Gilbert, C.; Tessier, P.A. Secretion of S100A8, S100A9, and S100A12 by Neutrophils Involves Reactive Oxygen Species and Potassium Efflux. J. Immunol. Res. 2015, 2015, 296149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef]
- van Gisbergen, K.P.; Sanchez-Hernandez, M.; Geijtenbeek, T.B.; van Kooyk, Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J. Exp. Med. 2005, 201, 1281–1292. [Google Scholar] [CrossRef]
- Krishnamoorthy, N.; Douda, D.N.; Bruggemann, T.R.; Ricklefs, I.; Duvall, M.G.; Abdulnour, R.E.; Martinod, K.; Tavares, L.; Wang, X.; Cernadas, M.; et al. Neutrophil cytoplasts induce TH17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Vono, M.; Lin, A.; Norrby-Teglund, A.; Koup, R.A.; Liang, F.; Lore, K. Neutrophils acquire the capacity for antigen presentation to memory CD4(+) T cells in vitro and ex vivo. Blood 2017, 129, 1991–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.W.; Strong, B.S.; Miller, M.J.; Unanue, E.R. Neutrophils influence the level of antigen presentation during the immune response to protein antigens in adjuvants. J. Immunol. 2010, 185, 2927–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charmoy, M.; Brunner-Agten, S.; Aebischer, D.; Auderset, F.; Launois, P.; Milon, G.; Proudfoot, A.E.; Tacchini-Cottier, F. Neutrophil-derived CCL3 is essential for the rapid recruitment of dendritic cells to the site of Leishmania major inoculation in resistant mice. PLoS Pathog. 2010, 6, e1000755. [Google Scholar] [CrossRef]
- Bennouna, S.; Bliss, S.K.; Curiel, T.J.; Denkers, E.Y. Cross-talk in the innate immune system: Neutrophils instruct recruitment and activation of dendritic cells during microbial infection. J. Immunol. 2003, 171, 6052–6058. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; de la Rosa, G.; Tewary, P.; Oppenheim, J.J. Alarmins link neutrophils and dendritic cells. Trends Immunol. 2009, 30, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Oppenheim, J.J. Alarmins and antimicrobial immunity. Med. Mycol. 2009, 47 (Suppl. S1), S146–S153. [Google Scholar] [CrossRef] [Green Version]
- Meinderts, S.M.; Baker, G.; van Wijk, S.; Beuger, B.M.; Geissler, J.; Jansen, M.H.; Saris, A.; Ten Brinke, A.; Kuijpers, T.W.; van den Berg, T.K.; et al. Neutrophils acquire antigen-presenting cell features after phagocytosis of IgG-opsonized erythrocytes. Blood Adv. 2019, 3, 1761–1773. [Google Scholar] [CrossRef]
- Lim, K.; Hyun, Y.M.; Lambert-Emo, K.; Capece, T.; Bae, S.; Miller, R.; Topham, D.J.; Kim, M. Neutrophil trails guide influenza-specific CD8(+) T cells in the airways. Science 2015, 349, aaa4352. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, M.; Maggi, L.; Micheletti, A.; Lazzeri, E.; Tamassia, N.; Costantini, C.; Cosmi, L.; Lunardi, C.; Annunziato, F.; Romagnani, S.; et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 2010, 115, 335–343. [Google Scholar] [CrossRef]
- Miller, E.K.; Gebretsadik, T.; Carroll, K.N.; Dupont, W.D.; Mohamed, Y.A.; Morin, L.L.; Heil, L.; Minton, P.A.; Woodward, K.; Liu, Z.; et al. Viral etiologies of infant bronchiolitis, croup and upper respiratory illness during 4 consecutive years. Pediatr. Infect. Dis. J. 2013, 32, 950–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef] [Green Version]
- Piedimonte, G.; Perez, M.K. Respiratory syncytial virus infection and bronchiolitis. Pediatr. Rev. 2014, 35, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.B.; Douglas, R.G., Jr.; Geiman, J.M. Respiratory syncytial virus infections in infants: Quantitation and duration of shedding. J. Pediatrics 1976, 89, 11–15. [Google Scholar] [CrossRef]
- Jha, A.; Jarvis, H.; Fraser, C.; Openshaw, P.J.M. Respiratory syncytial virus. In SARS, MERS and Other Viral Lung Infections; Hui, D.S., Rossi, G.A., Johnston, S.L., Eds.; European Respiratory Society: Lausanne, Switzerland, 2016. [Google Scholar]
- Wu, W.; Munday, D.C.; Howell, G.; Platt, G.; Barr, J.N.; Hiscox, J.A. Characterization of the interaction between human respiratory syncytial virus and the cell cycle in continuous cell culture and primary human airway epithelial cells. J. Virol. 2011, 85, 10300–10309. [Google Scholar] [CrossRef] [Green Version]
- Carroll, K.N.; Wu, P.; Gebretsadik, T.; Griffin, M.R.; Dupont, W.D.; Mitchel, E.F.; Hartert, T.V. The severity-dependent relationship of infant bronchiolitis on the risk and morbidity of early childhood asthma. J. Allergy Clin. Immunol. 2009, 123, 1055–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Harb, M.; Bell, F.; Finn, A.; Rao, W.H.; Nixon, L.; Shale, D.; Everard, M.L. IL-8 and neutrophil elastase levels in the respiratory tract of infants with RSV bronchiolitis. Eur. Respir. J. 1999, 14, 139–143. [Google Scholar] [CrossRef]
- Kirsebom, F.C.M.; Kausar, F.; Nuriev, R.; Makris, S.; Johansson, C. Neutrophil recruitment and activation are differentially dependent on MyD88/TRIF and MAVS signaling during RSV infection. Mucosal Immunol. 2019, 12, 1244–1255. [Google Scholar] [CrossRef] [Green Version]
- Stokes, K.L.; Currier, M.G.; Sakamoto, K.; Lee, S.; Collins, P.L.; Plemper, R.K.; Moore, M.L. The respiratory syncytial virus fusion protein and neutrophils mediate the airway mucin response to pathogenic respiratory syncytial virus infection. J. Virol. 2013, 87, 10070–10082. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.L.; Strieter, R.M.; Gruber, A.D.; Ho, S.B.; Lukacs, N.W. CXCR2 regulates respiratory syncytial virus-induced airway hyperreactivity and mucus overproduction. J. Immunol. 2003, 170, 3348–3356. [Google Scholar] [CrossRef]
- Deng, Y.; Herbert, J.A.; Smith, C.M.; Smyth, R.L. An in vitro transepithelial migration assay to evaluate the role of neutrophils in Respiratory Syncytial Virus (RSV) induced epithelial damage. Sci. Rep. 2018, 8, 6777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigurs, N.; Bjarnason, R.; Sigurbergsson, F.; Kjellman, B. Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am. J. Respir. Crit. Care Med. 2000, 161, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.T.; Sherrill, D.; Morgan, W.J.; Holberg, C.J.; Halonen, M.; Taussig, L.M.; Wright, A.L.; Martinez, F.D. Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet 1999, 354, 541–545. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E. HMGB1, IL-1alpha, IL-33 and S100 proteins: Dual-function alarmins. Cell Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef] [Green Version]
- Simpson, J.; Loh, Z.; Ullah, M.A.; Lynch, J.P.; Werder, R.B.; Collinson, N.; Zhang, V.; Dondelinger, Y.; Bertrand, M.J.M.; Everard, M.L.; et al. Respiratory syncytial virus infection promotes necroptosis and hmgb1 release by airway epithelial cells. Am. J. Respir. Crit. Care Med. 2020, 201, 1358–1371. [Google Scholar] [CrossRef]
- Bem, R.A.; Domachowske, J.B.; Rosenberg, H.F. Animal models of human respiratory syncytial virus disease. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2011, 301, L148–L156. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G. Animal models of respiratory syncytial virus infection. Vaccine 2017, 35, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Sacco, R.E.; Durbin, R.K.; Durbin, J.E. Animal models of respiratory syncytial virus infection and disease. Curr. Opin. Virol. 2015, 13, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Lynch, J.P.; Werder, R.B.; Loh, Z.; Sikder, M.A.A.; Curren, B.; Zhang, V.; Rogers, M.J.; Lane, K.; Simpson, J.; Mazzone, S.B.; et al. Plasmacytoid dendritic cells protect from viral bronchiolitis and asthma through semaphorin 4a-mediated T reg expansion. J. Exp. Med. 2018, 215, 537–557. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Loh, Z.; Spann, K.; Lynch, J.P.; Lalwani, A.; Zheng, Z.; Davidson, S.; Uematsu, S.; Akira, S.; Hayball, J.; et al. Toll-like receptor 7 gene deficiency and early-life Pneumovirus infection interact to predispose toward the development of asthma-like pathology in mice. J. Allergy Clin. Immunol. 2013, 131, 1331–1339.e10. [Google Scholar] [CrossRef]
- Davidson, S.; Kaiko, G.; Loh, Z.; Lalwani, A.; Zhang, V.; Spann, K.; Foo, S.Y.; Hansbro, N.; Uematsu, S.; Akira, S.; et al. Plasmacytoid dendritic cells promote host defense against acute pneumovirus infection via the TLR7-MyD88-dependent signaling pathway. J. Immunol. 2011, 186, 5938–5948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, Z.; Simpson, J.; Ullah, A.; Zhang, V.; Gan, W.J.; Lynch, J.P.; Werder, R.B.; Sikder, A.A.; Lane, K.; Sim, C.B.; et al. HMGB1 amplifies ILC2-induced type-2 inflammation and airway smooth muscle remodelling. PLoS Pathog. 2020, 16, e1008651. [Google Scholar] [CrossRef] [PubMed]
- Upham, J.W.; Zhang, G.; Rate, A.; Yerkovich, S.T.; Kusel, M.; Sly, P.D.; Holt, P.G. Plasmacytoid dendritic cells during infancy are inversely associated with childhood respiratory tract infections and wheezing. J. Allergy Clin. Immunol. 2009, 124, 707–713.e2. [Google Scholar] [CrossRef]
- de Oliveira, S.; Reyes-Aldasoro, C.C.; Candel, S.; Renshaw, S.A.; Mulero, V.; Calado, A. Cxcl8 (IL-8) mediates neutrophil recruitment and behavior in the zebrafish inflammatory response. J. Immunol. 2013, 190, 4349–4359. [Google Scholar] [CrossRef]
- Lee, E.K.S.; Gillrie, M.R.; Li, L.; Arnason, J.W.; Kim, J.H.; Babes, L.; Lou, Y.; Sanati-Nezhad, A.; Kyei, S.K.; Kelly, M.M.; et al. Leukotriene B4-mediated neutrophil recruitment causes pulmonary capillaritis during lethal fungal sepsis. Cell Host Microbe 2018, 23, 121–133.e4. [Google Scholar] [CrossRef]
- Pinto, L.A.; LA, D.E.A.L.; Mocellin, M.; Acosta, P.; Caballero, M.T.; Libster, R.; Vargas, J.E.; Polack, F.; Comaru, T.; Stein, R.T.; et al. IL-8/IL-17 gene variations and the susceptibility to severe viral bronchiolitis. Epidemiol Infect. 2017, 145, 642–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beigelman, A.; Isaacson-Schmid, M.; Sajol, G.; Baty, J.; Rodriguez, O.M.; Leege, E.; Lyons, K.; Schweiger, T.L.; Zheng, J.; Schechtman, K.B.; et al. Randomized trial to evaluate azithromycin’s effects on serum and upper airway IL-8 levels and recurrent wheezing in infants with respiratory syncytial virus bronchiolitis. J. Allergy Clin. Immunol. 2015, 135, 1171–1178.e1. [Google Scholar] [CrossRef] [Green Version]
- Sznajer, Y.; Westcott, J.Y.; Wenzel, S.E.; Mazer, B.; Tucci, M.; Toledano, B.J. Airway eicosanoids in acute severe respiratory syncytial virus bronchiolitis. J. Pediatr. 2004, 145, 115–118. [Google Scholar] [CrossRef]
- Lammermann, T.; Afonso, P.V.; Angermann, B.R.; Wang, J.M.; Kastenmuller, W.; Parent, C.A.; Germain, R.N. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 2013, 498, 371–375. [Google Scholar] [CrossRef]
- Welliver, R.C., 2nd; Hintz, K.H.; Glori, M.; Welliver, R.C., Sr. Zileuton reduces respiratory illness and lung inflammation, during respiratory syncytial virus infection, in mice. J. Infect. Dis. 2003, 187, 1773–1779. [Google Scholar] [CrossRef]
- Bonville, C.A.; Percopo, C.M.; Dyer, K.D.; Gao, J.; Prussin, C.; Foster, B.; Rosenberg, H.F.; Domachowske, J.B. Interferon-gamma coordinates CCL3-mediated neutrophil recruitment in vivo. BMC Immunol. 2009, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Domachowske, J.B.; Bonville, C.A.; Gao, J.L.; Murphy, P.M.; Easton, A.J.; Rosenberg, H.F. The chemokine macrophage-inflammatory protein-1 alpha and its receptor CCR1 control pulmonary inflammation and antiviral host defense in paramyxovirus infection. J. Immunol. 2000, 165, 2677–2682. [Google Scholar] [CrossRef] [Green Version]
- Bonville, C.A.; Easton, A.J.; Rosenberg, H.F.; Domachowske, J.B. Altered pathogenesis of severe pneumovirus infection in response to combined antiviral and specific immunomodulatory agents. J. Virol. 2003, 77, 1237–1244. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.A.; Revez, J.A.; Loh, Z.; Simpson, J.; Zhang, V.; Bain, L.; Varelias, A.; Rose-John, S.; Blumenthal, A.; Smyth, M.J.; et al. Allergen-induced IL-6 trans-signaling activates gammadelta T cells to promote type 2 and type 17 airway inflammation. J. Allergy Clin. Immunol. 2015, 136, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Li, J.; Zhu, H.; Li, P.; Zou, Z.; Xiao, Y. Hmgb1-IL-23-IL-17-IL-6-Stat3 axis promotes tumor growth in murine models of melanoma. Mediat. Inflamm. 2013, 2013, 713859. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.M.; Wang, S.; Shen, M.; Wang, Y.J.; Zhang, B.; Wu, Z.Q.; Lu, J.; Zheng, Y.L. S100A9 gene silencing inhibits the release of pro-inflammatory cytokines by blocking the IL-17 signalling pathway in mice with acute pancreatitis. J. Cell Mol. Med. 2018, 22, 2378–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Lindell, D.M.; Berlin, A.A.; Morris, S.B.; Shanley, T.P.; Hershenson, M.B.; Lukacs, N.W. IL-17-induced pulmonary pathogenesis during respiratory viral infection and exacerbation of allergic disease. Am. J. Pathol. 2011, 179, 248–258. [Google Scholar] [CrossRef]
- Stoppelenburg, A.J.; Salimi, V.; Hennus, M.; Plantinga, M.; Huis in‘t Veld, R.; Walk, J.; Meerding, J.; Coenjaerts, F.; Bont, L.; Boes, M. Local IL-17A potentiates early neutrophil recruitment to the respiratory tract during severe RSV infection. PLoS ONE 2013, 8, e78461. [Google Scholar] [CrossRef] [PubMed]
- Geerdink, R.J.; Pillay, J.; Meyaard, L.; Bont, L. Neutrophils in respiratory syncytial virus infection: A target for asthma prevention. J. Allergy Clin. Immunol. 2015, 136, 838–847. [Google Scholar] [CrossRef]
- Patel, J.A.; Kunimoto, M.; Sim, T.C.; Garofalo, R.; Eliott, T.; Baron, S.; Ruuskanen, O.; Chonmaitree, T.; Ogra, P.L.; Schmalstieg, F. Interleukin-1 alpha mediates the enhanced expression of intercellular adhesion molecule-1 in pulmonary epithelial cells infected with respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 1995, 13, 602–609. [Google Scholar] [CrossRef]
- Bolger, G.; Lapeyre, N.; Dansereau, N.; Lagace, L.; Berry, G.; Klosowski, K.; Mewhort, T.; Liuzzi, M. Primary infection of mice with high titer inoculum respiratory syncytial virus: Characterization and response to antiviral therapy. Can. J. Physiol. Pharmacol. 2005, 83, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Thwaites, R.S.; Coates, M.; Ito, K.; Ghazaly, M.; Feather, C.; Abdulla, F.; Tunstall, T.; Jain, P.; Cass, L.; Rapeport, G.; et al. Reduced Nasal Viral Load and IFN Responses in Infants with Respiratory Syncytial Virus Bronchiolitis and Respiratory Failure. Am. J. Respir. Crit. Care Med. 2018, 198, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Knudson, C.J.; Hartwig, S.M.; Meyerholz, D.K.; Varga, S.M. RSV vaccine-enhanced disease is orchestrated by the combined actions of distinct CD4 T cell subsets. PLoS Pathog. 2015, 11, e1004757. [Google Scholar] [CrossRef] [Green Version]
- Emboriadou, M.; Hatzistilianou, M.; Magnisali, C.; Sakelaropoulou, A.; Exintari, M.; Conti, P.; Aivazis, V. Human neutrophil elastase in RSV bronchiolitis. Ann. Clin. Lab. Sci. 2007, 37, 79–84. [Google Scholar] [PubMed]
- Bradley, L.M.; Douglass, M.F.; Chatterjee, D.; Akira, S.; Baaten, B.J. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog. 2012, 8, e1002641. [Google Scholar] [CrossRef] [Green Version]
- Hosakote, Y.M.; Liu, T.; Castro, S.M.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am. J. Respir. Cell Mol. Biol. 2009, 41, 348–357. [Google Scholar] [CrossRef]
- Cervantes-Ortiz, S.L.; Zamorano Cuervo, N.; Grandvaux, N. Respiratory Syncytial Virus and Cellular Stress Responses: Impact on Replication and Physiopathology. Viruses 2016, 8, 124. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Li, N.; Niu, F. Baicalein triazole prevents respiratory tract infection by RSV through suppression of oxidative damage. Microb. Pathog. 2019, 131, 227–233. [Google Scholar] [CrossRef]
- Cortjens, B.; de Boer, O.J.; de Jong, R.; Antonis, A.F.; Sabogal Pineros, Y.S.; Lutter, R.; van Woensel, J.B.; Bem, R.A. Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease. J. Pathol. 2016, 238, 401–411. [Google Scholar] [CrossRef]
- Beyrau, M.; Bodkin, J.V.; Nourshargh, S. Neutrophil heterogeneity in health and disease: A revitalized avenue in inflammation and immunity. Open Biol. 2012, 2, 120134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scapini, P.; Marini, O.; Tecchio, C.; Cassatella, M.A. Human neutrophils in the saga of cellular heterogeneity: Insights and open questions. Immunol. Rev. 2016, 273, 48–60. [Google Scholar] [CrossRef]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018, 48, 364–379.e8. [Google Scholar] [CrossRef] [PubMed]
- Cortjens, B.; Ingelse, S.A.; Calis, J.C.; Vlaar, A.P.; Koenderman, L.; Bem, R.A.; van Woensel, J.B. Neutrophil subset responses in infants with severe viral respiratory infection. Clin. Immunol. 2017, 176, 100–106. [Google Scholar] [CrossRef]
- Wang, S.Z.; Smith, P.K.; Lovejoy, M.; Bowden, J.J.; Alpers, J.H.; Forsyth, K.D. Shedding of L-selectin and PECAM-1 and upregulation of Mac-1 and ICAM-1 on neutrophils in RSV bronchiolitis. Am. J. Physiol. 1998, 275, L983–L989. [Google Scholar] [CrossRef]
- Geerdink, R.J.; Hennus, M.P.; Westerlaken, G.H.A.; Abrahams, A.C.; Albers, K.I.; Walk, J.; Wesselink, E.; Janssen, R.; Bont, L.; Meyaard, L. LAIR-1 limits neutrophil extracellular trap formation in viral bronchiolitis. J. Allergy Clin. Immunol. 2018, 141, 811–814. [Google Scholar] [CrossRef] [Green Version]
- Kumawat, K.; Geerdink, R.J.; Hennus, M.P.; Roda, M.A.; van Ark, I.; Leusink-Muis, T.; Folkerts, G.; van Oort-Jansen, A.; Mazharian, A.; Watson, S.P.; et al. LAIR-1 Limits Neutrophilic Airway Inflammation. Front. Immunol. 2019, 10, 842. [Google Scholar] [CrossRef] [PubMed]
- Stark, J.M.; Godding, V.; Sedgwick, J.B.; Busse, W.W. Respiratory syncytial virus infection enhances neutrophil and eosinophil adhesion to cultured respiratory epithelial cells. Roles of CD18 and intercellular adhesion molecule-1. J. Immunol. 1996, 156, 4774–4782. [Google Scholar] [PubMed]
- Heinonen, S.; Velazquez, V.M.; Ye, F.; Mertz, S.; Acero-Bedoya, S.; Smith, B.; Bunsow, E.; Garcia-Maurino, C.; Oliva, S.; Cohen, D.M.; et al. Immune profiles provide insights into respiratory syncytial virus disease severity in young children. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Funchal, G.A.; Jaeger, N.; Czepielewski, R.S.; Machado, M.S.; Muraro, S.P.; Stein, R.T.; Bonorino, C.B.; Porto, B.N. Respiratory syncytial virus fusion protein promotes TLR-4-dependent neutrophil extracellular trap formation by human neutrophils. PLoS ONE 2015, 10, e0124082. [Google Scholar] [CrossRef]
- Halfhide, C.P.; Brearey, S.P.; Flanagan, B.F.; Hunt, J.A.; Howarth, D.; Cummerson, J.; Edwards, S.; Hart, C.A.; Smyth, R.L. Neutrophil TLR4 expression is reduced in the airways of infants with severe bronchiolitis. Thorax 2009, 64, 798–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tal, G.; Mandelberg, A.; Dalal, I.; Cesar, K.; Somekh, E.; Tal, A.; Oron, A.; Itskovich, S.; Ballin, A.; Houri, S.; et al. Association between common Toll-like receptor 4 mutations and severe respiratory syncytial virus disease. J. Infect. Dis. 2004, 189, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Shirey, K.A.; Lai, W.; Scott, A.J.; Lipsky, M.; Mistry, P.; Pletneva, L.M.; Karp, C.L.; McAlees, J.; Gioannini, T.L.; Weiss, J.; et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 2013, 497, 498–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansbach, J.M.; Hasegawa, K.; Ajami, N.J.; Petrosino, J.F.; Piedra, P.A.; Tierney, C.N.; Espinola, J.A.; Camargo, C.A. Serum LL-37 Levels Associated With Severity of Bronchiolitis and Viral Etiology. Clin. Infect. Dis. 2017, 65, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Currie, S.M.; Findlay, E.G.; McHugh, B.J.; Mackellar, A.; Man, T.; Macmillan, D.; Wang, H.; Fitch, P.M.; Schwarze, J.; Davidson, D.J. The human cathelicidin LL-37 has antiviral activity against respiratory syncytial virus. PLoS ONE 2013, 8, e73659. [Google Scholar] [CrossRef]
- Harcourt, J.L.; McDonald, M.; Svoboda, P.; Pohl, J.; Tatti, K.; Haynes, L.M. Human cathelicidin, LL-37, inhibits respiratory syncytial virus infection in polarized airway epithelial cells. BMC Res. Notes 2016, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Currie, S.M.; Gwyer Findlay, E.; McFarlane, A.J.; Fitch, P.M.; Bottcher, B.; Colegrave, N.; Paras, A.; Jozwik, A.; Chiu, C.; Schwarze, J.; et al. Cathelicidins Have Direct Antiviral Activity against Respiratory Syncytial Virus In Vitro and Protective Function In Vivo in Mice and Humans. J. Immunol. 2016, 196, 2699–2710. [Google Scholar] [CrossRef]
- Lukens, M.V.; van de Pol, A.C.; Coenjaerts, F.E.; Jansen, N.J.; Kamp, V.M.; Kimpen, J.L.; Rossen, J.W.; Ulfman, L.H.; Tacke, C.E.; Viveen, M.C.; et al. A systemic neutrophil response precedes robust CD8(+) T-cell activation during natural respiratory syncytial virus infection in infants. J. Virol. 2010, 84, 2374–2383. [Google Scholar] [CrossRef] [Green Version]
- Yasui, K.; Baba, A.; Iwasaki, Y.; Kubo, T.; Aoyama, K.; Mori, T.; Yamazaki, T.; Kobayashi, N.; Ishiguro, A. Neutrophil-mediated inflammation in respiratory syncytial viral bronchiolitis. Pediatr. Int. 2005, 47, 190–195. [Google Scholar] [CrossRef]
- Muraro, S.P.; De Souza, G.F.; Gallo, S.W.; Da Silva, B.K.; De Oliveira, S.D.; Vinolo, M.A.R.; Saraiva, E.M.; Porto, B.N. Respiratory Syncytial Virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci. Rep. 2018, 8, 14166. [Google Scholar] [CrossRef]
- Cortjens, B.; van Woensel, J.B.; Bem, R.A. Neutrophil Extracellular Traps in Respiratory Disease: Guided anti-microbial traps or toxic webs? Paediatr. Respir. Rev. 2017, 21, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.Y.; Clancy, J.P.; Peng, N.; Li, Y.; Szul, T.J.; Xu, X.; Oster, R.; Sullender, W.; Ambalavanan, N.; Blalock, J.E.; et al. Pulmonary matrix metalloproteinase-9 activity in mechanically ventilated children with respiratory syncytial virus. Eur. Respir. J. 2014, 43, 1086–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, M.Y.; Whitley, R.J.; Peng, N.; Oster, R.; Schoeb, T.R.; Sullender, W.; Ambalavanan, N.; Clancy, J.P.; Gaggar, A.; Blalock, J.E. Matrix Metalloproteinase-9 Mediates RSV Infection in Vitro and in Vivo. Viruses 2015, 7, 4230–4253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, M.Y.; Li, Y.; Oster, R.; Gaggar, A.; Clancy, J.P. Early elevation of matrix metalloproteinase-8 and -9 in pediatric ARDS is associated with an increased risk of prolonged mechanical ventilation. PLoS ONE 2011, 6, e22596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beigelman, A.; Mikols, C.L.; Gunsten, S.P.; Cannon, C.L.; Brody, S.L.; Walter, M.J. Azithromycin attenuates airway inflammation in a mouse model of viral bronchiolitis. Respir. Res. 2010, 11, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, J.L.; Powell, H.; Boyle, M.J.; Scott, R.J.; Gibson, P.G. Clarithromycin targets neutrophilic airway inflammation in refractory asthma. Am. J. Respir. Crit. Care Med. 2008, 177, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Bacharier, L.B.; Isaacson-Schmid, M.; Baty, J.; Schechtman, K.B.; Sajol, G.; Wylie, K.; Storch, G.A.; Castro, M.; Beigelman, A. Azithromycin therapy during respiratory syncytial virus bronchiolitis: Upper airway microbiome alterations and subsequent recurrent wheeze. J. Allergy Clin. Immunol. 2016, 138, 1215–1219.e5. [Google Scholar] [CrossRef] [Green Version]
- Busch-Petersen, J.; Carpenter, D.C.; Burman, M.; Foley, J.; Hunsberger, G.E.; Kilian, D.J.; Salmon, M.; Mayer, R.J.; Yonchuk, J.G.; Tal-Singer, R. Danirixin: A Reversible and Selective Antagonist of the CXC Chemokine Receptor 2. J. Pharmacol. Exp. Ther. 2017, 362, 338–346. [Google Scholar] [CrossRef] [Green Version]
- Bruce, C.; Thomas, P.S. The effect of marimastat, a metalloprotease inhibitor, on allergen-induced asthmatic hyper-reactivity. Toxicol. Appl. Pharmacol. 2005, 205, 126–132. [Google Scholar] [CrossRef]
- Kumagai, K.; Ohno, I.; Okada, S.; Ohkawara, Y.; Suzuki, K.; Shinya, T.; Nagase, H.; Iwata, K.; Shirato, K. Inhibition of matrix metalloproteinases prevents allergen-induced airway inflammation in a murine model of asthma. J. Immunol. 1999, 162, 4212–4219. [Google Scholar] [PubMed]
- Stockley, R.; De Soyza, A.; Gunawardena, K.; Perrett, J.; Forsman-Semb, K.; Entwistle, N.; Snell, N. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir. Med. 2013, 107, 524–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Warner, R.; Ruggeri, R.; Su, C.; Cortes, C.; Skoura, A.; Ward, J.; Ahn, K.; Kalgutkar, A.; Sun, D.; et al. PF-1355, a mechanism-based myeloperoxidase inhibitor, prevents immune complex vasculitis and anti-glomerular basement membrane glomerulonephritis. J. Pharmacol. Exp. Ther. 2015, 353, 288–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussey, S.E.; Liang, H.; Costford, S.R.; Klip, A.; DeFronzo, R.A.; Sanchez-Avila, A.; Ely, B.; Musi, N. TAK-242, a small-molecule inhibitor of Toll-like receptor 4 signalling, unveils similarities and differences in lipopolysaccharide- and lipid-induced inflammation and insulin resistance in muscle cells. Biosci. Rep. 2012, 33, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Palanichamy, A.; Bauer, J.W.; Yalavarthi, S.; Meednu, N.; Barnard, J.; Owen, T.; Cistrone, C.; Bird, A.; Rabinovich, A.; Nevarez, S.; et al. Neutrophil-mediated IFN activation in the bone marrow alters B cell development in human and murine systemic lupus erythematosus. J. Immunol. 2014, 192, 906–918. [Google Scholar] [CrossRef]
- Radermecker, C.; Sabatel, C.; Vanwinge, C.; Ruscitti, C.; Marechal, P.; Perin, F.; Schyns, J.; Rocks, N.; Toussaint, M.; Cataldo, D.; et al. Locally instructed CXCR4(hi) neutrophils trigger environment-driven allergic asthma through the release of neutrophil extracellular traps. Nat. Immunol. 2019, 20, 1444–1455. [Google Scholar] [CrossRef]
- Sapey, E.; Patel, J.M.; Greenwood, H.; Walton, G.M.; Grudzinska, F.; Parekh, D.; Mahida, R.Y.; Dancer, R.C.A.; Lugg, S.T.; Howells, P.A.; et al. Simvastatin Improves Neutrophil Function and Clinical Outcomes in Pneumonia. A Pilot Randomized Controlled Clinical Trial. Am. J. Respir. Crit. Care Med. 2019, 200, 1282–1293. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Protein Name | Function |
|---|---|
| Azurophil (primary) neutrophil granules | |
| Azurocidin | Antibacterial activity (in particular, specific to Gram-bacteria) [16,18] |
| Neutrophil defensins | Antibacterial, fungicidal, and antiviral activities [19] |
| Myeloblastin | Serine protease; facilitates transendothelial neutrophil migration [16] |
| CD63 antigen | Cell surface receptor for TIMP1; activates cellular signalling cascades [20] |
| Cathepsin G | Serine protease, cleaves complement C3 and has antibacterial activity [21] |
| Neutrophil elastase (NE) | Modifies the functions of NK cells, monocytes, and granulocytes; inhibits C5a-dependent neutrophil enzyme release and chemotaxis [16,22] |
| Myeloperoxidase (MPO) | Microbicidal activity against a wide range of organisms [23] |
| Cap57 | Antibacterial activity (Specific to Gram-bacteria) [16] |
| Specific (secondary) neutrophil granules | |
| Chitinase-3-like protein 1 | Important for inflammation [18] |
| Lipocalin 2 | Iron-trafficking; involved in apoptosis, innate immunity, and renal development; limits bacterial proliferation [24] |
| Lactoferrin | Antimicrobial activity; stimulates TLR4 signalling, binds heparin [16] |
| Gelatinase (tertiary) neutrophil granules | |
| Matrix metalloproteinase-9 (MMP-9) | Cleaves gelatin types I and V and collagen types IV and V; important roles in leukocyte migration [25] |
| Ficolin-1 | Anti-microbial pattern-recognition receptor |
| Cathelicidin antimicrobial peptide | Antibacterial activity; cleaved into 2 antimicrobial peptides FALL-39 and LL-37 [26] |
| Neutrophil collagenase | Degrades fibrillar collagens (type I, II, and III) [27] |
| Mediators | Examples | Potential Pathogenic Effects during Bronchiolitis |
|---|---|---|
| Cytokines | 1L-1α | Enhances ICAM-1 expression on AECs [102] |
| IL-1β | Pro-inflammatory, cell death [103] | |
| IL-6 | Pro-inflammatory, induces fever, induces AEC damage [104,105] | |
| TNFα | Pro-inflammatory, induces fever, induces AEC damage [69,105] | |
| IFNγ | Pro-inflammatory, induces fever, induces AEC damage [99] | |
| IL-17A/F | Pro-inflammatory, augments neutrophil recruitment and activation [100,101] | |
| Chemokines | IL-8 | Augments neutrophil chemotaxis to the lung [85] |
| CCL3 | Recruitment of innate and adaptive leukocytes to the lung, activation of DCs [92,93] | |
| CXCL12 | Recruitment of CD8 T-cells [59] | |
| Neutrophil Granules | MPO | Induces mucus production, oedema and AEC death |
| NE | Induces mucus production, oedema and AEC death [68,106] | |
| MMP-9 | Induces lung inflammation [107] | |
| Others | ROS mediators | Induces oxidative stress, AEC death, augment NETosis formation [108,109,110] |
| NETosis | Induces mucus hypersecretion and airway obstruction [111] | |
| DAMPs (e.g., HMGB1) | Induce secretion of pro-inflammatory cytokines, drive ILC2 responses, induce necroptosis and AEC death [76,83] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sebina, I.; Phipps, S. The Contribution of Neutrophils to the Pathogenesis of RSV Bronchiolitis. Viruses 2020, 12, 808. https://doi.org/10.3390/v12080808
Sebina I, Phipps S. The Contribution of Neutrophils to the Pathogenesis of RSV Bronchiolitis. Viruses. 2020; 12(8):808. https://doi.org/10.3390/v12080808
Chicago/Turabian StyleSebina, Ismail, and Simon Phipps. 2020. "The Contribution of Neutrophils to the Pathogenesis of RSV Bronchiolitis" Viruses 12, no. 8: 808. https://doi.org/10.3390/v12080808
APA StyleSebina, I., & Phipps, S. (2020). The Contribution of Neutrophils to the Pathogenesis of RSV Bronchiolitis. Viruses, 12(8), 808. https://doi.org/10.3390/v12080808