Characterization of vB_StuS_MMDA13, a Newly Discovered Bacteriophage Infecting the Agar-Degrading Species Sphingomonas turrisvirgatae

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Host and Growth Conditions
2.2. Isolation, Purification and Large-Scale Production of Bacteriophage
2.3. Transmission Electron Microscopy (TEM)
2.4. Determination of Bacteriophage Host Range
2.5. One-Step Growth Curve
2.6. Influence of pH and Temperature on Phage Viability
2.7. Lysogeny Tests
2.8. Extraction of Bacteriophage vB_StuS_MMDA13 DNA
2.9. Genome Sequencing and Bioinformatics Analysis of vB_StuS_MMDA13
3. Results and Discussion
3.1. Phage Isolation and Morphological Features
3.2. Host-Range Determination
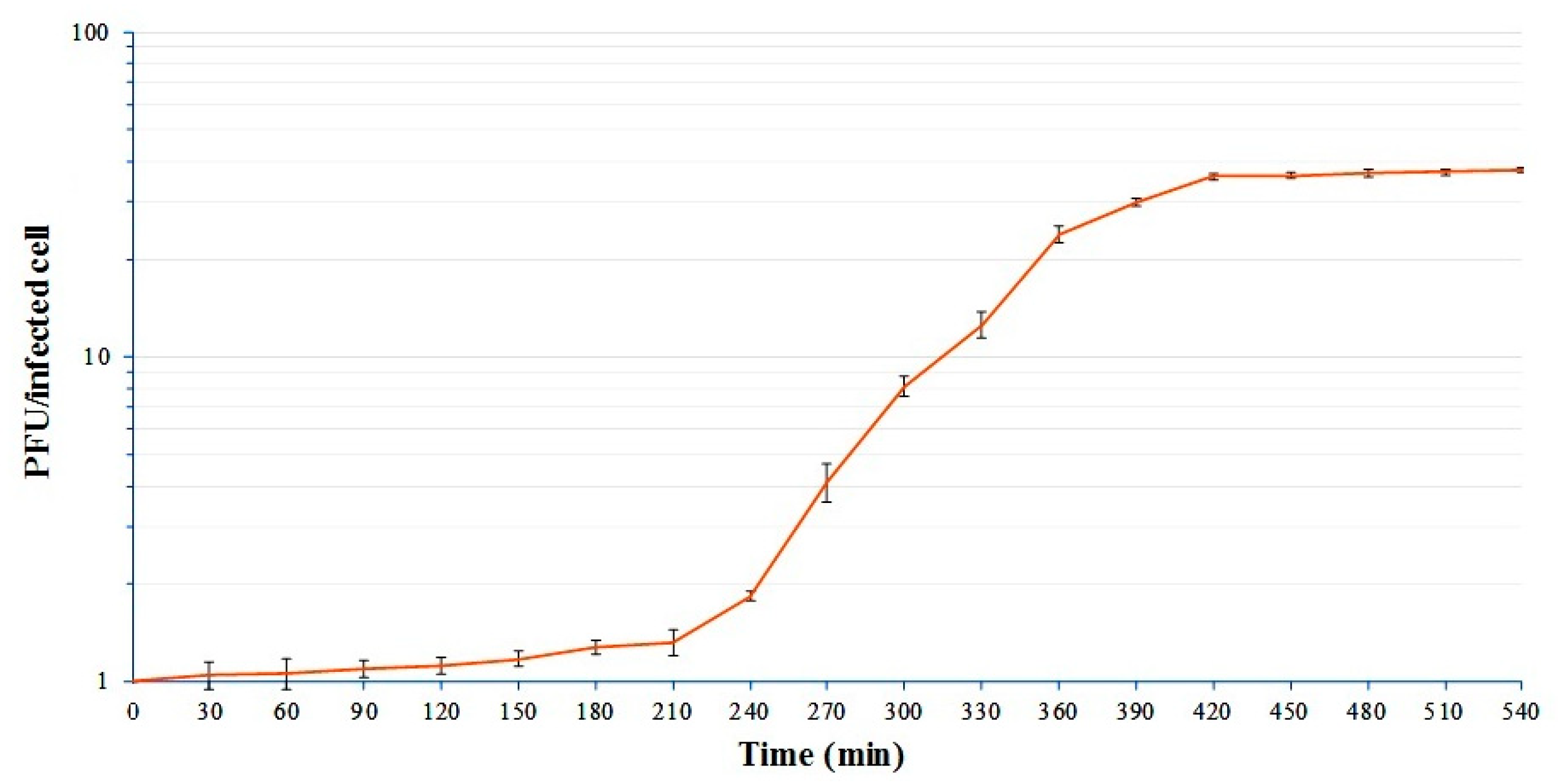
3.3. One-Step Growth Curve
3.4. Phage Stability to pH and Temperature
3.5. Lysogeny Analysis
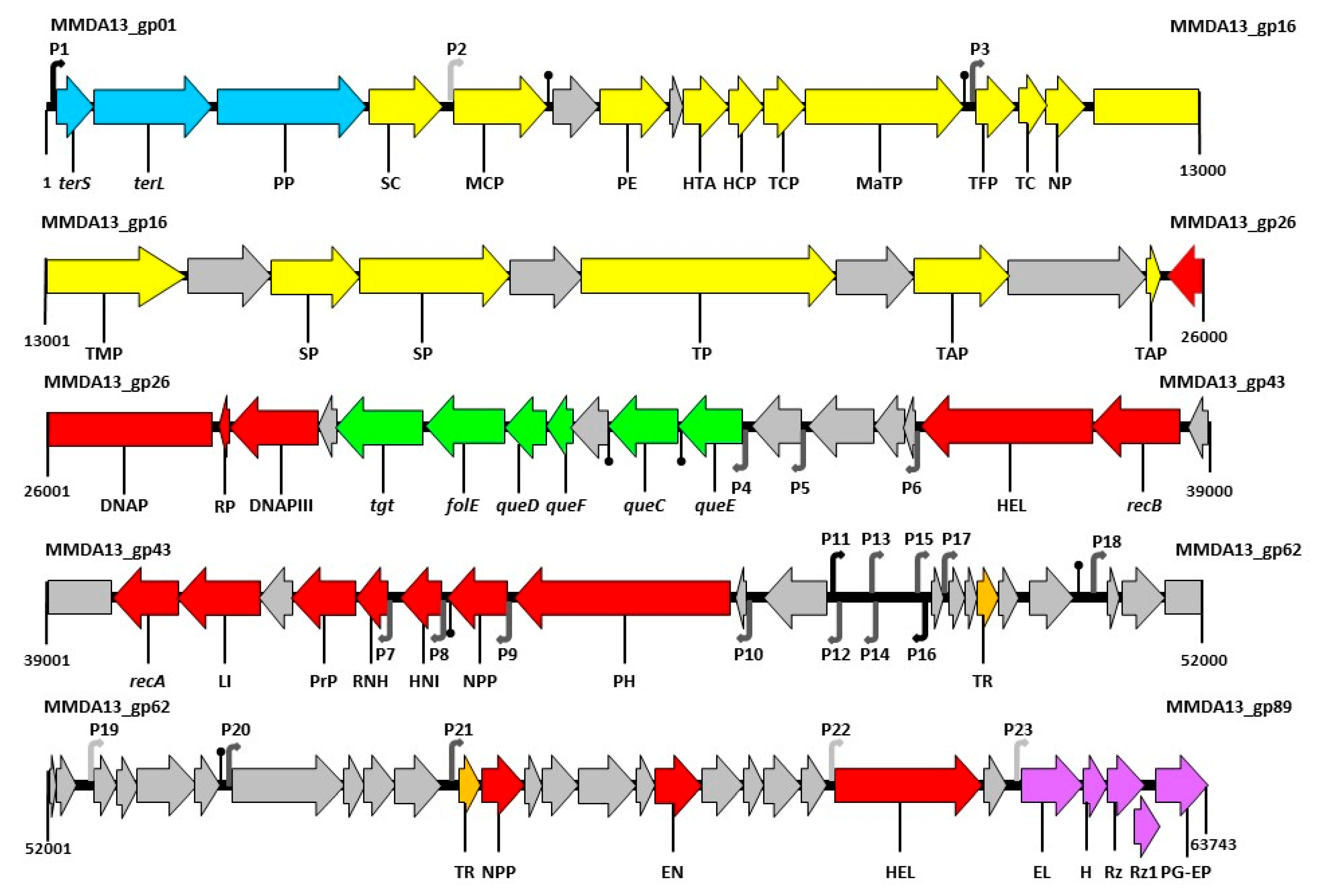
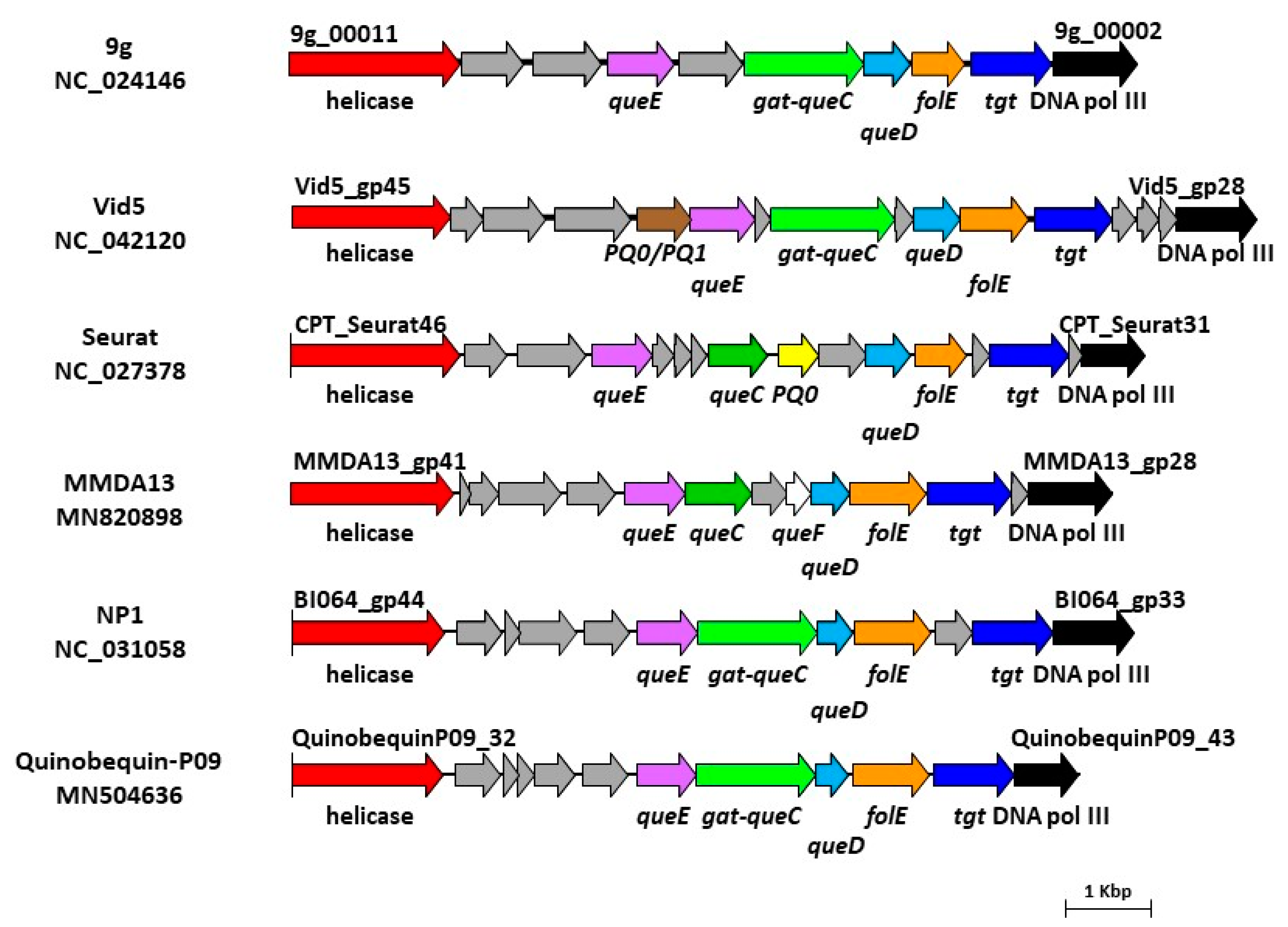
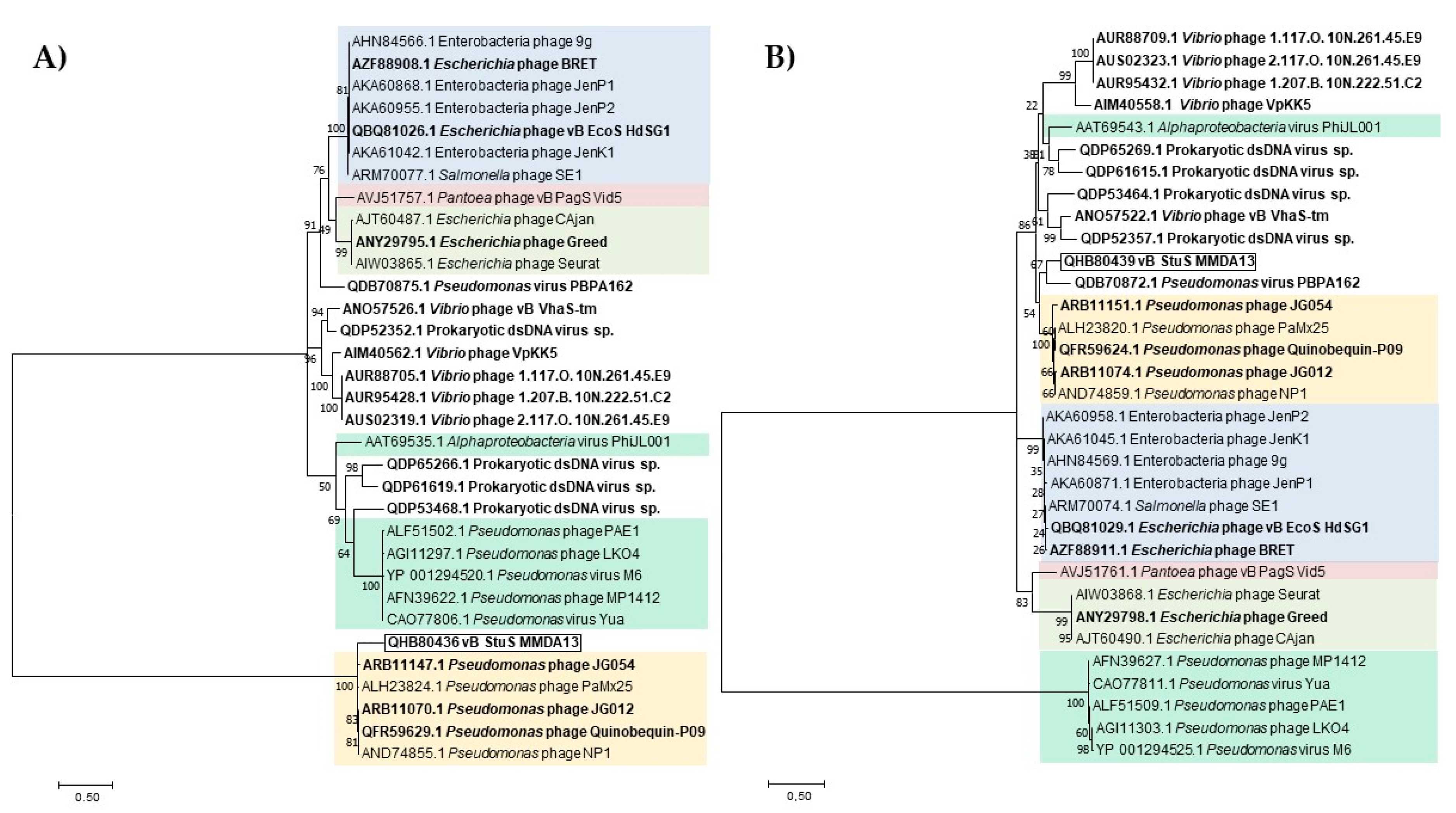
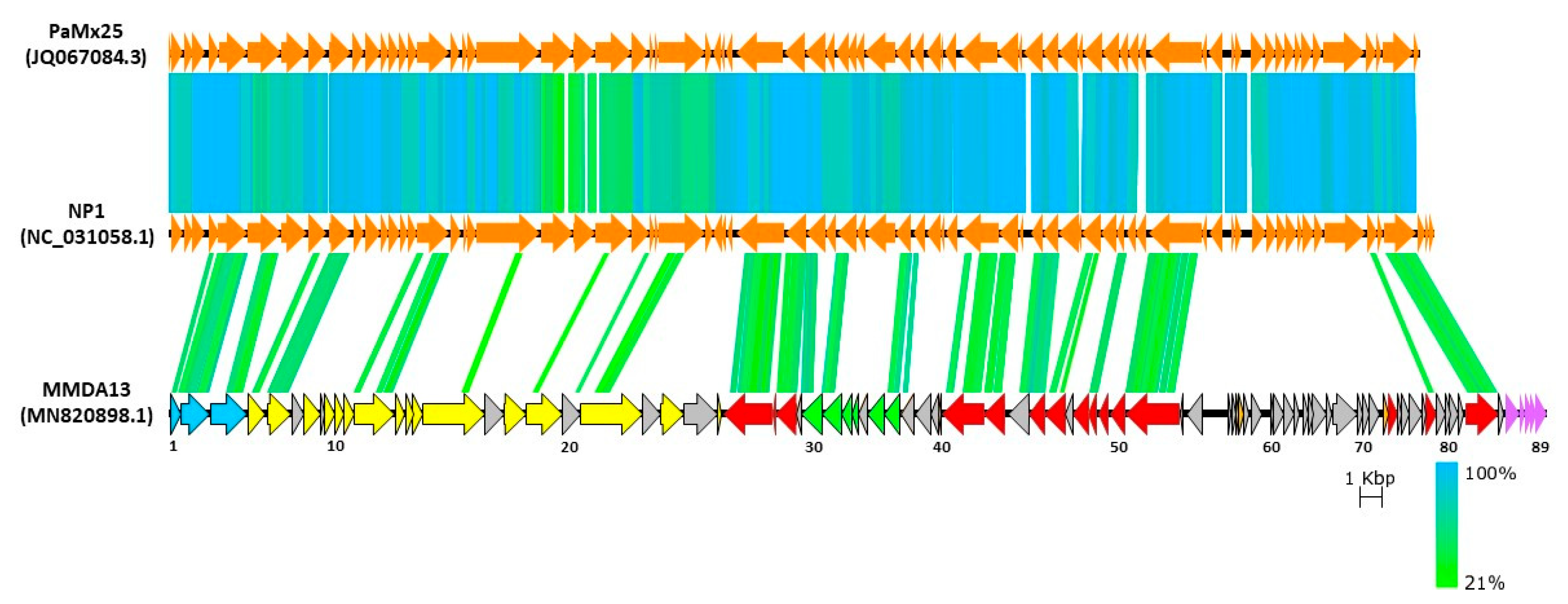
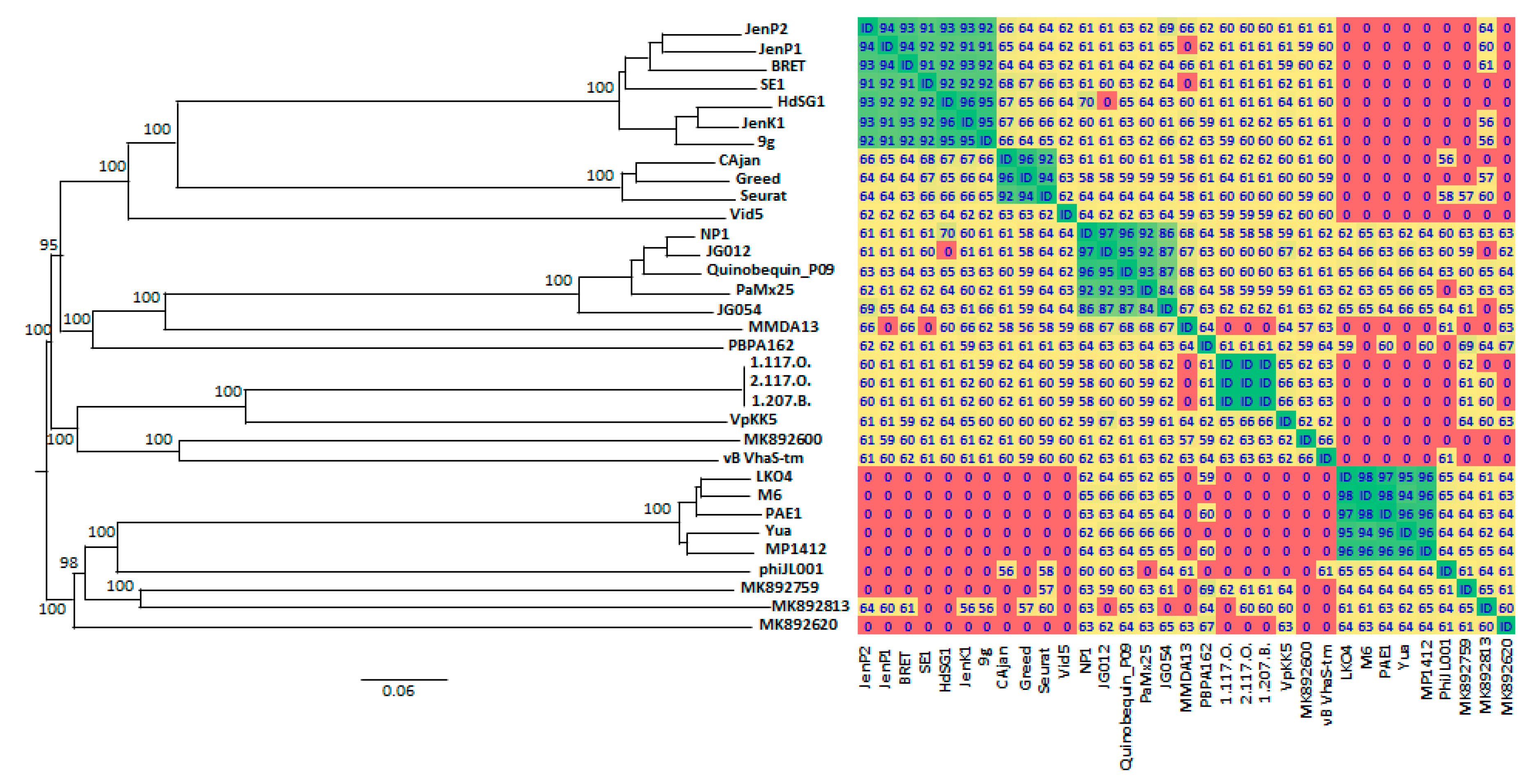
3.6. Bioinformatics Analysis of vB_StuS_MMDA13 Genome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef]
- Hendrix, R.W. Bacteriophages: Evolution of the Majority. Theor. Popul. Biol. 2002, 61, 471–480. [Google Scholar] [CrossRef]
- Hanlon, G.W. Bacteriophages: An appraisal of their role in the treatment of bacterial infections. Int. J. Antimicrob. Agents 2007, 30, 118–128. [Google Scholar] [CrossRef]
- Zulkarneev, E.R.; Aleshkin, A.V.; Kiseleva, I.A.; Rubalsky, E.O.; Rubalsky, O.V. Bacteriophage Cocktail Effectively Prolonging the Shelf-Life of Chilled Fish. Bull. Exp. Biol. Med. 2019, 167, 818–822. [Google Scholar] [CrossRef]
- Abedon, S.T.; García, P.; Mullany, P.; Aminov, R. Editorial: Phage Therapy: Past, Present and Future. Front. Microbiol. 2017, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Mertz, L. Battling Superbugs: How Phage Therapy Went From Obscure to Promising. IEEE Pulse 2019, 10, 3–9. [Google Scholar] [CrossRef]
- Frenkel, D.; Solomon, B. Filamentous phage as vector-mediated antibody delivery to the brain. Proc. Natl. Acad. Sci. USA 2002, 99, 5675–5679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhu, S.S. Phage display in pharmaceutical biotechnology. Curr. Opin. Biotechnol. 2000, 11, 610–616. [Google Scholar] [CrossRef]
- Rodríguez-Rubio, L.; Gutiérrez, D.; Donovan, D.M.; Martínez, B.; Rodríguez, A.; García, P. Phage lytic proteins: biotechnological applications beyond clinical antimicrobials. Crit. Rev. Biotechnol. 2016, 36, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altamirano, F.L.G.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef] [Green Version]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and their Genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, H.W. 5500 Phages examined in the electron microscope. Arch. Virol. 2007, 152, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Wittmann, J.; Kuhn, J.H.; Turner, D.; Sullivan, M.B.; Dutilh, B.E.; Jang, H.B.; Van Zyl, L.J.; Klumpp, J.; Lobocka, M.; et al. Taxonomy of prokaryotic viruses: 2017 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2018, 163, 1125–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barylski, J.; Enault, F.; Dutilh, B.E.; Schuller, M.B.; Edwards, R.A.; Gillis, A.; Klumpp, J.; Knezevic, P.; Krupovic, M.; Kuhn, J.H.; et al. Taxonomy proposal To create one (1) new family, Herelleviridae, in the order Caudovirales. In ICTV Online: International Committee on Taxonomy of Viruses (ICTV); International Committee on Taxonomy of Viruses (ICTV): London, UK, 2018; p. 2018.118B. [Google Scholar]
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; van Zyl, L.J.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Łobocka, M.; Tong, Y.; Brister, J.R.; et al. Taxonomy of prokaryotic viruses: 2018–2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020, 165, 1253–1260. [Google Scholar] [CrossRef] [Green Version]
- Leys, N.M.E.J.; Ryngaert, A.; Bastiaens, L.; Verstraete, W.; Top, E.M.; Springael, D. Occurrence and Phylogenetic Diversity of Sphingomonas Strains in Soils Contaminated with Polycyclic Aromatic Hydrocarbons. Appl. Environ. Microbiol. 2004, 70, 1944–1955. [Google Scholar] [CrossRef] [Green Version]
- Festa, S.; Macchi, M.; Cortés, F.; Morelli, I.S.; Coppotelli, B.M. Monitoring the Impact of Bioaugmentation with a PAH-degrading Strain on Different Soil Microbiomes Using Pyrosequencing. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef]
- Zhou, N.A.; Gough, H.L. Enhanced Biological Trace Organic Contaminant Removal: A Lab-Scale Demonstration with Bisphenol A-Degrading Bacteria Sphingobium sp. BiD32. Environ. Sci. Technol. 2016, 50, 8057–8066. [Google Scholar] [CrossRef]
- Ramos, C.; Suárez-Ojeda, M.E.; Carrera, J. Long-term impact of salinity on the performance and microbial population of an aerobic granular reactor treating a high-strength aromatic wastewater. Bioresour. Technol. 2015, 198, 844–851. [Google Scholar] [CrossRef]
- Feld, L.; Nielsen, T.K.; Hansen, L.H.; Aamand, J.; Albers, C.N. Establishment of Bacterial Herbicide Degraders in a Rapid Sand Filter for Bioremediation of Phenoxypropionate-Polluted Groundwater. Appl. Environ. Microbiol. 2015, 82, 878–887. [Google Scholar] [CrossRef] [Green Version]
- Cycoń, M.; Mrozik, A.; Piotrowska-Seget, Z. Bioaugmentation as a strategy for the remediation of pesticide-polluted soil: A review. Chemosphere 2017, 172, 52–71. [Google Scholar] [CrossRef]
- Lu, L.; Cai, L.; Jiao, N.; Zhang, R. Isolation and characterization of the first phage infecting ecologically important marine bacteria Erythrobacter. Virol. J. 2017, 14, 104. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, N.; Tsuge, T.; Asakawa, S.; Kimura, M. Morphology, host range and phylogenetic diversity of Sphingomonas phages in the floodwater of a Japanese paddy field. Soil Sci. Plant Nutr. 2009, 55, 53–64. [Google Scholar] [CrossRef]
- Nielsen, T.K.; Carstens, A.B.; Browne, P.; Lametsch, R.; Neve, H.; Kot, W.; Hansen, L.H. The first characterized phage against a member of the ecologically important sphingomonads reveals high dissimilarity against all other known phages. Sci. Rep. 2017, 7, 13566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Q.; Chen, Q.; Xu, Y.; Suttle, C.A.; Jiao, N. A Virus Infecting Marine Photoheterotrophic Alphaproteobacteria (Citromicrobium spp.) Defines a New Lineage of ssdna Viruses. Front. Microbiol. 2018, 9, 1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jost, G.; Wiese, J. Temporal variations in the concentrations of bacteria and their lytic phages: an example of an indigenous phage host system in Lake Plußsee, Germany. Fundam. Appl. Limnol. 2013, 182, 183–190. [Google Scholar] [CrossRef]
- Wolf, A.; Wiese, J.; Jost, G.; Witzel, K.P. Wide Geographic Distribution of Bacteriophages That Lyse the Same Indigenous Freshwater Isolate (Sphingomonas sp. Strain B18). Appl. Environ. Microbiol. 2003, 69, 2395–2398. [Google Scholar] [CrossRef] [Green Version]
- Thaller, M.C.; D’Andrea, M.M.; Marmo, P.; Civitareale, C.; Casu, F.; Migliore, L. Sphingomonas turrisvirgatae sp. nov., an agar-degrading species isolated from freshwater. Int. J. Syst. Evol. Microbiol. 2018, 68, 2794–2799. [Google Scholar] [CrossRef]
- Zobell, C.E. Studies on marine bacteria. The cultural requirements of heterotrophic aerobes. J. Mar. Res. 1941, 4, 42–75. [Google Scholar]
- Di Lallo, G.; Evangelisti, M.; Mancuso, F.; Ferrante, P.; Marcelletti, S.; Tinari, A.; Superti, F.; Migliore, L.; D’Addabbo, P.; Frezza, D.; et al. Isolation and partial characterization of bacteriophages infecting Pseudomonas syringae pv. actinidiae, causal agent of kiwifruit bacterial canker. J. Basic Microbiol. 2014, 54, 1210–1221. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Shin, Y.K.; Yoon, J.H.; Takeuchi, M.; Pyun, Y.R.; Park, Y.H. Sphingomonas aquatilis sp. nov., Sphingomonas koreensis sp. nov., and Sphingomonas taejonensis sp. nov., yellow-pigmented bacteria isolated from natural mineral water. Int. J. Syst. Evol. Microbiol. 2001, 51, 1491–1498. [Google Scholar] [CrossRef]
- Talà, A.; Lenucci, M.; Gaballo, A.; Durante, M.; Tredici, S.M.; Debowles, D.A.; Pizzolante, G.; Marcuccio, C.; Carata, E.; Piro, G.; et al. Sphingomonas cynarae sp. nov., a proteobacterium that produces an unusual type of sphingan. Int. J. Syst. Evol. Microbiol. 2013, 63, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Park, S.; Kang, S.J.; Kim, W.; Oh, T.K. Sphingomonas hankookensis sp. nov., isolated from wastewater. Int. J. Syst. Evol. Microbiol. 2009, 59, 2788–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.H.; Kang, S.J.; Lee, S.Y.; Oh, T.K. Sphingomonas insulae sp. nov., isolated from soil. Int. J. Syst. Evol. Microbiol. 2008, 58, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Kämpfer, P.; Meurer, U.; Esser, M.; Hirsch, T.; Busse, H.J. Sphingomonas pseudosanguinis sp. nov., isolated from the water reservoir of an air humidifier. Int. J. Syst. Evol. Microbiol. 2007, 57, 1342–1345. [Google Scholar] [CrossRef] [PubMed]
- Busse, H.J.; Hauser, E.; Kämpfer, P. Description of two novel species, Sphingomonas abaci sp. nov. and Sphingomonas panni sp. nov. Int. J. Syst. Evol. Microbiol. 2005, 55, 2565–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.C.; Im, W.T.; Kim, M.K.; Ohta, H.; Lee, S.T. Sphingomonas soli gen. nov., a beta-glucosidase-producing bacterium in the family Sphingomonadaceae in the alpha-4 subgroup of the Proteobacteria. Int. J. Syst. Evol. Microbiol. 2006, 56, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Moon, J.Y.; Lim, J.M.; Ahn, J.H.; Weon, H.Y.; Ahn, T.Y; Kwon, S.W. Sphingomonas aerophila sp. nov. and Sphingomonas naasensis sp. nov., isolated from air and soil, respectively. Int. J. Syst. Evol. Microbiol. 2014, 64, 926–932. [Google Scholar] [CrossRef]
- Takeuchi, M.; Hamana, K.; Hiraishi, A. Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int. J. Syst. Bacteriol. 2001, 51, 1405–1417. [Google Scholar] [CrossRef]
- Balkwill, D.L.; Drake, G.R.; Reeves, R.H.; Fredrickson, J.K.; White, D.C.; Ringelberg, D.B.; Chandler, D.P.; Romine, M.F.; Kennedy, D.W.; Spadoni, C.M. Taxonomic study of aromatic-degrading bacteria from deep-terrestrial-subsurface sediments and description of Sphingomonas aromaticivorans sp. nov., Sphingomonas subterranea sp. nov., and Sphingomonas stygia sp. nov. Int. J. Syst. Bacteriol. 1997, 47, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Vancanneyt, M.; Schut, F.; Snauwaert, C.; Goris, J.; Swings, J.; Gottschal, J.C. Sphingomonas alaskensis sp. nov., a dominant bacterium from a marine oligotrophic environment. Int. J. Syst. Evol. Microbiol. 2001, 51, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Holloway, B.W. Genetic Recombination in Pseudomonas aeruginosa. J. Gen. Microbiol. 1955, 13, 572–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govan, J.R.; Brown, P.H.; Maddison, J.; Doherty, C.J.; Nelson, J.W.; Dodd, M.; Greening, A.P.; Webb, A.K. Evidence for transmission of Pseudomonas cepacia by social contact in cystic fibrosis. Lancet 1993, 34, 15–19. [Google Scholar] [CrossRef]
- Adams, M.H. Bacteriophages; Interscience Publishers Inc.: New York, NY, USA, 1959; pp. 450–456. [Google Scholar]
- Ellis, E.L.; Delbrück, M. The Growth of Bacteriophage. J. Gen. Physiol. 1939, 22, 365–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, A.; Tavares, P.; Petit, M.A.; Guérois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genom. 2014, 15, 1027. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2015, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: a genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Zafar, N.; Mazumder, R.; Seto, D. CoreGenes: A computational tool for identifying and cataloging “core” genes in a set of small genomes. BMC Bioinform. 2002, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W.; Eisenstark, A. The present state of phage taxonomy. Intervirology 1974, 3, 201–219. [Google Scholar] [CrossRef]
- Yang, Y.; Cai, L.; Ma, R.; Xu, Y.; Tong, Y.; Huang, Y.; Jiao, N.; Zhang, R. A Novel Roseosiphophage Isolated from the Oligotrophic South China Sea. Viruses 2017, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Casjens, S.R.; Gilcrease, E.B. Determining DNA Packaging Strategy by Analysis of the Termini of the Chromosomes in Tailed-Bacteriophage Virions. Methods Mol. Biol. 2009, 502, 91–111. [Google Scholar] [CrossRef] [Green Version]
- Thiaville, J.J.; Kellner, S.M.; Yuan, Y.; Hutinet, G.; Thiaville, P.C.; Jumpathong, W.; Mohapatra, S.; Brochier-Armanet, C.; Letarov, A.V.; Hillebrand, R.; et al. Novel genomic island modifies DNA with 7-deazaguanine derivatives. Proc. Natl. Acad. Sci. USA 2016, 113, E1452–E1459. [Google Scholar] [CrossRef] [Green Version]
- Hutinet, G.; Kot, W.; Cui, L.; Hillebrand, R.; Balamkundu, S.; Gnanakalai, S.; Neelakandan, R.; Carstens, A.B.; Fa Lui, C.; Tremblay, D.; et al. 7-Deazaguanine modifications protect phage DNA from host restriction systems. Nat. Commun. 2019, 10, 5442. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, E.E.; Golomidova, A.K.; Letarova, M.A.; Kostryukova, E.S.; Zelenin, A.S.; Prokhorov, N.S.; Letarov, A.V. Genomic Sequencing and Biological Characteristics of a Novel Escherichia coli Bacteriophage 9g, a Putative Representative of a New Siphoviridae Genus. Viruses 2014, 6, 5077–5092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, V.; Sepúlveda-Robles, O.; Cazares, A.; Kameyama, L.; Guarneros, G. Comparative genomic analysis of Pseudomonas aeruginosa phage PaMx25 reveals a novel siphovirus group related to phages infecting hosts of different taxonomic classes. Arch. Virol. 2017, 162, 2345–2355. [Google Scholar] [CrossRef] [PubMed]
- Šimoliūnas, E.; Šimoliūnienė, M.; Kaliniene, L.; Zajančkauskaitė, A.; Skapas, M.; Meškys, R.; Kaupinis, A.; Valius, M.; Truncaitė, L. Pantoea Bacteriophage vB_PagS_Vid5: A Low-Temperature Siphovirus That Harbors a Cluster of Genes Involved in the Biosynthesis of Archaeosine. Viruses 2018, 10, 583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lood, R.; Winer, B.Y.; Pelzek, A.J.; Diez-Martinez, R.; Thandar, M.; Euler, C.W.; Schuch, R.; Fischetti, V.A. Novel phage lysin capable of killing the multidrug-resistant gram-negative bacterium Acinetobacter baumannii in a mouse bacteremia model. Antimicrob. Agents Chemother. 2015, 59, 1983–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R. Bacteriophage holins: deadly diversity. J. Mol. Microbiol. Biotechnol. 2002, 4, 21–36. [Google Scholar]
- Summer, E.J.; Berry, J.; Tran, T.A.; Niu, L.; Struck, D.K.; Young, R. Rz/Rz1 Lysis Gene Equivalents in Phages of Gram-negative Hosts. J. Mol. Biol. 2007, 373, 1098–1112. [Google Scholar] [CrossRef]
- Hett, E.C.; Chao, M.C.; Steyn, A.J.; Fortune, S.M.; Deng, L.L.; Rubin, E.J. A partner for the resuscitation-promoting factors of Mycobacterium tuberculosis. Mol. Microbiol. 2007, 66, 658–668. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Edwards, R.; Nash, J.H.E.; Mahadevan, P.; Seto, D.; Ackermann, H.W.; Lavigne, R.; Kropinski, A.M. Integration of genomic and proteomic analyses in the classification of the Siphoviridae family. Virology 2015, 477, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: the viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Growth Medium a | Susceptibility to vB_StuS_MMDA13 | Reference |
|---|---|---|---|
| Sphingomonas turrisvirgatae MCT13T | ZBB | + | [28] |
| Sphingomonas koreensis NBRC_16723 | ZBB | − | [31] |
| Sphingomonas cynarae DSM 25525 | SPB | − | [32] |
| Sphingomonas hankookensis DSM 23329 | ZBB | − | [33] |
| Sphingomonas insulae DSM 21792 | ZBB | − | [34] |
| Sphingomonas pseudosanguinis DSM 19512 | ZBB | − | [35] |
| Sphingomonas panni DSM 15761 | ZBB | − | [36] |
| Sphingomonas soli DSM 18313 | ZBB | − | [37] |
| Sphingomonas naasensis DSM 100060 | ZBB | − | [38] |
| Sphingobium yanoikuyae DSM 7462 | TSB | − | [39] |
| Sphingobium chlorophenolicum DSM 7098 | NB | − | [39] |
| Novosphingobium aromaticivorans DSM 12444 | PCA | − | [40] |
| Novosphingobium capsulatum DSM 30196 | NB | − | [39] |
| Sphingopyxis alaskensis DSM 13593 | TSB | − | [41] |
| Sphingopyxis macrogoltabida DSM 8826 | NB | − | [39] |
| Pseudomonas aeruginosa PAO1 | ZBB | − | [42] |
| Pseudomonas aeruginosa PAO4028 | ZBB | − | - |
| Pseudomonas aeruginosa PA 661 | ZBB | − | - |
| Pseudomonas aeruginosa ATCC 27853 | ZBB | − | - |
| Pseudomonas aeruginosa ATCC 15442 | ZBB | − | - |
| Burkholderia cenocepacia J2315 (CF5610) | ZBB | − | [43] |
| Agrobacterium tumefaciens DSM 5172 | ZBB | − | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marmo, P.; Thaller, M.C.; Di Lallo, G.; Henrici De Angelis, L.; Poerio, N.; De Santis, F.; Fraziano, M.; Migliore, L.; D’Andrea, M.M. Characterization of vB_StuS_MMDA13, a Newly Discovered Bacteriophage Infecting the Agar-Degrading Species Sphingomonas turrisvirgatae. Viruses 2020, 12, 894. https://doi.org/10.3390/v12080894
Marmo P, Thaller MC, Di Lallo G, Henrici De Angelis L, Poerio N, De Santis F, Fraziano M, Migliore L, D’Andrea MM. Characterization of vB_StuS_MMDA13, a Newly Discovered Bacteriophage Infecting the Agar-Degrading Species Sphingomonas turrisvirgatae. Viruses. 2020; 12(8):894. https://doi.org/10.3390/v12080894
Chicago/Turabian StyleMarmo, Pasquale, Maria Cristina Thaller, Gustavo Di Lallo, Lucia Henrici De Angelis, Noemi Poerio, Federica De Santis, Maurizio Fraziano, Luciana Migliore, and Marco Maria D’Andrea. 2020. "Characterization of vB_StuS_MMDA13, a Newly Discovered Bacteriophage Infecting the Agar-Degrading Species Sphingomonas turrisvirgatae" Viruses 12, no. 8: 894. https://doi.org/10.3390/v12080894