Convolutional Neural Network Based Approach to In Silico Non-Anticipating Prediction of Antigenic Distance for Influenza Virus


Abstract
1. Introduction
1.1. Convolutional Neural Networks
1.2. Related Work
- (i)
- They deal with a point mutation as a single event, while it is widely known that amino acids located at some specific position affects its close, and even not so close (due to the folding) neighbors in the protein sequence.
- (ii)
- Despite the wide engagement of the deep learning principle in biological research, all the known models of the antigenic evolution rely on manual feature engineering.
- (iii)
- Previous research did explicitly take into account the temporal factor, that is, the date/time when a certain virus strain was isolated for the first time. Therefore, all of them were not non-anticipating, since they relied on measurements describing future substitutions.
1.3. Our Contribution
- (i)
- We propose a novel approach for prediction of the antigenic distance based on convolutional neural networks trained in a few-dimensional physicochemical feature space of amino acids, constituting HA sequences of the compared strains of the influenza virus.
- (ii)
- By employing the Grid-Search method for tuning the hyper-parameters of a neural network, we choose the best CNN architecture, and the performance of the obtained model exceeds the well-known SqueezeNet CNN model [49] taken as a baseline both by the performance and number of learnable parameters.
- (iii)
- In addition, relying on experiment scenarios proposed in [18], we evaluate the performance of our best CNN model and show that it provides quite an acceptable prediction quality. All the source code, auxiliary scripts, trained networks, and figures are freely available at https://github.com/ForghaniM/FLU.
2. Materials and Methods
2.1. Data Collection
2.1.1. HI Assay Dataset
2.1.2. Hemagglutinin Sequence
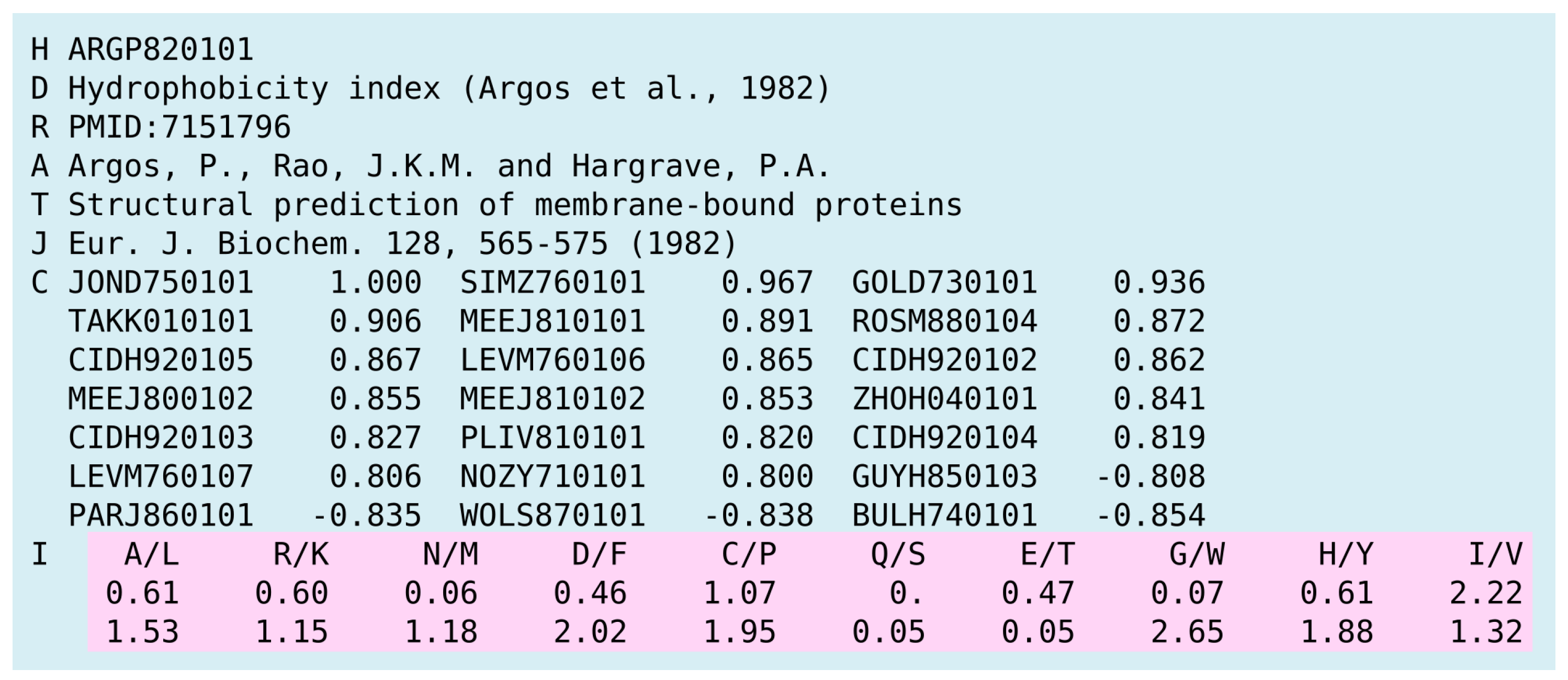
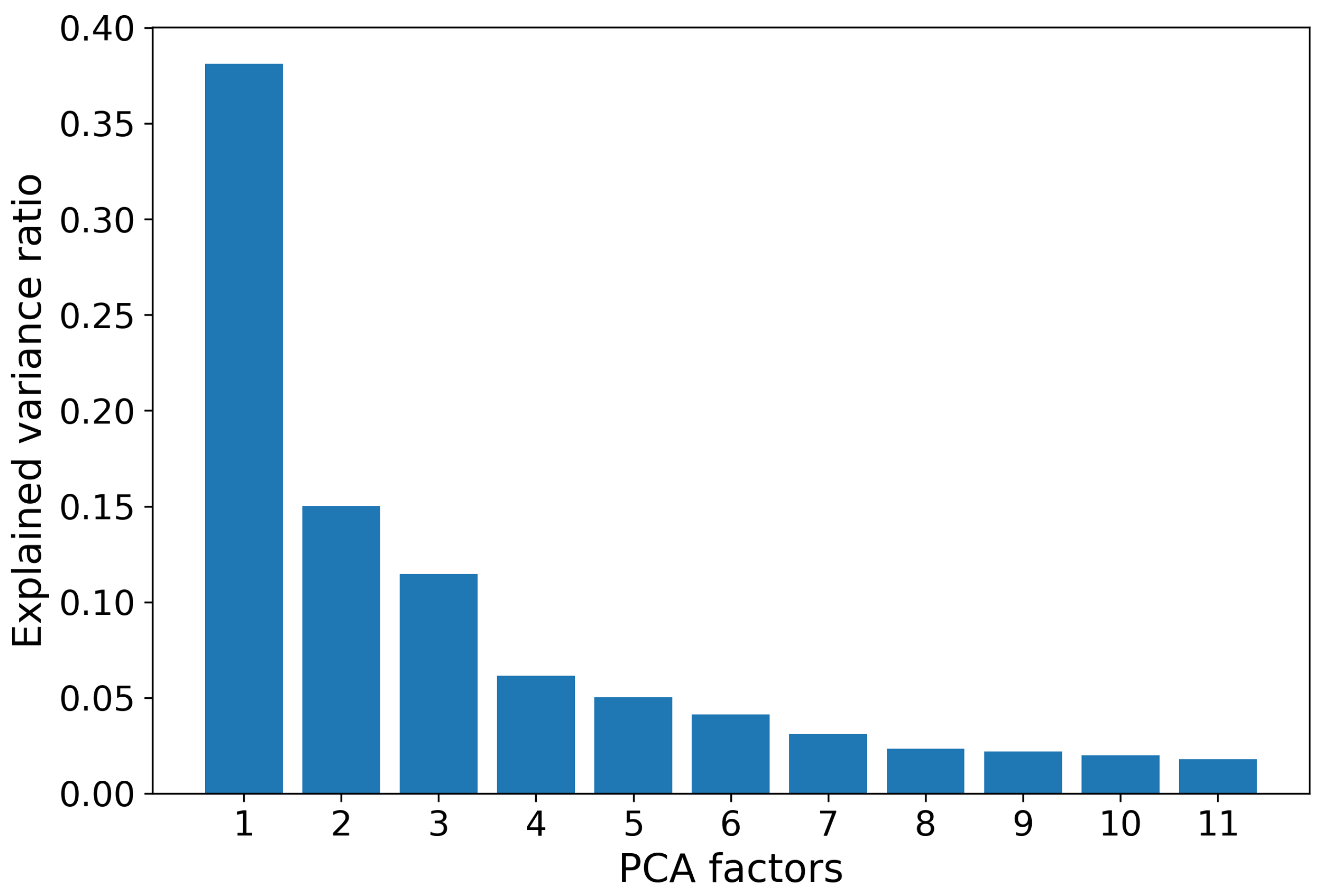
2.2. Amino Acid Sequence Encoding
2.3. Input Tensor Structure
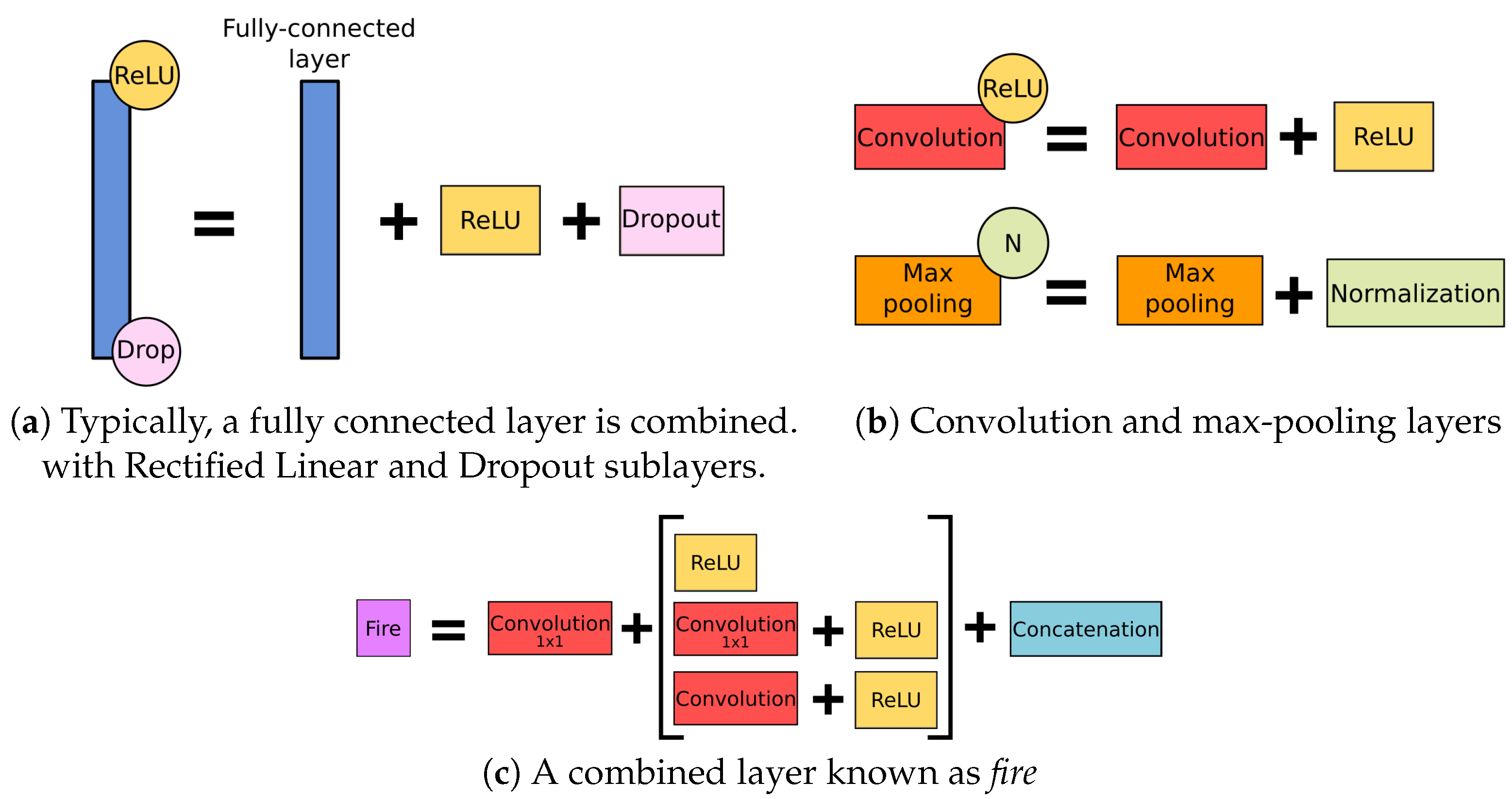
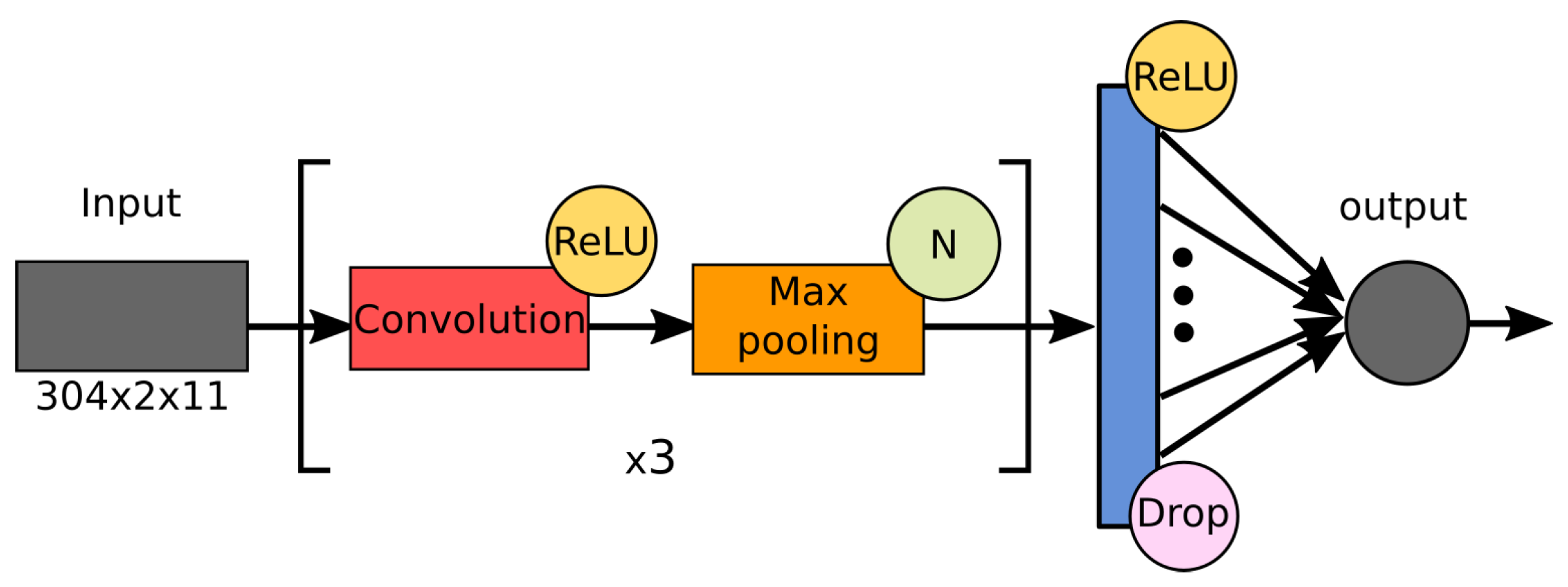
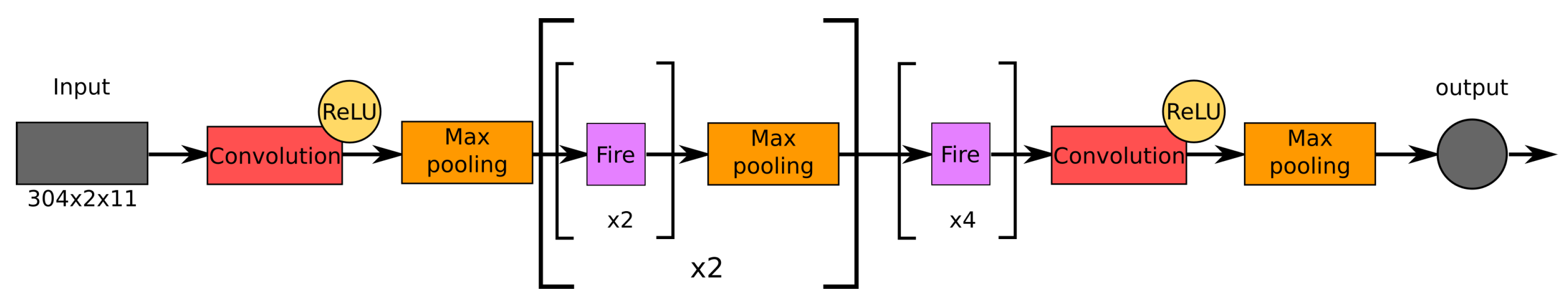
2.4. Architectures of the Examined Networks
2.5. Experimental Design
2.5.1. Temporal Experiments
2.5.2. Static Experiments
3. Results
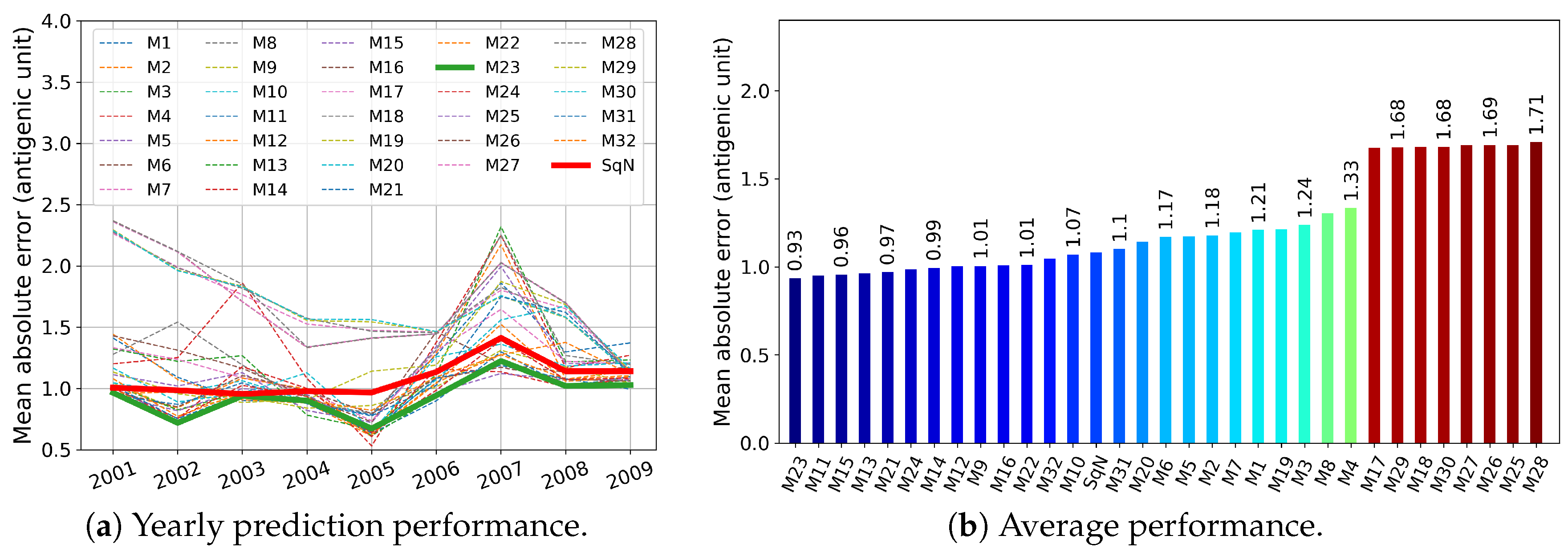
3.1. Temporal Experiments
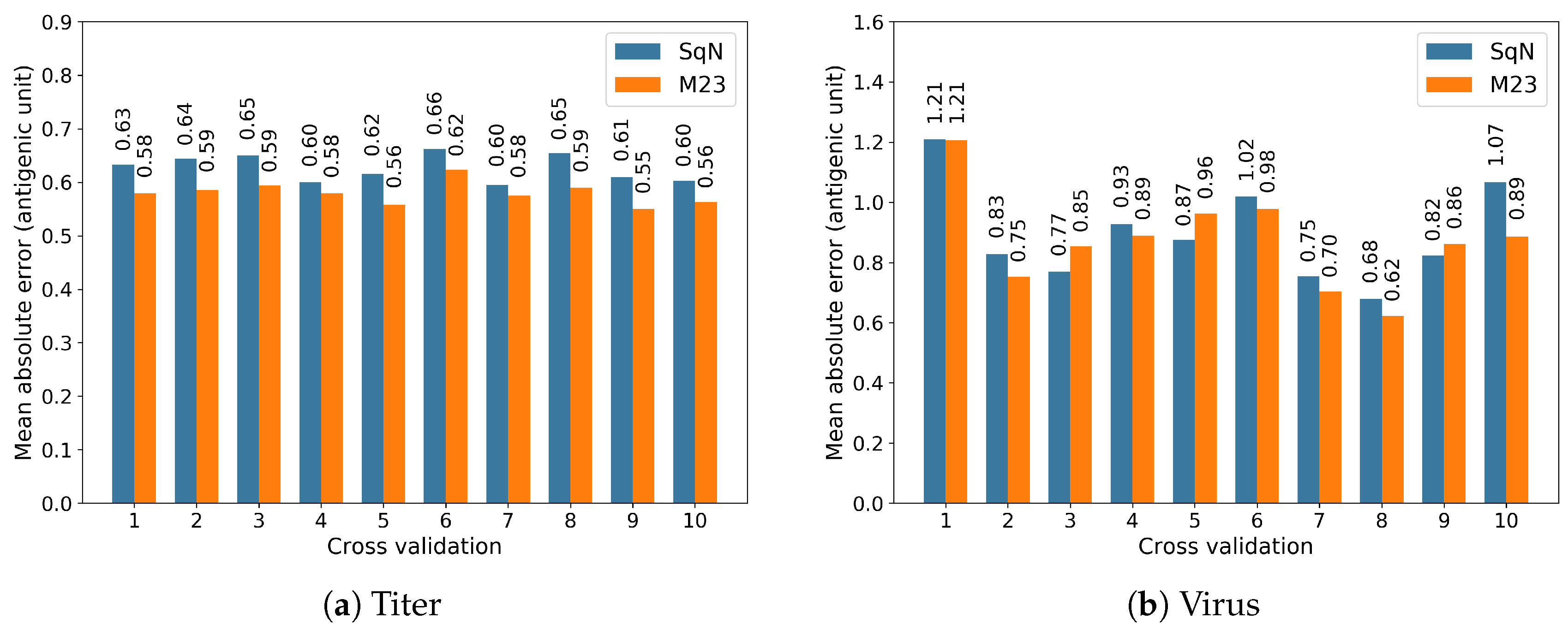
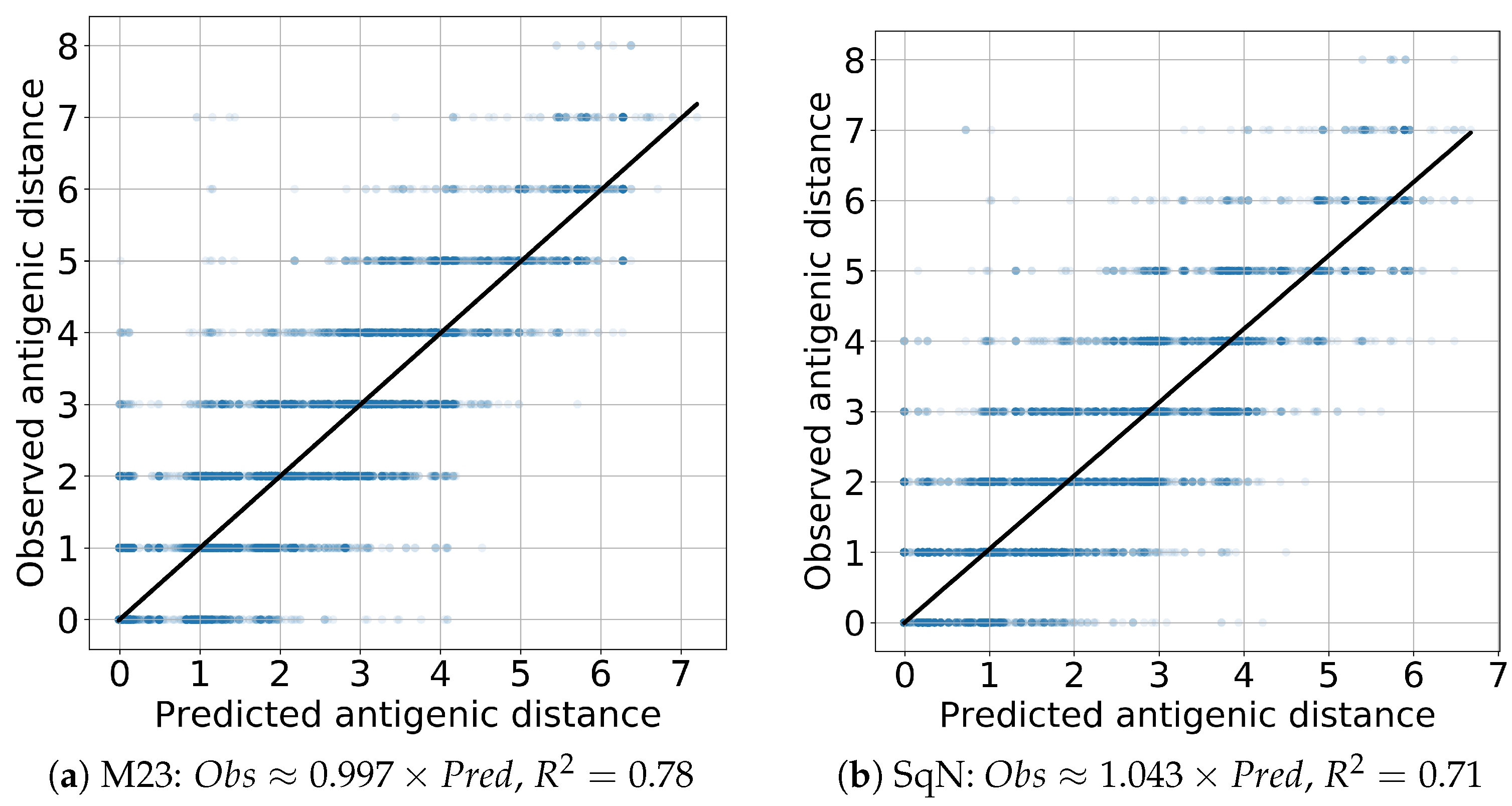
3.2. Static Experiments
Comparison with Previous Results
4. Discussion
- All the models proposed in this paper are fully non-anticipating, that is, they were trained to predict antigenic distances for a given year without taking into account any information concerning future events, such as high-impact substitutions or test virus relationships in a phylogenetic tree. Therefore, all predictions were carried out on the basis of the prehistory exclusively.
- Unless something was a major part of conventional research [18,62], tackling the protein sequences as alphabetic strings, we used a number of physicochemical properties of the constituent amino acids presented in the AAindex1 dataset to encode the HA protein sequence that provides a multi-representation of input genetic data and specifies mutation patterns in a more descriptive way.
- Unlike most papers which adopted manual feature engineering [35,62], for example, those based on prior knowledge about antigenic sites and receptor-binding, our approach relies on the advantage of the convolutional neural network framework to provide fully automatic feature extraction by automatically assigning the most relevant features for prediction of the antigenic distance along with the model training.
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CNN | Convolutional neural network |
| FE | Feature engineering |
| GISAID | Global Initiative on Sharing All Influenza Data |
| GISRS | Global Influenza Surveillance and Response System |
| HA | Hemagglutinin |
| HI | Hemagglutination inhibition |
| MAE | Mean absolute error |
| NA | Neuraminidase |
| PCA | Principal component analysis |
| WHO | World Health Organization |
References
- Agor, J.K.; Özaltın, O.Y. Models for predicting the evolution of influenza to inform vaccine strain selection. Hum. Vaccines Immunother. 2018, 14, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Neher, R.A.; Bedford, T. Nextflu: Real-time tracking of seasonal influenza virus evolution in humans. Bioinformatics 2015, 31, 3546–3548. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Carney, P.J.; Chang, J.C.; Guo, Z.; Villanueva, J.M.; Stevens, J. Structure and receptor-binding preferences of recombinant human A (H3N2) virus hemagglutinins. Virology 2015, 477, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.L.; Yang, J.; Cai, Z.; Zhang, T.; Wan, X.F. AntigenMap 3D: An online antigenic cartography resource. Bioinformatics 2012, 28, 1292–1293. [Google Scholar] [CrossRef]
- Klingen, T.R.; Reimering, S.; Guzmán, C.A.; McHardy, A.C. In silico vaccine strain prediction for human influenza viruses. Trends Microbiol. 2018, 26, 119–131. [Google Scholar] [CrossRef]
- Castro, L.A.; Bedford, T.; Ancel Meyers, L. Early prediction of antigenic transitions for influenza A/H3N2. PLoS Comput. Biol. 2020, 16, 1–23. [Google Scholar] [CrossRef]
- Sylte, M.J.; Suarez, D.L. Influenza neuraminidase as a vaccine antigen. In Vaccines for Pandemic Influenza; Springer: Berlin, Germany, 2009; pp. 227–241. [Google Scholar]
- Ellebedy, A.; Webby, R. Influenza vaccines. Vaccine 2009, 27, D65–D68. [Google Scholar] [CrossRef]
- Smith, D.J.; Lapedes, A.S.; de Jong, J.C.; Bestebroer, T.M.; Rimmelzwaan, G.F.; Osterhaus, A.D.; Fouchier, R.A. Mapping the antigenic and genetic evolution of influenza virus. Science 2004, 305, 371–376. [Google Scholar] [CrossRef]
- Forghani, M.; Khachay, M. Feature Extraction Technique for Prediction the Antigenic Variants of the Influenza Virus. World Acad. Sci. Eng. Technol. Int. Sci. Index 2018, 143, 525–530. [Google Scholar]
- Larson, G.; Thorne, J.L.; Schmidler, S. Incorporating Nearest-Neighbor Site Dependence into Protein Evolution Models. J. Comput. Biol. 2020, 27, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.C. Hemagglutination-inhibition assay for influenza virus subtype identification and the detection and quantitation of serum antibodies to influenza virus. Methods Mol. Biol. 2014, 1161, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Zand, M.S.; Wang, J.; Hilchey, S. Graphical representation of proximity measures for multidimensional data: Classical and metric multidimensional scaling. Math. J. 2015, 17, 7. [Google Scholar] [PubMed]
- Kratsch, C.; Klingen, T.R.; Mümken, L.; Steinbrück, L.; McHardy, A.C. Determination of antigenicity-altering patches on the major surface protein of human influenza A/H3N2 viruses. Virus Evol. 2016, 2. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smith, D.J.; Forrest, S.; Ackley, D.H.; Perelson, A.S. Variable efficacy of repeated annual influenza vaccination. Proc. Natl. Acad. Sci. USA 1999, 96, 14001–14006. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Chen, J.S.E. Predicting antigenic variants of influenza A/H3N2 viruses. Emerg. Infect. Dis. 2004, 10, 1385. [Google Scholar] [CrossRef]
- Neher, R.A.; Bedford, T.; Daniels, R.S.; Russell, C.A.; Shraiman, B.I. Prediction, dynamics, and visualization of antigenic phenotypes of seasonal influenza viruses. Proc. Natl. Acad. Sci. USA 2016, 113, E1701–E1709. [Google Scholar] [CrossRef] [PubMed]
- De Jong, J.; Smith, D.J.; Lapedes, A.; Donatelli, I.; Campitelli, L.; Barigazzi, G.; Van Reeth, K.; Jones, T.; Rimmelzwaan, G.; Osterhaus, A.; et al. Antigenic and genetic evolution of swine influenza A (H3N2) viruses in Europe. J. Virol. 2007, 81, 4315–4322. [Google Scholar] [CrossRef]
- Gupta, V.; Earl, D.J.; Deem, M.W. Quantifying influenza vaccine efficacy and antigenic distance. Vaccine 2006, 24, 3881–3888. [Google Scholar] [CrossRef]
- Lecun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef]
- Hubel, D.H.; Wiesel, T.N. Receptive fields and functional architecture of monkey striate cortex. J. Physiol. 1968, 195, 215–243. [Google Scholar] [CrossRef] [PubMed]
- Rawat, W.; Wang, Z. Deep convolutional neural networks for image classification: A comprehensive review. Neural Comput. 2017, 29, 2352–2449. [Google Scholar] [CrossRef] [PubMed]
- Mazurov, V.; Khachai, M. Committees of systems of linear inequalities. Autom. Remote Control 2004, 65, 193–203. [Google Scholar] [CrossRef]
- Khachai, M. Computational and approximational complexity of combinatorial problems related to the committee polyhedral separability of finite sets. Pattern Recognit. Image Anal. 2008, 18, 236–242. [Google Scholar] [CrossRef]
- Khachay, M. Committee polyhedral separability: Complexity and polynomial approximation. Mach. Learn. 2015, 101, 231–251. [Google Scholar] [CrossRef]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; Adaptive Computation and Machine Learning Series; The MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- He, J.; Deem, M.W. Low-dimensional clustering detects incipient dominant influenza strain clusters. Protein Eng. Des. Sel. 2010, 23, 935–946. [Google Scholar] [CrossRef]
- Pan, K.; Subieta, K.C.; Deem, M.W. A novel sequence-based antigenic distance measure for H1N1, with application to vaccine effectiveness and the selection of vaccine strains. Protein Eng. Des. Sel. 2011, 24, 291–299. [Google Scholar] [CrossRef]
- Skarlupka, A.L.; Handel, A.; Ross, T.M. Influenza hemagglutinin antigenic distance measures capture trends in HAI differences and infection outcomes, but are not suitable predictive tools. Vaccine 2020, 38, 5822–5830. [Google Scholar] [CrossRef]
- Du, X.; Dong, L.; Lan, Y.; Peng, Y.; Wu, A.; Zhang, Y.; Huang, W.; Wang, D.; Wang, M.; Guo, Y.; et al. Mapping of H3N2 influenza antigenic evolution in China reveals a strategy for vaccine strain recommendation. Nat. Commun. 2012, 3, 1–9. [Google Scholar] [CrossRef]
- Liu, M.; Zhao, X.; Hua, S.; Du, X.; Peng, Y.; Li, X.; Lan, Y.; Wang, D.; Wu, A.; Shu, Y.; et al. Antigenic patterns and evolution of the human influenza A (H1N1) virus. Sci. Rep. 2015, 5, 14171. [Google Scholar] [CrossRef]
- Yao, Y.; Li, X.; Liao, B.; Huang, L.; He, P.; Wang, F.; Yang, J.; Sun, H.; Zhao, Y.; Yang, J. Predicting influenza antigenicity from Hemagglutintin sequence data based on a joint random forest method. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhu, W.; Liao, B.; Cai, L.; Peng, L.; Yang, J. Predicting Influenza Antigenicity by Matrix Completion With Antigen and Antiserum Similarity. Front. Microbiol. 2018, 9, 2500. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Wei, X.; Huang, Y.; Hu, B.; Fang, Y.; Wang, J. Using multiple linear regression and physicochemical changes of amino acid mutations to predict antigenic variants of influenza A/H3N2 viruses. Bio-Med. Mater. Eng. 2014, 24, 3729–3735. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2007, 36, D202–D205. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y. Selecting vaccine strains for H3N2 human influenza A virus. Meta Gene 2015, 4, 64–72. [Google Scholar] [CrossRef]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Briefings Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef]
- Spencer, M.; Eickholt, J.; Cheng, J. A deep learning network approach to ab initio protein secondary structure prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 12, 103–112. [Google Scholar] [CrossRef]
- Lee, B.; Lee, T.; Na, B.; Yoon, S. DNA-level splice junction prediction using deep recurrent neural networks. arXiv 2015, arXiv:1512.05135. [Google Scholar]
- Park, S.; Min, S.; Choi, H.; Yoon, S. deepMiRGene: Deep neural network based precursor microrna prediction. arXiv 2016, arXiv:1605.00017. [Google Scholar]
- Lee, B.; Baek, J.; Park, S.; Yoon, S. deepTarget: End-to-end learning framework for microRNA target prediction using deep recurrent neural networks. In Proceedings of the 7th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Washington, DC, USA, 2–5 October 2016; pp. 434–442. [Google Scholar]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef]
- Lanchantin, J.; Singh, R.; Lin, Z.; Qi, Y. Deep motif: Visualizing genomic sequence classifications. arXiv 2016, arXiv:1605.01133. [Google Scholar]
- Yin, R.; Luusua, E.; Dabrowski, J.; Zhang, Y.; Kwoh, C.K. Tempel: Time-series mutation prediction of influenza A viruses via attention-based recurrent neural networks. Bioinformatics 2020, 36, 2697–2704. [Google Scholar] [CrossRef] [PubMed]
- Chadha, A.; Dara, R.; Poljak, Z. Convolutional Classification of Pathogenicity in H5 Avian Influenza Strains. In Proceedings of the 2019 18th IEEE International Conference On Machine Learning And Applications (ICMLA), Boca Raton, FL, USA, 16–19 December 2019; pp. 1570–1577. [Google Scholar]
- Papastefanopoulos, V.; Linardatos, P.; Kotsiantis, S. COVID-19: A Comparison of Time Series Methods to Forecast Percentage of Active Cases per Population. Appl. Sci. 2020, 10, 3880. [Google Scholar] [CrossRef]
- Cai, Y.d.; Lin, S.L. Support vector machines for predicting rRNA-, RNA-, and DNA-binding proteins from amino acid sequence. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2003, 1648, 127–133. [Google Scholar] [CrossRef]
- Iandola, F.N.; Moskewicz, M.W.; Ashraf, K.; Han, S.; Dally, W.J.; Keutzer, K. SqueezeNet: AlexNet-level accuracy with 50x fewer parameters and <1MB model size. arXiv 2016, arXiv:1602.07360. [Google Scholar]
- Gregory, V.; Harvey, W.; Daniels, R.S.; Reeve, R.; Whittaker, L.; Halai, C.; Douglas, A.; Gonsalves, R.; Skehel, J.J.; Hay, A.J.; et al. Human former Seasonal Influenza A (H1N1) Haemagglutination Inhibition Data 1977–2009 from the WHO Collaborating Centre for Reference and Research on Influenza, London, UK. 2016. [Google Scholar]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Harvey, W.T. Quantifying the Genetic Basis of Antigenic Variation among Human Influenza A Viruses. Ph.D. Thesis, University of Glasgow, Glasgow, UK, 2016. [Google Scholar]
- Shi, W.; Bao, S.; Tan, D. FFESSD: An Accurate and Efficient Single-Shot Detector for Target Detection. Appl. Sci. 2019, 9, 4276. [Google Scholar] [CrossRef]
- Shelhamer, E.; Long, J.; Darrell, T. Fully Convolutional Networks for Semantic Segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 2017, 39, 640–651. [Google Scholar] [CrossRef]
- Baklanov, A.; Khachay, M.; Pasynkov, M. Application of Fully Convolutional Neural Networks to Mapping Industrial Oil Palm Plantations. In Analysis of Images, Social Networks and Texts; van der Aalst, W.M.P., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 155–167. [Google Scholar]
- Long, W.; Lu, Z.; Cui, L. Deep learning-based feature engineering for stock price movement prediction. Knowl. Based Syst. 2019, 164, 163–173. [Google Scholar] [CrossRef]
- Alom, M.Z.; Taha, T.M.; Yakopcic, C.; Westberg, S.; Sidike, P.; Nasrin, M.S.; Van Esesn, B.C.; Awwal, A.A.S.; Asari, V.K. The history began from alexnet: A comprehensive survey on deep learning approaches. arXiv 2018, arXiv:1803.01164. [Google Scholar]
- Russakovsky, O.; Deng, J.; Su, H.; Krause, J.; Satheesh, S.; Ma, S.; Huang, Z.; Karpathy, A.; Khosla, A.; Bernstein, M.; et al. ImageNet Large Scale Visual Recognition Challenge. Int. J. Comput. Vis. (IJCV) 2015, 115, 211–252. [Google Scholar] [CrossRef]
- Jia, Y.; Shelhamer, E.; Donahue, J.; Karayev, S.; Long, J.; Girshick, R.; Guadarrama, S.; Darrell, T. Caffe: Convolutional Architecture for Fast Feature Embedding. In Proceedings of the 22nd ACM International Conference on Multimedia; Association for Computing Machinery: New York, NY, USA, 2014; pp. 675–678. [Google Scholar] [CrossRef]
- Bedford, T.; Suchard, M.A.; Lemey, P.; Dudas, G.; Gregory, V.; Hay, A.J.; McCauley, J.W.; Russell, C.A.; Smith, D.J.; Rambaut, A. Integrating influenza antigenic dynamics with molecular evolution. eLife 2014, 3, e01914. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Benton, D.J.; Gregory, V.; Hall, J.P.; Daniels, R.S.; Bedford, T.; Haydon, D.T.; Hay, A.J.; McCauley, J.W.; Reeve, R. Identification of low-and high-impact hemagglutinin amino acid substitutions that drive antigenic drift of influenza A (H1N1) viruses. PLoS Pathog. 2016, 12, e1005526. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Huang, Z.; Mao, Y.; Tao, S. Neighbor preferences of amino acids and context-dependent effects of amino acid substitutions in human, mouse, and dog. Int. J. Mol. Sci. 2014, 15, 15963–15980. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F. Vaccination and antigenic drift in influenza. Vaccine 2008, 26, C8–C14. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
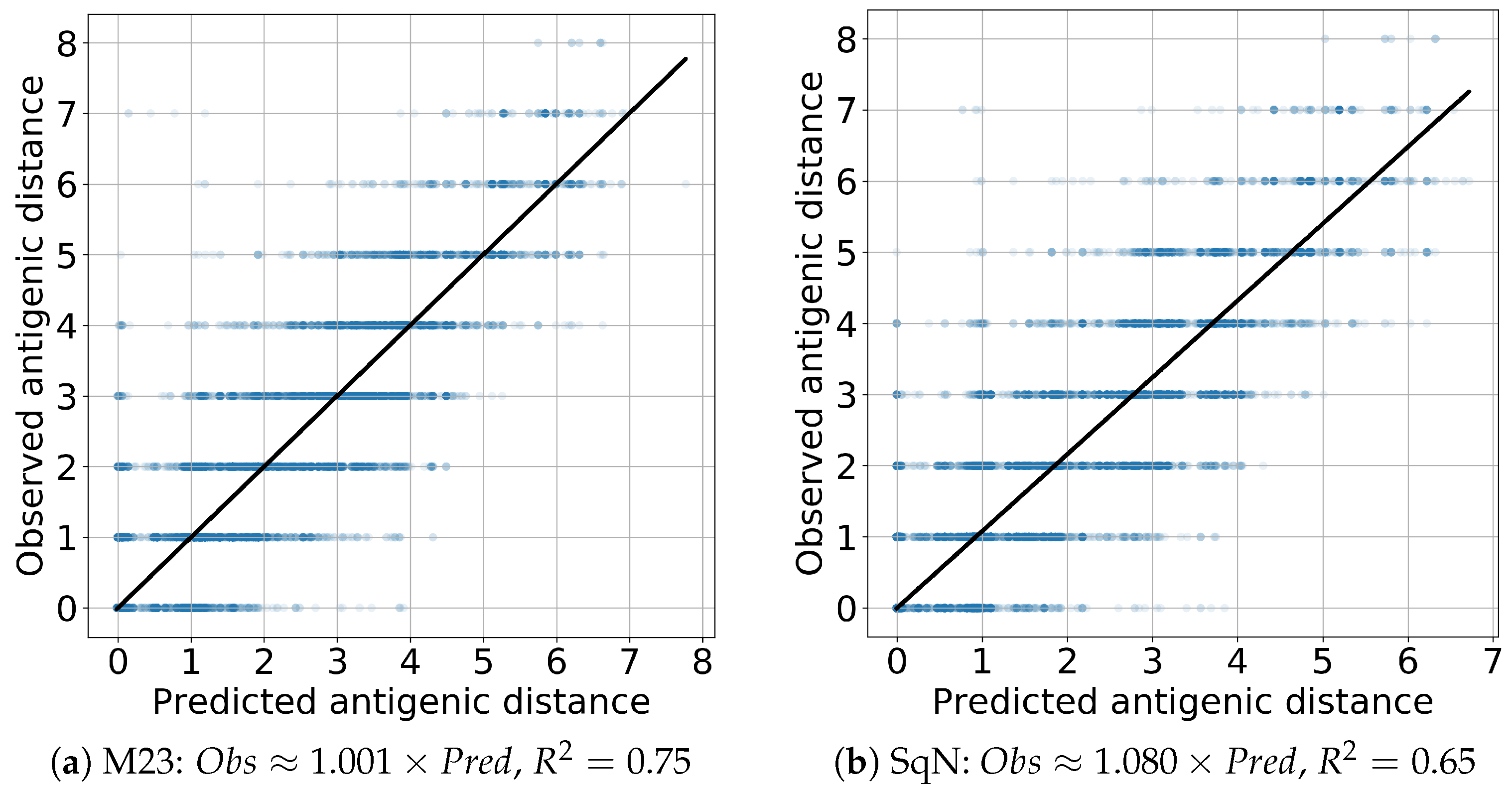{kind=link}
| Epitope Name | Sub-Domain |
|---|---|
| antigenic site Ca | 137, 138, 139, 140, 141, 142, 166, 167, 168, 169, 170, 203, 204, 205, 221, 222, 235, 236, 237 |
| antigenic site Cb | 69, 70, 71, 72, 73, 74 |
| antigenic site Sa | 124, 125, 153, 154, 155, 156, 157, 159, 160, 161, 162, 163, 164 |
| antigenic site Sb | 184, 185, 186, 187, 188, 189, 190, 191, 192, 193, 194, 195 |
| receptor-binding site | 94, 131, 133, 150, 152, 180, 187, 191, 223, 225 |
| Model Name | Number of Convolution Layers | Number of Kernels | Kernel Size | Total Number of Parameters (K = 1000, M = 1,000,000) |
|---|---|---|---|---|
| M1 | 1 | 32 | 109 K | |
| M2 | 1 | 32 | 72 K | |
| M3 | 1 | 64 | 428 K | |
| M4 | 1 | 64 | 274 K | |
| M5 | 1 | 128 | 1.7 M | |
| M6 | 1 | 128 | 1.1 M | |
| M7 | 1 | 256 | 6.7 M | |
| M8 | 1 | 256 | 4.2 M | |
| M9 | 2 | 32 | , | 29 K |
| M10 | 2 | 32 | , | 24 K |
| M11 | 2 | 64 | , | 104 K |
| M12 | 2 | 64 | , | 81 K |
| M13 | 2 | 128 | , | 397 K |
| M14 | 2 | 128 | , | 293 K |
| M15 | 2 | 256 | , | 1.5 M |
| M16 | 2 | 256 | , | 1.1 M |
| M17 | 3 | 32 | , | 19 K |
| M18 | 3 | 32 | , | 23 K |
| M19 | 3 | 64 | , | 67 K |
| M20 | 3 | 64 | , | 77 K |
| M21 | 3 | 128 | , | 250 K |
| M22 | 3 | 128 | , | 277 K |
| M23 | 3 | 256 | , | 957 K |
| M24 | 3 | 256 | , | 1 M |
| M25 | 4 | 32 | , , | 19 K |
| M26 | 4 | 32 | , , | 26 K |
| M27 | 4 | 64 | , , | 67 K |
| M28 | 4 | 64 | , , | 89 K |
| M29 | 4 | 128 | , , | 249 K |
| M30 | 4 | 128 | , , | 326 K |
| M31 | 4 | 256 | , , | 957 K |
| M32 | 4 | 256 | , , | 1.2 M |
| Model Name | 2001 | 2002 | 2003 | 2004 | 2005 | 2006 | 2007 | 2008 | 2009 | Mean | STD |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Experiment : full prehistory | |||||||||||
| M23 | 0.965 | 0.719 | 0.939 | 0.900 | 0.673 | 0.943 | 1.224 | 1.020 | 1.280 | 0.935 | 0.165 |
| M11 | 1.030 | 0.713 | 0.961 | 0.910 | 0.633 | 0.900 | 1.299 | 1.013 | 1.097 | 0.951 | 0.198 |
| M15 | 0.947 | 0.818 | 0.999 | 0.950 | 0.655 | 0.967 | 1.122 | 1.068 | 1.070 | 0.955 | 0.143 |
| M13 | 1.027 | 0.825 | 0.971 | 0.948 | 0.615 | 0.935 | 1.241 | 1.034 | 1.065 | 0.962 | 0.172 |
| M21 | 0.945 | 0.870 | 1.046 | 0.876 | 0.645 | 1.084 | 1.187 | 1.079 | 0.994 | 0.970 | 0.159 |
| SqN | 1.007 | 0.985 | 0.955 | 0.978 | 0.967 | 1.135 | 1.413 | 1.140 | 1.141 | 1.080 | 0.139 |
| Experiment : five years | |||||||||||
| M23 | 0.890 | 0.836 | 0.964 | 0.924 | 0.657 | 1.039 | 1.186 | 1.092 | 1.134 | 0.970 | 0.165 |
| M13 | 1.032 | 0.887 | 1.063 | 0.919 | 0.656 | 0.941 | 1.135 | 1.074 | 1.080 | 0.976 | 0.146 |
| M10 | 1.004 | 1.034 | 1.008 | 0.962 | 0.528 | 0.943 | 1.197 | 1.119 | 1.005 | 0.978 | 0.186 |
| M15 | 0.962 | 0.975 | 1.034 | 0.911 | 0.633 | 0.990 | 1.170 | 1.082 | 1.127 | 0.987 | 0.157 |
| SqN | 0.870 | 0.830 | 0.994 | 0.985 | 0.590 | 1.048 | 1.447 | 1.061 | 1.128 | 0.995 | 0.220 |
| Experiment : four years | |||||||||||
| SqN | 0.877 | 1.110 | 0.949 | 0.887 | 0.618 | 0.882 | 1.330 | 1.133 | 1.038 | 0.981 | 0.191 |
| M24 | 0.992 | 0.798 | 0.975 | 0.921 | 0.893 | 1.054 | 1.519 | 1.077 | 1.129 | 1.040 | 0.206 |
| M10 | 0.992 | 0.986 | 1.102 | 0.926 | 0.644 | 1.090 | 1.556 | 1.066 | 1.025 | 1.043 | 0.237 |
| M16 | 1.029 | 0.780 | 1.067 | 0.933 | 0.871 | 1.054 | 1.465 | 1.105 | 1.147 | 1.050 | 0.195 |
| M15 | 1.003 | 0.834 | 0.957 | 0.944 | 0.771 | 0.954 | 1.796 | 1.090 | 1.143 | 1.055 | 0.301 |
| Experiment : three years | |||||||||||
| SqN | 1.008 | 0.961 | 0.982 | 0.978 | 0.631 | 1.166 | 1.549 | 1.251 | 1.084 | 1.068 | 0.235 |
| M16 | 0.945 | 0.748 | 0.985 | 0.934 | 0.850 | 1.281 | 1.618 | 1.149 | 1.125 | 1.070 | 0.262 |
| M24 | 0.950 | 0.830 | 0.959 | 0.942 | 1.044 | 1.301 | 1.476 | 1.067 | 1.072 | 1.071 | 0.200 |
| M23 | 0.924 | 0.807 | 0.928 | 0.945 | 0.869 | 1.290 | 1.671 | 1.114 | 1.166 | 1.079 | 0.271 |
| M9 | 1.015 | 0.813 | 0.102 | 0.959 | 0.643 | 0.319 | 1.788 | 1.080 | 1.025 | 1.085 | 0.323 |
| Model Name | Average MAE | STD |
|---|---|---|
| Titer | ||
| M23 | 0.58 | 0.020 |
| SqN | 0.627 | 0.024 |
| Virus | ||
| M23 | 0.871 | 0.154 |
| SqN | 0.895 | 0.154 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forghani, M.; Khachay, M. Convolutional Neural Network Based Approach to In Silico Non-Anticipating Prediction of Antigenic Distance for Influenza Virus. Viruses 2020, 12, 1019. https://doi.org/10.3390/v12091019
Forghani M, Khachay M. Convolutional Neural Network Based Approach to In Silico Non-Anticipating Prediction of Antigenic Distance for Influenza Virus. Viruses. 2020; 12(9):1019. https://doi.org/10.3390/v12091019
Chicago/Turabian StyleForghani, Majid, and Michael Khachay. 2020. "Convolutional Neural Network Based Approach to In Silico Non-Anticipating Prediction of Antigenic Distance for Influenza Virus" Viruses 12, no. 9: 1019. https://doi.org/10.3390/v12091019
APA StyleForghani, M., & Khachay, M. (2020). Convolutional Neural Network Based Approach to In Silico Non-Anticipating Prediction of Antigenic Distance for Influenza Virus. Viruses, 12(9), 1019. https://doi.org/10.3390/v12091019