Development of a Dual Fluorescent Microsphere Immunological Assay for Detection of Pseudorabies Virus gE and gB IgG Antibodies


Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Strain, Serum Samples, and Antibodies
2.2. Expression of Recombinant PRV gE and gB Protein
2.3. Coupling of Recombinant Proteins to Fluorescent-Encoded Microspheres
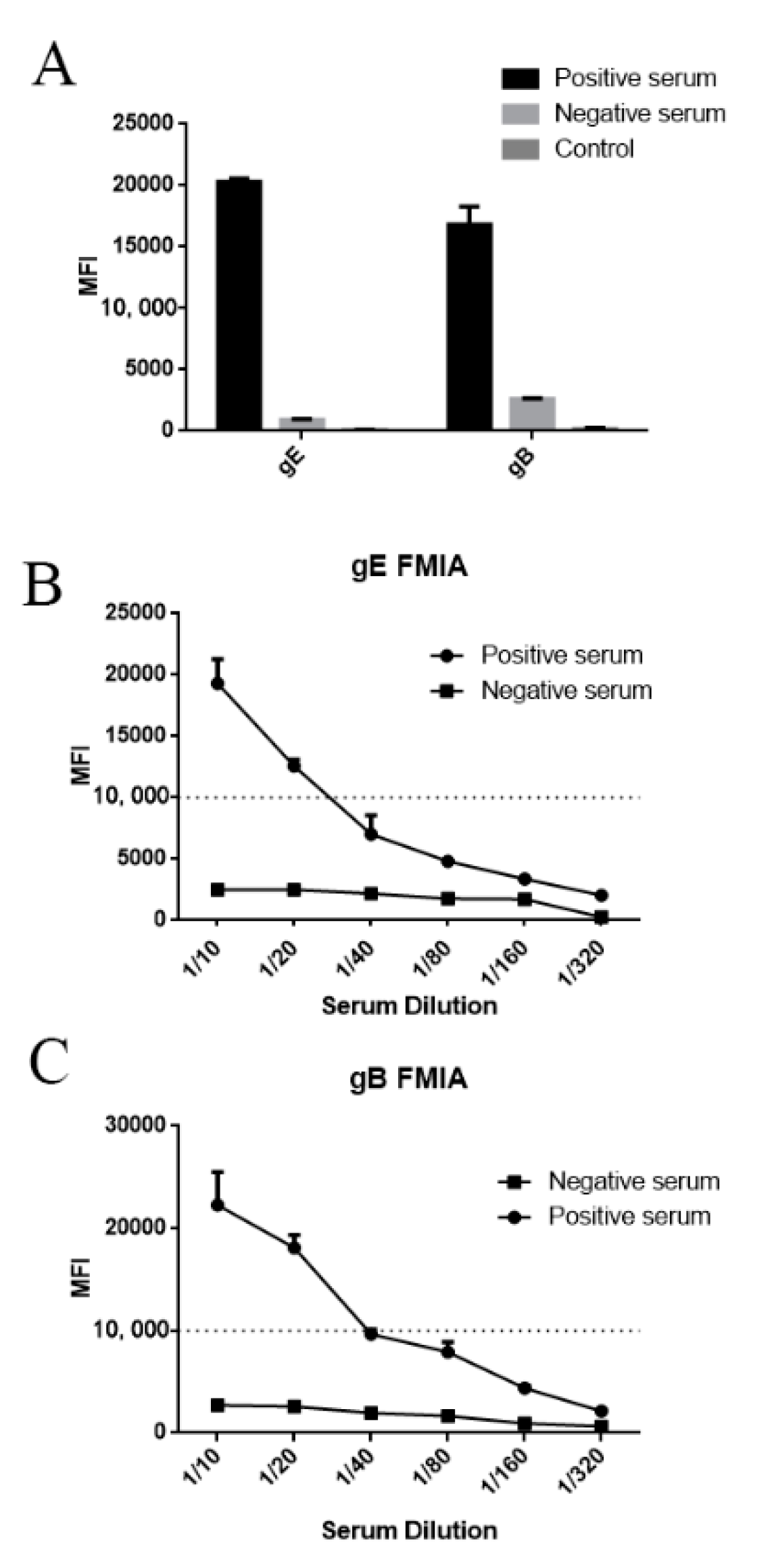
2.4. Selection of Optimal Serum Dilution
2.5. Establishment and Evaluation of Dual FMIA
2.6. Comparison with ELISA
2.7. Statistical Analysis
3. Results
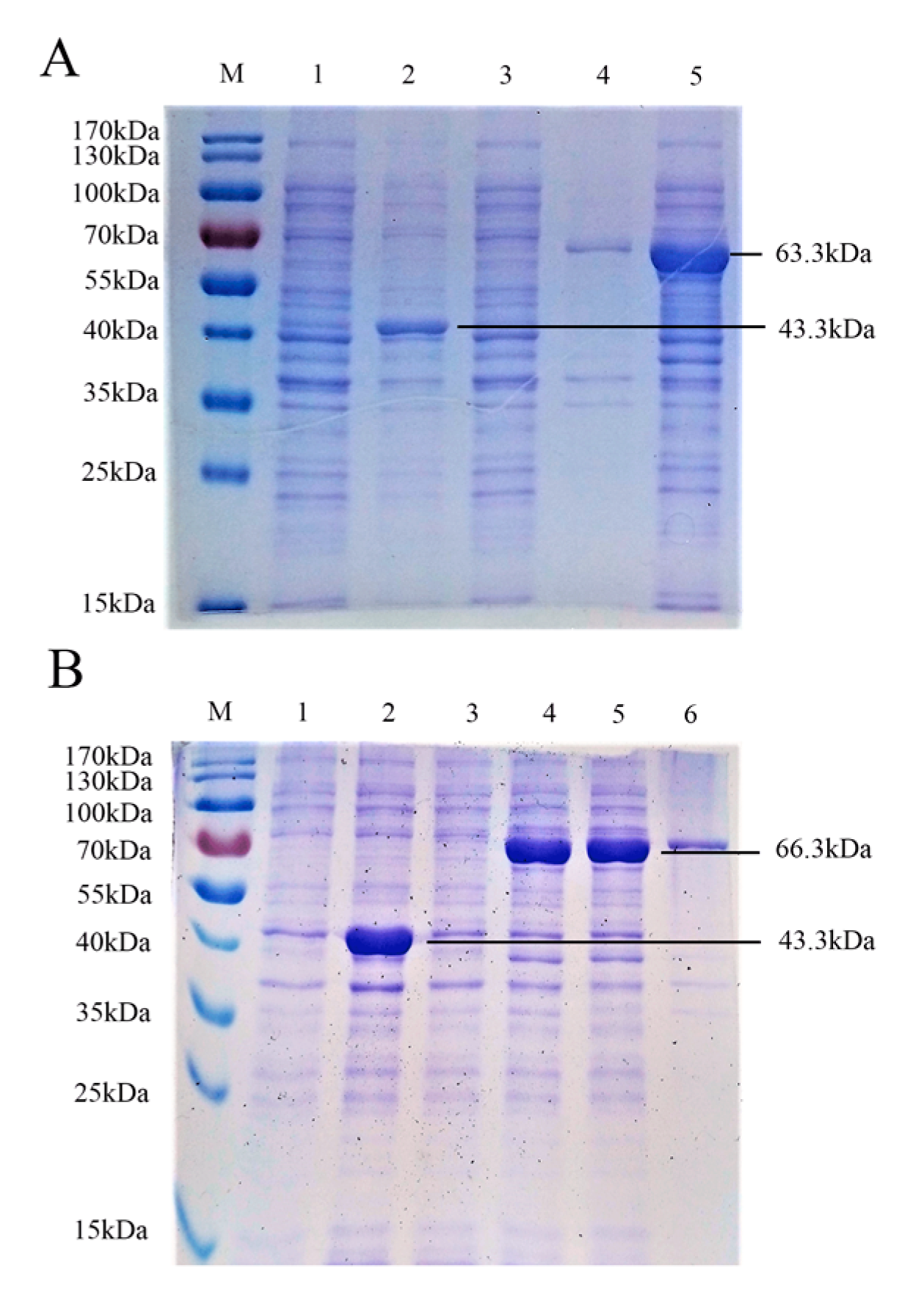
3.1. Expression and Verification of Recombinant PRV gE and gB Proteins
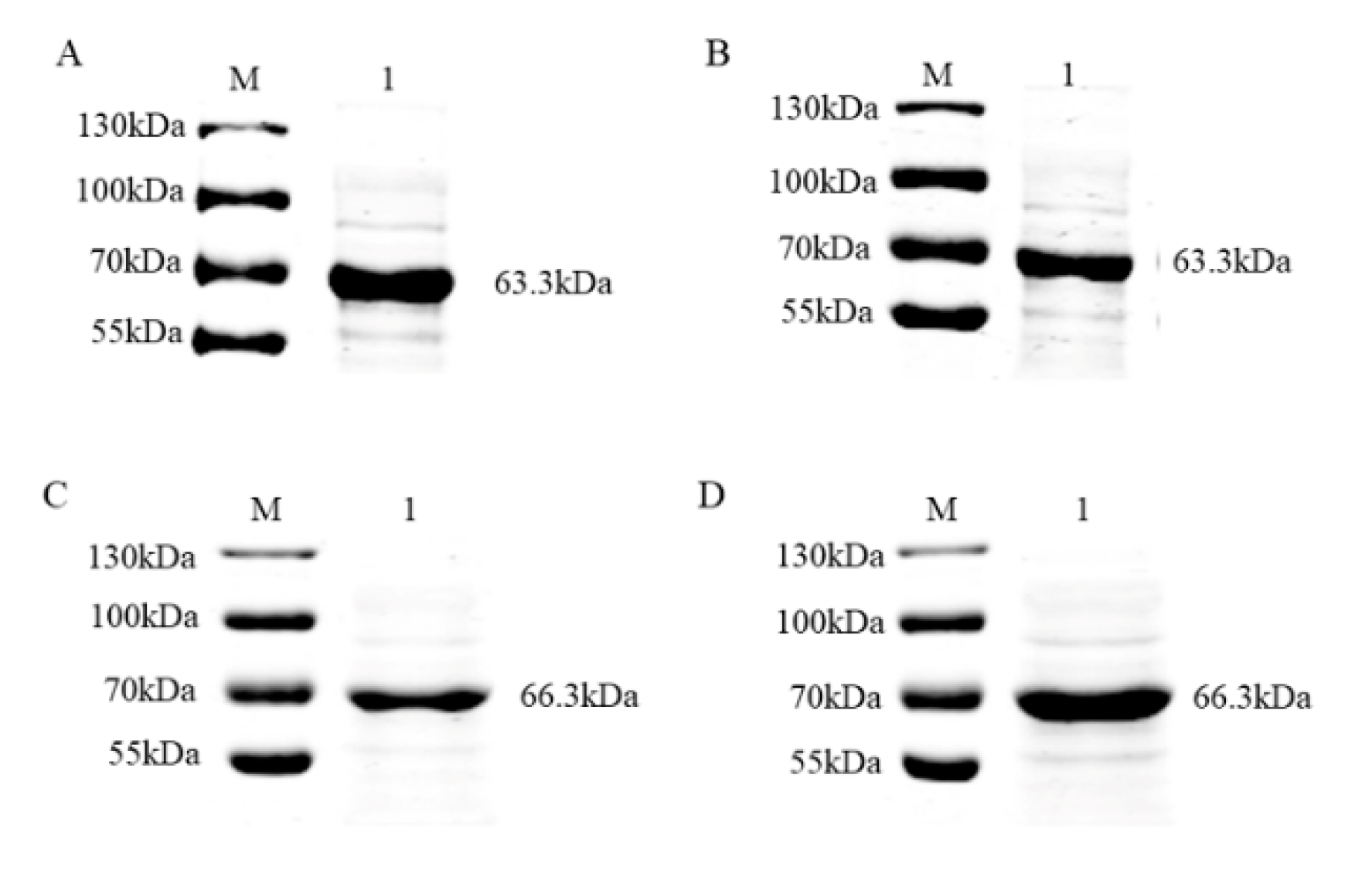
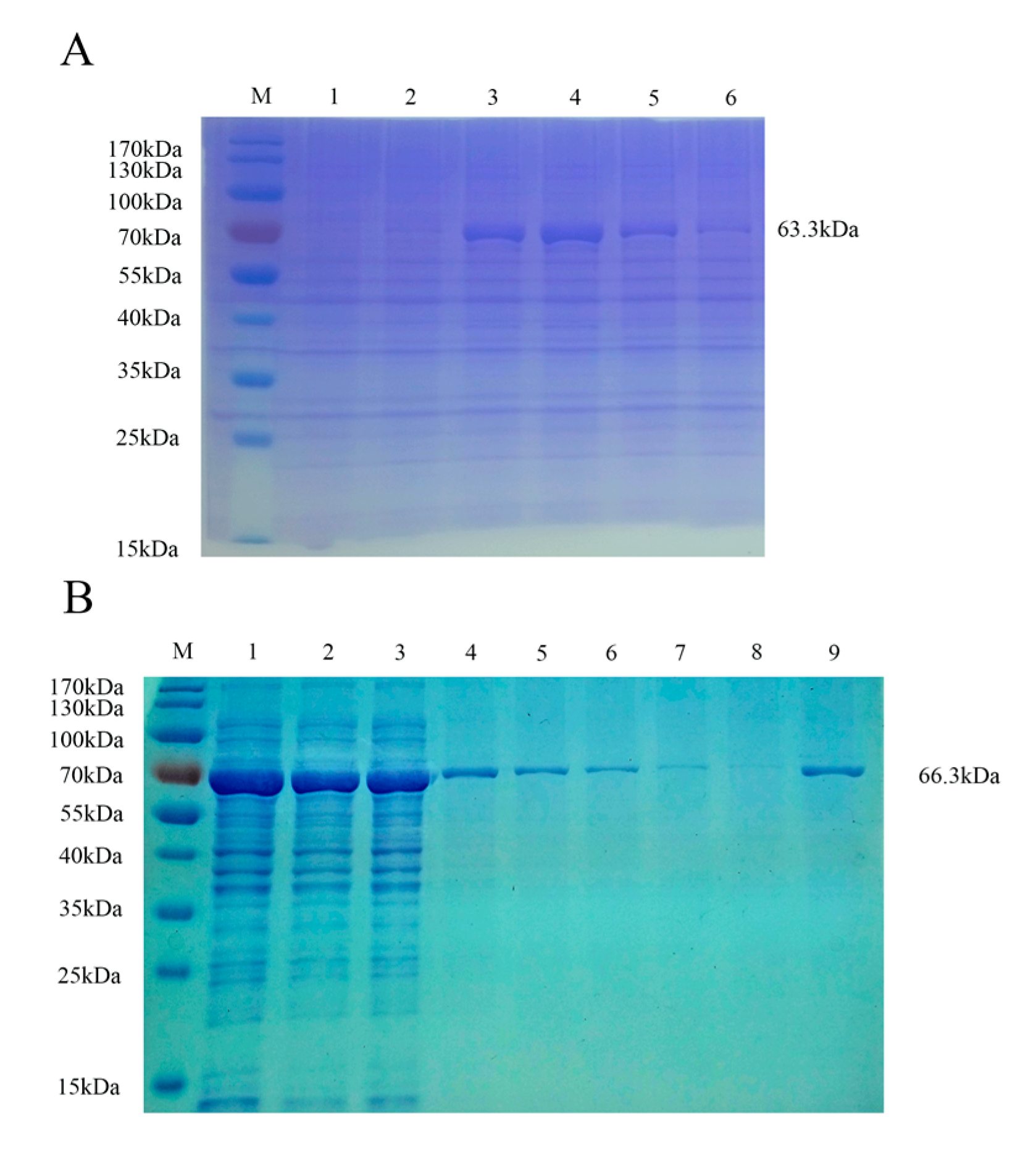
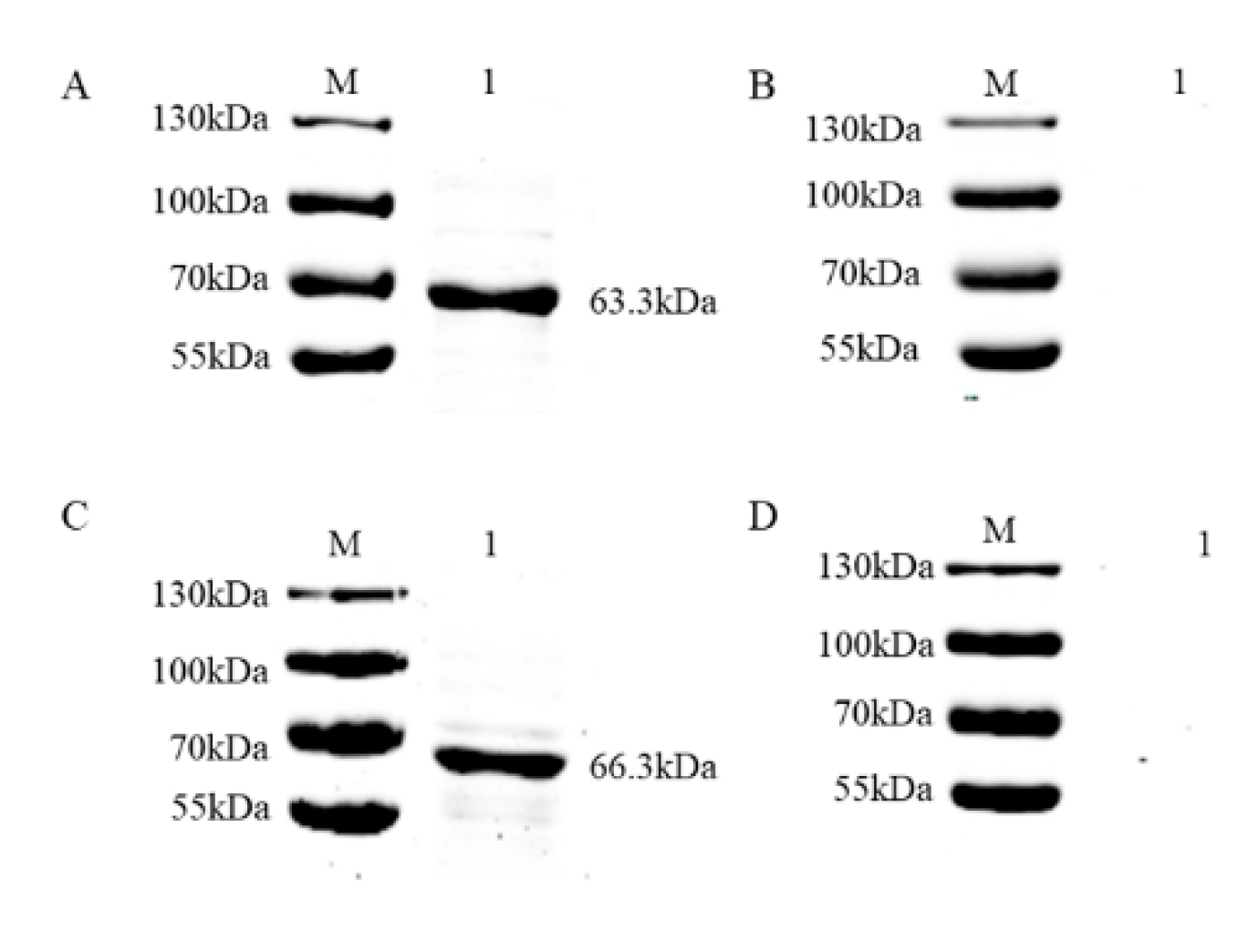
3.2. Purification and Verification of the Antigenicity of the Recombinant Proteins
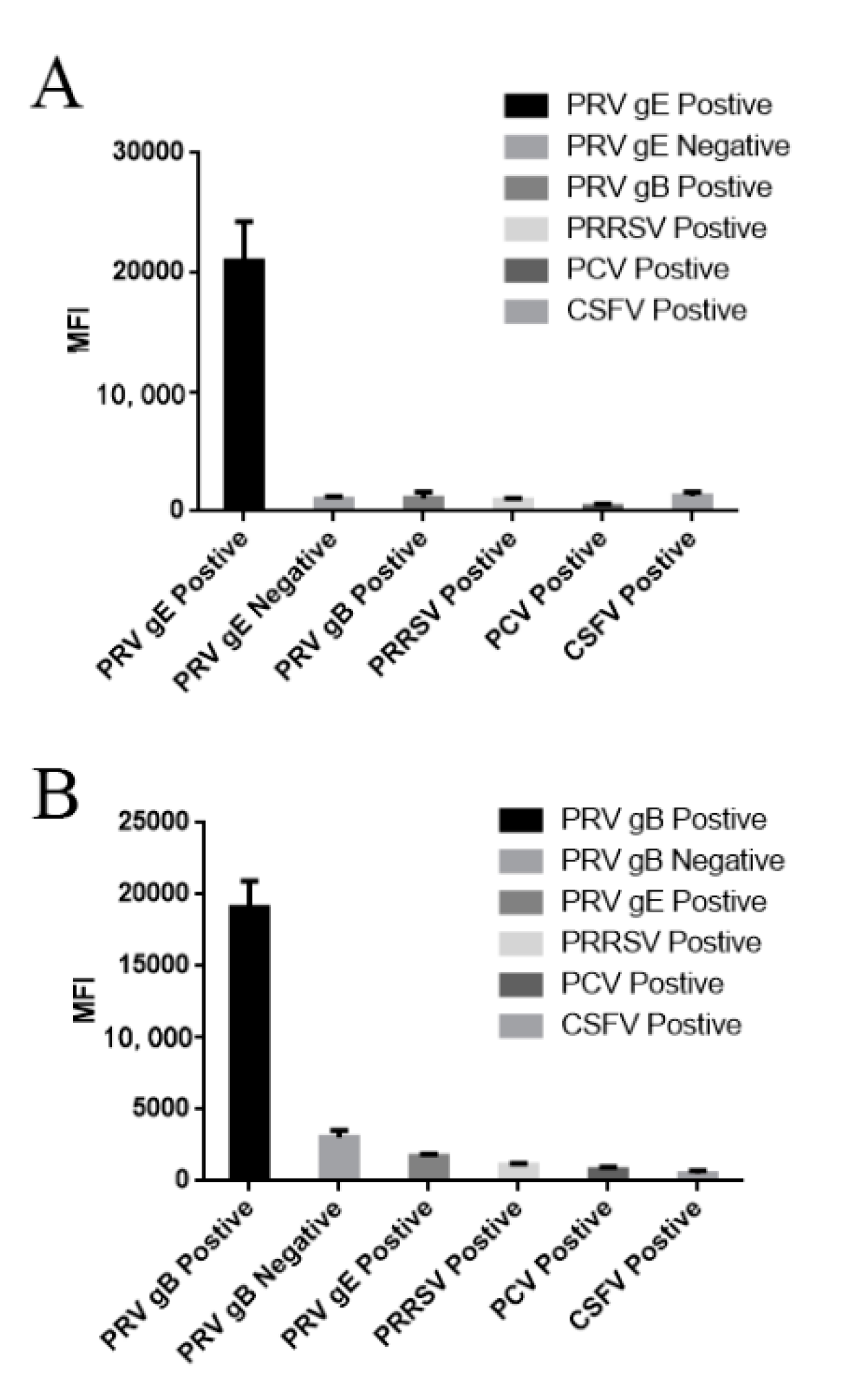
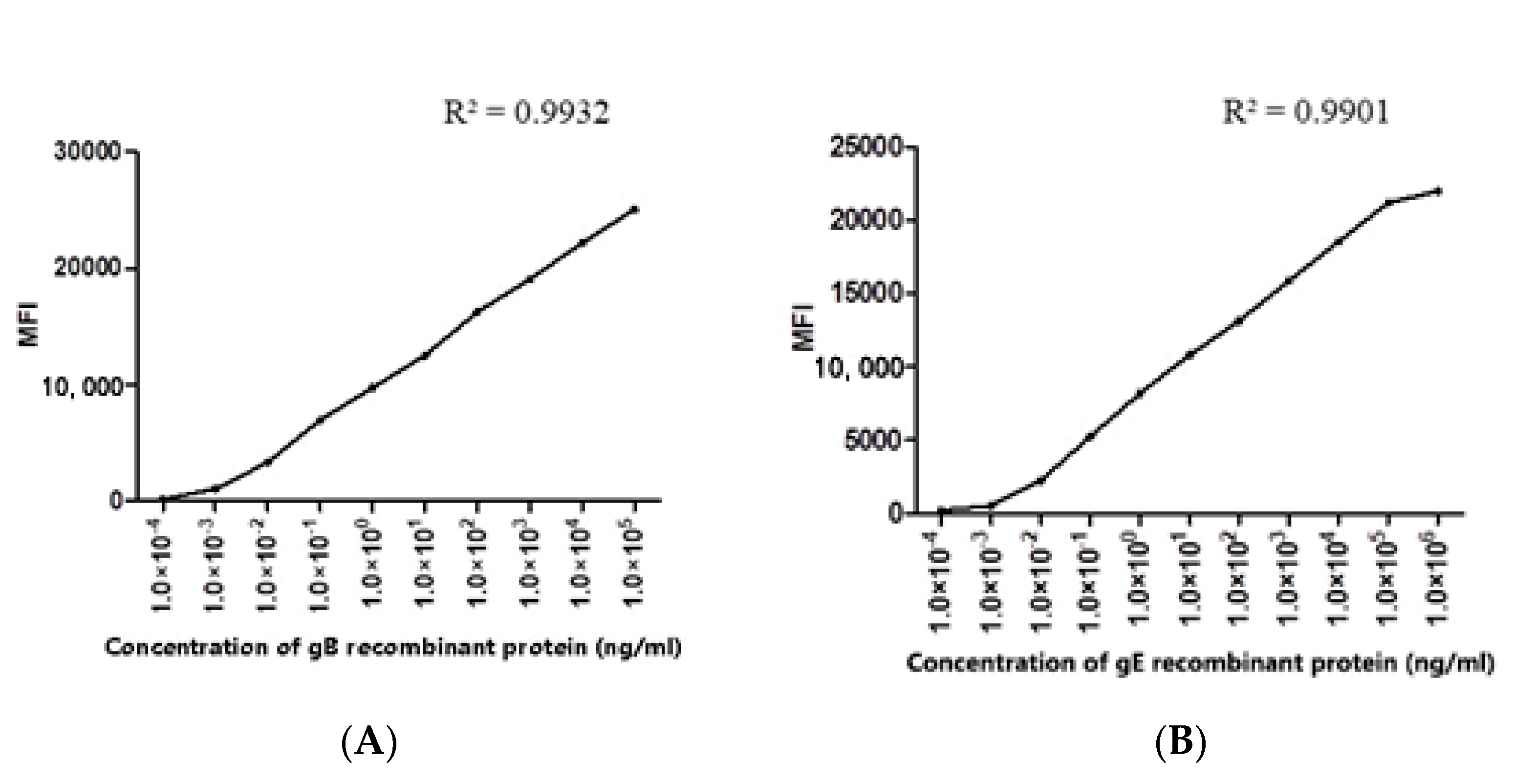
3.3. Evaluation of Specificity, Coupling Efficiency, Serum Dilution and Determination of the Optimal Concentration of Recombinant Protein Coupling
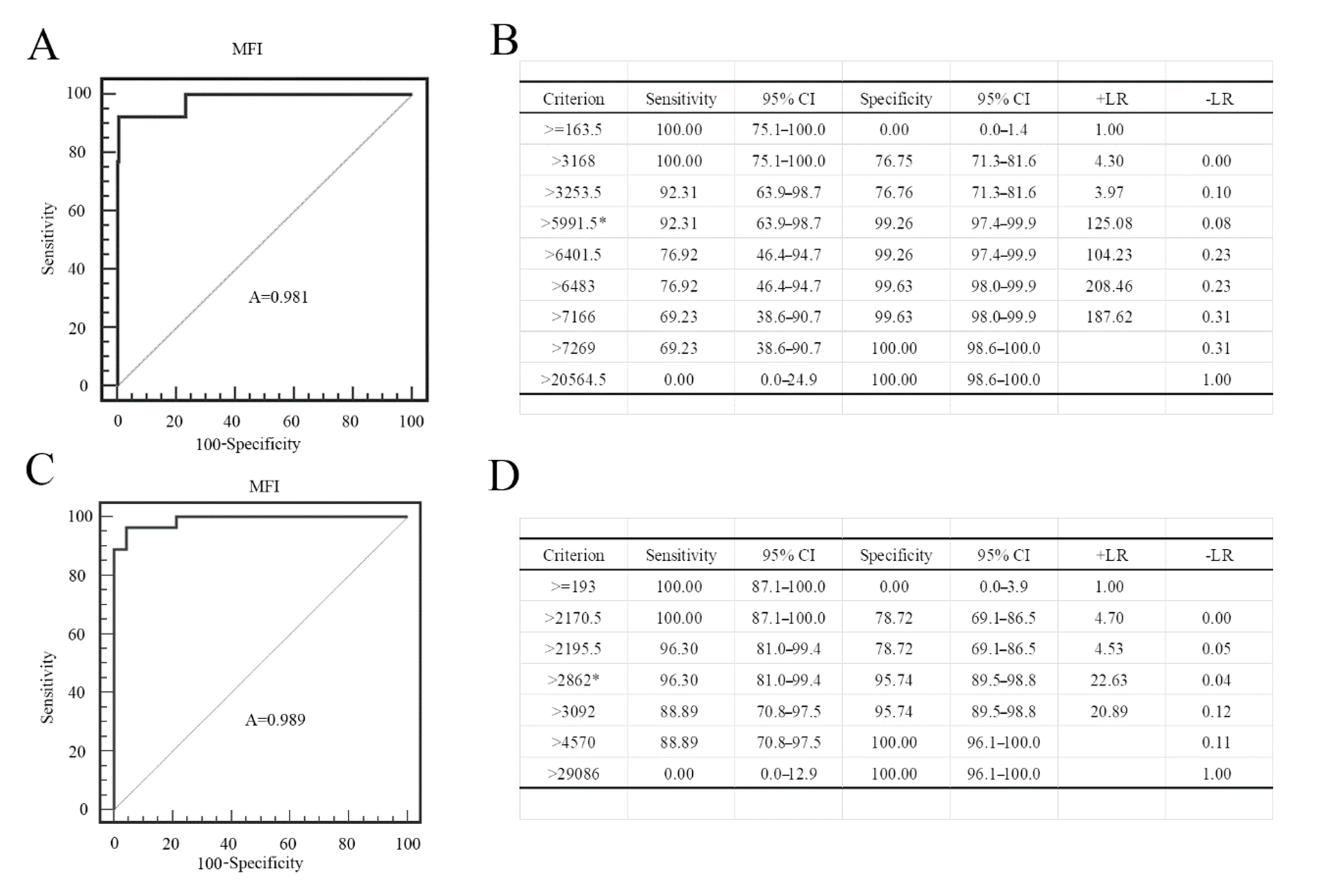
3.4. Establishment of PRV gE and gB FMIA
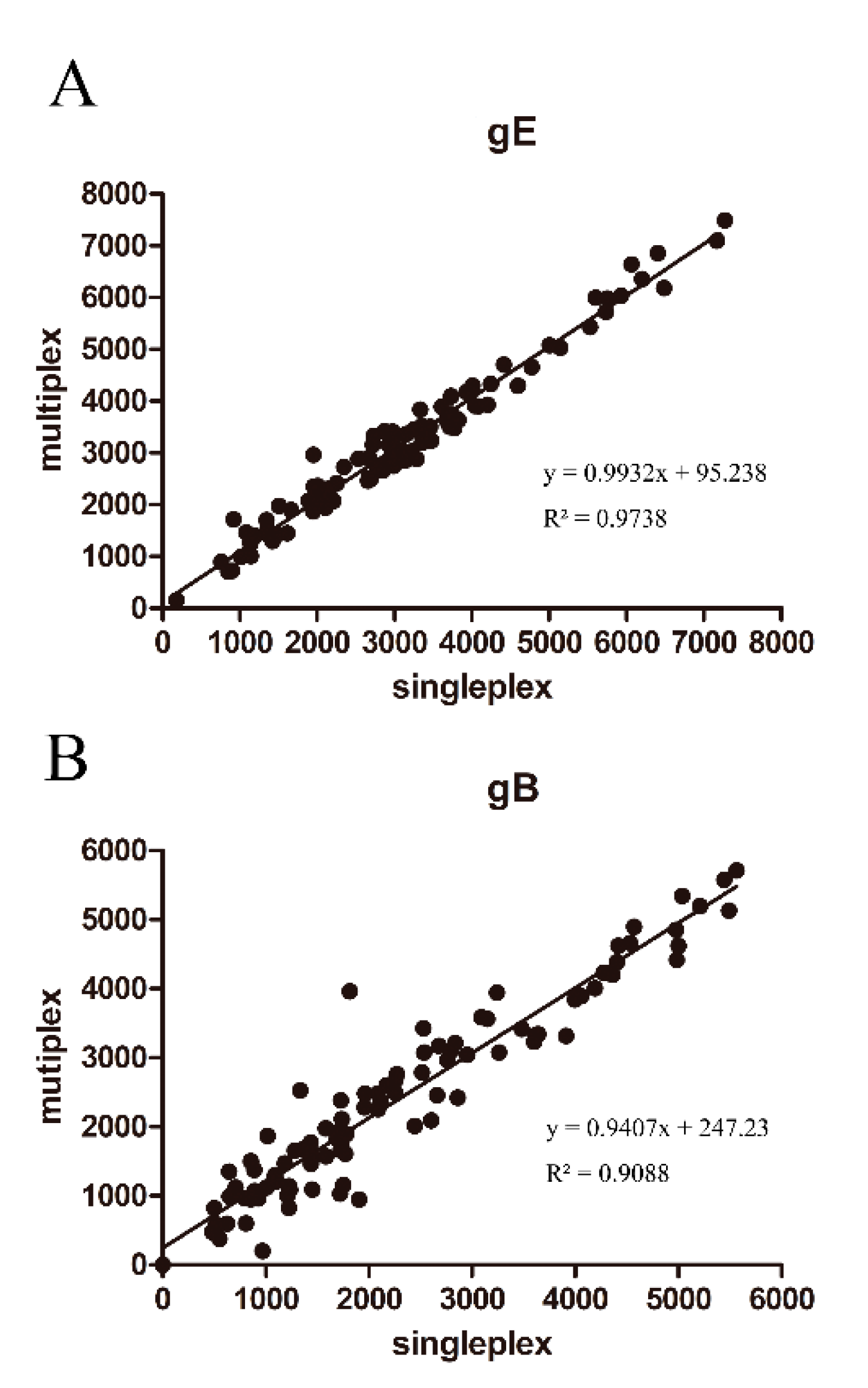
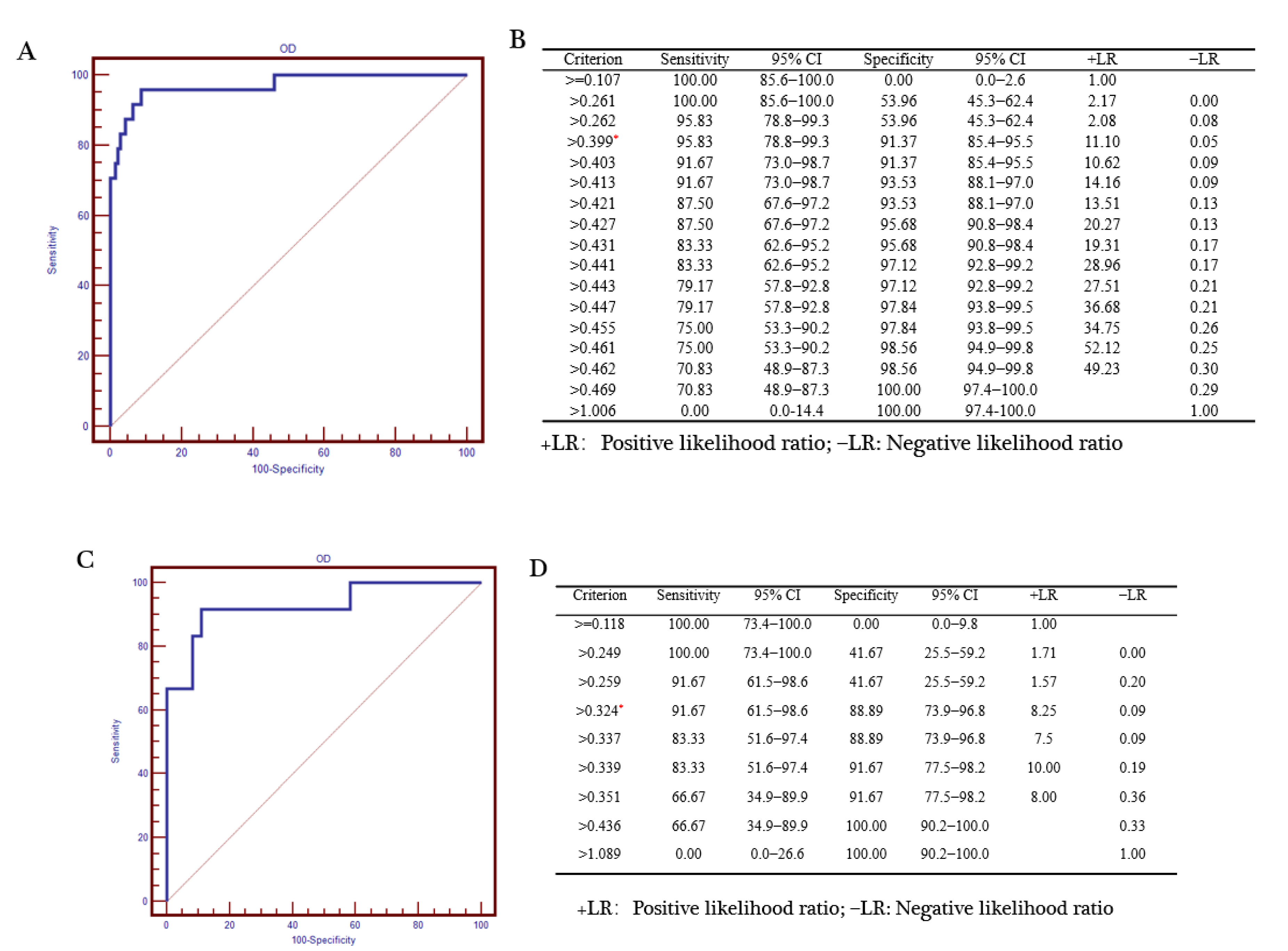
3.5. Comparison of ELISA and FMIA
3.5.1. Comparison of ELISA and FMIA Chi-Squared Tests
3.5.2. Repetitive Experiment
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Bouma, A. Determination of the effectiveness of Pseudorabies marker vaccines in experiments and field trials. Biologicals 2005, 33, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhou, Z.; Hu, D.; Zhang, Q.; Han, T.; Li, X.; Gu, X.; Yuan, L.; Zhang, S.; Wang, B.; et al. Pathogenic Pseudorabies Virus, China, 2012. Emerg. Infect. Dis. 2014, 20, 102–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qiao, S.; Li, X.; Xie, W.; Guo, J.; Li, Q.; Liu, X.; Hou, J.; Xu, Y.; Wang, L.; et al. Molecular epidemiology of outbreak-associated pseudorabies virus (PRV) strains in central China. Virus Genes 2015, 50, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Bai, C.; Sun, J.; Chang, S.; Zhang, X. Emergence of virulent pseudorabies virus infection in Northern China. J. Vet. Sci. 2013, 14, 363–365. [Google Scholar] [CrossRef]
- Luo, Y.; Li, N.; Cong, X.; Wang, C.; Du, M.; Li, L.; Zhao, B.; Yuan, J.; Liu, D.; Li, S.; et al. Pathogenicity and genomic characterization of a pseudorabies virus variant isolated from Bartha-K61-vaccinated swine population in China. Vet. Microbiol. 2014, 174, 107–115. [Google Scholar] [CrossRef]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biololgy of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. R 2005, 69, 462. [Google Scholar] [CrossRef]
- Klupp, B.G.; Hengartner, C.J.; Mettenleiter, T.C.; Enquist, L.W. Complete, annotated sequence of the pseudorabies virus genome. J. Virol. 2004, 78, 424–440. [Google Scholar] [CrossRef]
- Kratchmarov, R.; Kramer, T.; Greco, T.M.; Taylor, M.P.; Ch’Ng, T.H.; Cristea, I.M.; Enquist, L.W. Glycoproteins gE and gI Are Required for Efficient KIF1A-Dependent Anterograde Axonal Transport of Alphaherpesvirus Particles in Neurons. J. Virol. 2013, 87, 9431–9440. [Google Scholar] [CrossRef]
- Szpara, M.L.; Tafuri, Y.R.; Parsons, L.; Shamim, S.R.; Verstrepen, K.J.; Legendre, M.; Enquist, L.W. A Wide Extent of Inter-Strain Diversity in Virulent and Vaccine Strains of Alphaherpesviruses. PLoS. Pathog. 2011, 7. [Google Scholar] [CrossRef]
- Xiao, S.B.; Chen, H.C.; Fang, L.R.; Liu, C.S.; Zhang, H.; Jiang, Y.B.; Hong, W.Z. Comparison of immune responses and protective efficacy of suicidal DNA vaccine and conventional DNA vaccine encoding glycoprotein C of pseudorabies virus in mice. Vaccine 2004, 22, 345–351. [Google Scholar] [CrossRef]
- Grabowska, A.K.; Lipinska, A.D.; Rohde, J.; Szewczyk, B.; Bienkowska-Szewczyk, K.; Rziha, H. New baculovirus recombinants expressing Pseudorabies virus (PRV) glycoproteins protect mice against lethal challenge infection. Vaccine 2009, 27, 3584–3591. [Google Scholar] [CrossRef] [PubMed]
- Babic, N.; Klupp, B.; Brack, A.; Mettenleiter, T.C.; Ugolini, G.; Flamand, A. Deletion of glycoprotein gE reduces the propagation of pseudorabies virus in the nervous system of mice after intranasal inoculation. Virology 1996, 219, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Pontes, M.S.; Devriendt, B.; Favoreel, H.W. Pseudorabies Virus Triggers Glycoprotein gE-Mediated ERK1/2 Activation and ERK1/2-Dependent Migratory Behavior in T Cells. J. Virol. 2015, 89, 2149–2156. [Google Scholar] [CrossRef] [PubMed]
- Zaripov, M.M.; Morenkov, O.S.; Fodor, N.; Brown, A.; Schmatchenko, V.V.; Fodor, I. Distribution of B-cell epitopes on the pseudorabies virus glycoprotein B. J. Gen. Virol. 1999, 80, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Yong, T.; Chen, H.C.; Xiao, S.B.; Qin, Y.L.; He, Q.G.; Ren, Y.Q. Development of a latex agglutination test using the major epitope domain of glycoprotein E of pseudorabies virus expressed in E-coli to differentiate between immune responses in pigs naturally infected or vaccinated with pseudorabies virus. Vet. Res. Commun. 2005, 29, 487–497. [Google Scholar] [CrossRef]
- An, T.; Peng, J.; Tian, Z.; Zhao, H.; Li, N.; Liu, Y.; Chen, J.; Leng, C.; Sun, Y.; Chang, D.; et al. Pseudorabies Virus Variant in Bartha-K61-Vaccinated Pigs, China, 2012. Emerg. Infect. Dis. 2013, 19, 1749–1755. [Google Scholar] [CrossRef]
- Gu, Z.; Dong, J.; Wang, J.; Hou, C.; Sun, H.; Yang, W.; Bai, J.; Jiang, P. A novel inactivated gE/gI deleted pseudorabies virus (PRV) vaccine completely protects pigs from an emerged variant PRV challenge. Virus Res. 2015, 195, 57–63. [Google Scholar] [CrossRef]
- Zhang, C.; Guo, L.; Jia, X.; Wang, T.; Wang, J.; Sun, Z.; Wang, L.; Li, X.; Tan, F.; Tian, K. Construction of a triple gene-deleted Chinese Pseudorabies virus variant and its efficacy study as a vaccine candidate on suckling piglets. Vaccine 2015, 33, 2432–2437. [Google Scholar] [CrossRef]
- Pikalo, J.; Zani, L.; Huehr, J.; Beer, M.; Biome, S. Pathogenesis of African swine fever in domestic pigs and European wild boar—Lessons learned from recent animal trials. Virus Res. 2019, 271. [Google Scholar] [CrossRef]
- Mueller, T.; Hahn, E.C.; Tottewitz, F.; Kramer, M.; Klupp, B.G.; Mettenleiter, T.C.; Freuling, C. Pseudorabies virus in wild swine: A global perspective. Arch. Virol. 2011, 156, 1691–1705. [Google Scholar] [CrossRef]
- Wood, W.I.; Gitschier, J.; Lasky, L.A.; Lawn, R.M. Base composition-independent hybridization in tetramethylammonium chloride: A method for oligonucleotide screening of highly complex gene libraries. Proc. Natl. Acad. Sci. USA 1985, 82, 1585–1588. [Google Scholar] [CrossRef] [PubMed]
- van Gageldonk, P.G.M.; van Schaijk, F.G.; van der Klis, F.R.; Berbers, G.A.M. Development and validation of a multiplex immunoassay for the simultaneous determination of serum antibodies to Bordetella pertussis, diphtheria and tetanus. J. Immunol. Methods 2008, 335, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, F.; Williams, A.; Johnson, P. Validation and comparison of two multiplex technologies, Luminex (R) and Mesoscale Discovery, for human cytokine profiling. J. Immunol. Methods 2009, 340, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Clavijo, A.; Hole, K.; Li, M.Y.; Collignon, B. Simultaneous detection of antibodies to foot-and-mouth disease non-structural proteins 3ABC, 3D, 3A and 3B by a multiplexed Luminex assay to differentiate infected from vaccinated cattle. Vaccine 2006, 24, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Balasuriya, U.B.R.; Shi, P.; Wong, S.J.; Demarest, V.L.; Gardner, I.A.; Hullinger, P.J.; Ferraro, G.L.; Boone, J.D.; De Cino, C.L.; Glaser, A.L.; et al. Detection of antibodies to West Nile virus in equine sera using microsphere immunoassay. J. Vet. Diagn. Investig. 2006, 18, 392–395. [Google Scholar] [CrossRef]
- Watson, D.S.; Reddy, S.M.; Brahmakshatriya, V.; Lupiani, B. A multiplexed immunoassay for detection of antibodies against avian influenza virus. J. Immunol. Methods 2009, 340, 123–131. [Google Scholar] [CrossRef]
- Lindahl, J.F.; Ragan, I.K.; Rowland, R.R.; Wainaina, M.; Mbotha, D.; Wilson, W. A multiplex fluorescence microsphere immunoassay for increased understanding of Rift Valley fever immune responses in ruminants in Kenya. J. Virol. Methods 2019, 269, 70–76. [Google Scholar] [CrossRef]
- Korppi, M.; Leinonen, M.; Ruuskanen, O. Pneumococcal serology in children’s respiratory infections. Eur. J. Clin. Microbiol. 2008, 27, 167–175. [Google Scholar] [CrossRef]
- Wu, C.Y.; Wu, C.W.; Liao, C.M.; Chien, M.S.; Huang, C. Enhancing expression of the pseudorabies virus glycoprotein E in yeast and its application in an indirect sandwich ELISA. J. Appl. Microbiol. 2017, 123, 594–601. [Google Scholar] [CrossRef]
- Li, X.; Sun, Y.; Yang, S.; Wang, Y.; Yang, J.; Liu, Y.; Jin, Q.; Li, X.; Guo, C.; Zhang, G. Development of an immunochromatographic strip for antibody detection of pseudorabies virus in swine. J. Vet. Diagn. Investig. 2015, 27, 739–742. [Google Scholar] [CrossRef]
- Feichtner, F.; Schachner, A.; Berger, E.; Hess, M. Fiber-based fluorescent microsphere immunoassay (FMIA) as a novel multiplex serodiagnostic tool for simultaneous detection and differentiation of all clinically relevant fowl adenovirus (FAdV) serotypes. J. Immunol. Methods 2018, 458, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Pinette, M.M.; Rodriguez-Lecompte, J.C.; Pasick, J.; Ojkic, D.; Leith, M.; Suderman, M.; Berhane, Y. Development of a duplex Fluorescent Microsphere Immunoassay (FMIA) for the detection of antibody responses to influenza A and newcastle disease viruses. J. Immunol. Methods 2014, 405, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Langenhorst, R.J.; Lawson, S.; Kittawornrat, A.; Zimmerman, J.J.; Sun, Z.; Li, Y.; Christopher-Hennings, J.; Nelson, E.A.; Fang, Y. Development of a Fluorescent Microsphere Immunoassay for Detection of Antibodies against Porcine Reproductive and Respiratory Syndrome Virus Using Oral Fluid Samples as an Alternative to Serum-Based Assays. Clin. Vaccine Immunol. 2012, 19, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Xu, Z.; Liu, X.; Li, P.; Yang, F.; Zhao, J.; Fan, Y.; Sun, X.; Zhu, L. A live gI/gE-deleted pseudorabies virus (PRV) protects weaned piglets against lethal variant PRV challenge. Virus Genes 2017, 53, 565–572. [Google Scholar] [CrossRef]
- Liang, X.; Sun, L.; Yu, T.; Pan, Y.; Wang, D.; Hu, X.; Fu, Z.; He, Q.; Cao, G. A CRISPR/Cas9 and Cre/Lox system-based express vaccine development strategy against re-emerging Pseudorabies virus. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Balada-Llasat, J.; LaRue, H.; Kamboj, K.; Rigali, L.; Smith, D.; Thomas, K.; Pancholi, P. Detection of Yeasts in Blood Cultures by the Luminex xTAG Fungal Assay. J. Clin. Microbiol. 2012, 50, 492–494. [Google Scholar] [CrossRef][Green Version]
- Gimenez-Lirola, L.G.; Xiao, C.T.; Halbur, P.G.; Opriessnig, T. Development of a novel fluorescent microbead-based immunoassay and comparison with three enzyme-linked immunoassays for detection of anti-Erysipelothrix spp. IgG antibodies in pigs with known and unknown exposure. J. Microbiol. Meth. 2012, 91, 73–79. [Google Scholar] [CrossRef]
- Shen, H.; Chen, H.; Cheng, Z.; Ma, L.; Huang, L.; Xiao, M.; Xiao, W.; Xie, K.; Tang, Y. A novel fluorescent immunochromatographic strip combined with pocket fluorescence observation instrument for rapid detection of PRV. Anal. Bioanal. Chem. 2018, 410, 7655–7661. [Google Scholar] [CrossRef]
- Han, X.; Wang, H.; Chen, H.; Mei, L.; Wu, S.; Jia, G.; Cheng, T.; Zhu, S.; Lin, X. Development and primary application of a fluorescent liquid bead array for the simultaneous identification of multiple genetically modified maize. Biosens. Bioelectron. 2013, 49, 360–366. [Google Scholar] [CrossRef]
- Anderson, S.; Wakeley, P.; Wibberley, G.; Webster, K.; Sawyer, J. Development and evaluation of a Luminex multiplex serology assay to detect antibodies to bovine herpes virus 1, parainfluenza 3 virus, bovine viral diarrhoea virus, and bovine respiratory syncytial virus, with comparison to existing ELISA detection methods. J. Immunol. Methods 2011, 366, 79–88. [Google Scholar] [CrossRef]
- Wagner, B.; Freer, H.; Rollins, A.; Erb, H.N.; Lu, Z.; Groehn, Y. Development of a multiplex assay for the detection of antibodies to Borrelia burgdorferi in horses and its validation using Bayesian and conventional statistical methods. Vet. Immunol. Immunopathol. 2011, 144, 374–381. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Paired Chi Square Test | ELISA | |||
|---|---|---|---|---|
| + | − | Total | ||
| FMIA | + | 33 | 1 | 34 |
| − | 1 | 57 | 58 | |
| total | 34 | 58 | 92 | |
| Statistical results | Correlation test | Superiority test | ||
| Chi square value | p | Chi square value | p | |
| 79.57 | 0.000 | 0.50 | 0.480 | |
| Relevance significance | : there was no significant difference in advantage | |||
| Paired Chi Square Test | ELISA | |||
|---|---|---|---|---|
| + | − | Total | ||
| FMIA | + | 32 | 2 | 34 |
| − | 0 | 58 | 58 | |
| total | 32 | 60 | 92 | |
| Statistical results | Correlation test | Superiority test | ||
| Chi square value | p | Chi square value | p | |
| 79.60 | 0.000 | 0.50 | 0.480 | |
| Relevance significance | : there was no significant difference in advantage | |||
| Sample Number | ± s(MFI) | CV% |
|---|---|---|
| 1 | 491.55 ± 37.16 | 7.6 |
| 2 | 17,165.80 ± 456.90 | 2.7 |
| 3 | 25,527.70 ± 1139.50 | 4.4 |
| Sample Number | ± s(MFI) | CV% |
|---|---|---|
| 1 | 420.20 ± 31.85 | 7.6 |
| 2 | 19,980.50 ± 516.40 | 2.6 |
| 3 | 14,108.9 ± 1089.7 | 7.8 |
| Sample Number | ± s(MFI) | CV% | Sample Number | ± s(MFI) | CV% |
|---|---|---|---|---|---|
| 1 | 6348.67 ± 199.80 | 3.1 | 7 | 11,071.10 ± 958.10 | 8.7 |
| 2 | 36,623.80 ± 1407.00 | 3.8 | 8 | 26,685.90 ± 2263.70 | 8.5 |
| 3 | 34,446.20 ± 1843.2 | 5.4 | 9 | 22,252.30 ± 501.40 | 2.3 |
| 4 | 10,220.17 ± 542.52 | 5.3 | 10 | 6835.00 ± 863.20 | 12.6 |
| 5 | 7480.00 ± 431.28 | 5.8 | 11 | 42,555.30 ± 3882.30 | 9.1 |
| 6 | 17,935.30 ± 1191.50 | 6.6 | 12 | 735.33 ± 41.02 | 5.6 |
| Sample Number | ± s(MFI) | CV% | Sample Number | ± s(MFI) | CV% |
|---|---|---|---|---|---|
| 1 | 17,935.30 ± 1191.50 | 6.6 | 7 | 26,685.90 ± 2263.70 | 8.5 |
| 2 | 10,273.33 ± 546.50 | 5.3 | 8 | 10,304.83 ± 1011.01 | 9.8 |
| 3 | 8949.17 ± 374.49 | 4.2 | 9 | 39,946.60 ± 2393.60 | 6.0 |
| 4 | 6685.67 ± 596.60 | 8.9 | 10 | 24,298.17 ± 2322.32 | 9.6 |
| 5 | 16,384.83 ± 915.12 | 5.6 | 11 | 8701.00 ± 474.37 | 5.5 |
| 6 | 31,847.90 ± 2571.4 | 8.1 | 12 | 450.00 ± 10.00 | 2.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, C.; Wei, Y.; Wang, J.; Zeng, Y.; Pan, H.; Liang, G.; Ma, J.; Gong, L.; Zhang, W.; Zhang, G.; et al. Development of a Dual Fluorescent Microsphere Immunological Assay for Detection of Pseudorabies Virus gE and gB IgG Antibodies. Viruses 2020, 12, 912. https://doi.org/10.3390/v12090912
Ji C, Wei Y, Wang J, Zeng Y, Pan H, Liang G, Ma J, Gong L, Zhang W, Zhang G, et al. Development of a Dual Fluorescent Microsphere Immunological Assay for Detection of Pseudorabies Virus gE and gB IgG Antibodies. Viruses. 2020; 12(9):912. https://doi.org/10.3390/v12090912
Chicago/Turabian StyleJi, Chihai, Yingfang Wei, Jingyu Wang, Yuchen Zeng, Haoming Pan, Guan Liang, Jun Ma, Lang Gong, Wei Zhang, Guihong Zhang, and et al. 2020. "Development of a Dual Fluorescent Microsphere Immunological Assay for Detection of Pseudorabies Virus gE and gB IgG Antibodies" Viruses 12, no. 9: 912. https://doi.org/10.3390/v12090912
APA StyleJi, C., Wei, Y., Wang, J., Zeng, Y., Pan, H., Liang, G., Ma, J., Gong, L., Zhang, W., Zhang, G., & Wang, H. (2020). Development of a Dual Fluorescent Microsphere Immunological Assay for Detection of Pseudorabies Virus gE and gB IgG Antibodies. Viruses, 12(9), 912. https://doi.org/10.3390/v12090912