Abstract
Cells have evolved highly specialized sentinels that detect viral infection and elicit an antiviral response. Among these, the stress-sensing protein kinase R, which is activated by double-stranded RNA, mediates suppression of the host translation machinery as a strategy to limit viral replication. Non-translating mRNAs rapidly condensate by phase separation into cytosolic stress granules, together with numerous RNA-binding proteins and components of signal transduction pathways. Growing evidence suggests that the integrated stress response, and stress granules in particular, contribute to antiviral defense. This review summarizes the current understanding of how stress and innate immune signaling act in concert to mount an effective response against virus infection, with a particular focus on the potential role of stress granules in the coordination of antiviral signaling cascades.
Keywords:
virus; stress granules; stress response; innate immune response; PKR; G3BP1; antiviral signaling 1. Introduction
Viruses depend on the host translation apparatus to express viral proteins. By hijacking and redirecting ribosomes, translation factors, and RNA binding proteins (RBPs), viruses modulate the cellular translatome in favor of their needs and virus progeny production. In rare cases, viruses such as hantaviruses, myoviruses, haloarchaeal viruses, and giant viruses encode viral proteins that substitute for translation initiation, elongation, and termination factors, transfer RNAs (tRNAs), aminoacyl-tRNA synthetases [1,2,3,4,5,6], and—as lately identified via metagenome analysis—also ribosomal proteins [7]. Although the use of the host-encoded translation machinery saves coding capacity, the ensuing dependency on host factors to accomplish this critical step of the viral life cycle makes viruses vulnerable. To counteract virus reproduction, cells have evolved highly specialized stress sensors that detect viral products and actively suppress both host and viral translation. Hence, limiting the access, availability, or activity of the translation apparatus can be considered an effective defense mechanism and integral component of the antiviral response.
Immune sensors detect viral nucleic acids as non-self through specific features, termed pathogen-associated molecular patterns (PAMPs), among these incoming viral DNA and RNA genomes and viral replication intermediates. The activation of immune sensors elicits an antiviral state through the production of interferons (IFNs) and pro-inflammatory cytokines [8,9,10]. In addition, the accumulation of viral double-stranded (ds) RNA in the cytosol and of viral proteins in the endoplasmic reticulum (ER) triggers stress sensors such as protein kinase R (PKR) and PKR-like endoplasmic reticulum kinase (PERK), respectively [11]. Once activated, these kinases initiate the integrated stress response (ISR) by phosphorylating the alpha subunit of the eukaryotic translation initiation factor-2 (eIF2α), which delivers the Methionine initiator tRNA (tRNAiMet) to the small 40S ribosomal subunit. As a consequence, translation initiation is stalled and polysomes disassemble [12].
The assembly of stress granules (SGs) is an integral part of host stress responses, frequently observed upon infection with DNA or RNA viruses. SGs form by a cytosolic phase separation event, through which stalled mRNAs are separated from the remaining cytosol together with translation factors. SGs were primarily suggested to serve as storage and triage areas for mRNAs that can easily be reengaged with polysomes in order to resume translation once the stress is resolved [13,14]. However, the abundance of RBPs and signal transducing factors in SGs has broadened our perspective on the role of SGs in recent years. SGs are now considered to be signaling platforms that contribute to the coordination of cellular processes during stress, including apoptosis, cell growth, metabolic control, and antiviral defense [15,16,17,18]. It is therefore not surprising that viruses not only escape translational repression through various mechanisms, but also interfere with the assembly of SGs and repurpose SG proteins for their own replication [19,20,21,22]. The diversity of these viral strategies highlights the importance of the stress response, and SGs in particular, in antiviral defense.
Whether SGs as a whole or single proteins within SGs contribute to the antiviral response is still an open question whose answer might depend on the virus type, the cell type, and the resulting host–virus interactions. Recent advances in the purification of SG components have opened the route to investigate which proteins and mRNAs are selectively recruited to SGs during viral infections. Here, we aim to summarize the current understanding of the translational stress response in the context of virus infection and cover the various connections between the stress response and innate immune response, which are intertwined events that act in concert to establish an antiviral state. Emphasis will be given to the potential role of SGs in the initiation and coordination of antiviral signaling events.
2. General Mechanisms of Translation Control under Homeostasis and Virus Infection
Regulation of gene expression at the level of translation is a central control mechanism that enables rapid and reversible changes in protein levels, both spatially and temporally. In addition to essential roles in several biological processes, including cell growth, differentiation, and apoptosis [23,24,25], translational control plays a major role in the host stress response to virus infection [26,27].
2.1. Translation Initiation, a Key Step in the Regulation of Protein Synthesis
Mammalian protein synthesis is primarily regulated at the level of translation initiation, which requires recruitment of the small ribosomal subunit to the mRNA, scanning along the mRNA 5′ untranslated region (UTR), recognition of the start codon, and subsequent joining of the large ribosomal subunit [28]. These steps are regulated by upstream open reading frames (uORFs), RNA modifications, RNA structures and different RBPs, which together dictate mRNA translation initiation efficiency [29,30].
Most cellular mRNAs contain a cap structure at their 5′ end, composed of an inverted 7-methylguanosine connected to the mRNA via a 5′-5′-triphosphate bridge. Most 5′ ends are additionally methylated at the ribose 2′O position of the +1 ribonucleotide (cap1), and in many cases, also of the +2 ribonucleotide (cap2). This feature is not only important for mRNA translation and stability, but also for the distinction between self from non-self mRNAs [31]. Indeed, several single-stranded (ss) RNA viruses that replicate in the cytosol, including flaviviruses, picornaviruses, coronaviruses, and poxviruses, encode their own 2′O-methytransferases to escape recognition by host immune sensors [32]. Initially, the 5′ cap is bound by the eIF4F complex, which consists of the cap-binding protein eIF4E, the RNA helicase eIF4A, and the scaffold protein eIF4G. Subsequently, the 40S small ribosomal subunit together with the initiation factors eIF1, eIF1A, eIF5, eIF3, and the eIF2–GTP–tRNAiMet ternary complex is loaded onto the mRNA 5′ end, forming a scanning-competent 48S preinitiation complex (PIC). The PIC then scans the 5′ UTR until it recognizes a start codon within a favorable sequence context, leading to hydrolysis of the eIF2-bound GTP. This crucial step results in the release of eIF2-GDP, leaving the initiator tRNAiMet base-paired with the AUG start codon [33].
2.2. Repression of Translation Initiation upon Environmental Stress
Cells respond to unfavorable conditions such as amino acid starvation, ER stress, oxidative stress, hypoxia, UV irradiation, and virus infection by rapid attenuation of translation initiation rates, mainly through controlling (i) the eIF2–GTP–tRNAiMet ternary complex and (ii) the cap-binding complex. Thereby, the translation of mRNAs not critical for cell survival, e.g., housekeeping mRNAs, is strongly repressed during stress in favor of mRNAs encoding survival factors, repair enzymes, and stress-regulatory proteins [23,24].
The first mechanism involves four eIF2α-kinases, which are activated by a wide range of extrinsic and intrinsic stressors, and phosphorylate the α subunit of eIF2 at Ser51 [12]. This prevents release of eIF2 from the GTP-GDP exchange factor eIF2B, thereby impairing regeneration of the ternary complex and slowing down protein synthesis [34]. The heme-regulated inhibitor (HRI) senses oxidative stress, osmotic stress, heat shock, and heme-depletion. PKR is activated by cellular and viral dsRNAs. PERK, a transducer of the unfolded protein response (UPR), is activated by the accumulation of misfolded proteins in the ER. Finally, the general control nonderepressible 2 (GCN2) is mainly activated by limited amino acid availability and UV stress. As a common feature, eIF2α-kinases were found to be activated by stress-induced oligo- or dimerization and autophosphorylation [11]. The pool of the GTP-bound ternary complex is regenerated by dephosphorylation of eIF2α through protein phosphatase 1 (PP1), whose catalytic subunit is recruited to eIF2α via a specific regulatory subunit. In unstressed cells, the constitutive repressor of eIF2α phosphorylation (CReP), a constitutively expressed regulatory subunit of PP1, maintains low levels of eIF2α phosphorylation [35], especially at the ER [36]. Under stress conditions, the PP1 regulatory subunit growth arrest and DNA damage inducible protein 34 (GADD34) is both transcriptionally and translationally upregulated, antagonizing eIF2α phosphorylation and translational arrest imposed by eIF2α-kinase activation [37,38,39,40].
The second mechanism is based on regulating the activity of the eIF4F cap-binding complex, which controls cap-dependent translation. Certain types of stress, such as nutrient deprivation and hypoxia, lead to inhibition of the mammalian target of rapamycin (mTOR) complex 1 (mTORC1), the key kinase that regulates the phosphorylation state of 4E binding proteins (4E-BPs) [24]. In their unphosphorylated form, 4E-BPs have an increased affinity for the cap-binding protein eIF4E and outcompete eIF4G, preventing efficient recruitment of eIF3 and the 40S small ribosomal subunit to the 5′ end of the mRNA [41]. Thereby, inhibition of mTORC1 leads to a reduction in cap-dependent translation.
A subset of cellular mRNAs contains linear motifs and secondary structures that enable protein synthesis under stress conditions through alternative modes of translation initiation, which are independent of eIF2α and/or eIF4F. These include (i) transcripts containing short uORFs [42], mostly encoding for survival-related and stress-effector proteins, e.g., the transcription factors activating transcription factor 4 (ATF4) and the CCAAT/enhancer-binding protein homology protein (CHOP), as well as GADD34. (ii) Approximately 10% of all cellular mRNAs are suggested to contain internal ribosome entry sites (IRES) and hence, do not require eIF4F binding, but instead, rely on IRES trans-acting factors (ITAFs) for efficient translation initiation [43,44,45]. IRES elements were originally discovered in viruses belonging to the Picornaviridae family [46,47]. Even though their activity and efficiency is still under debate, numerous cellular IRES elements have been reported and characterized, particularly in genes involved in apoptosis such as the X-linked inhibitor of apoptosis protein (XIAP) [48] and B-cell lymphoma-2 (Bcl-2) [49], and more recently, in genes involved in cell growth and proliferation such as mTOR [50] and c-Src [51]. The existence and involvement of a growing number of cap-independent initiation mechanisms in eukaryotes, including (iii) N6-methyladenosine (m6A)-dependent translation [52,53,54], and (iv) the use of alternative cap-binding complexes such as eIF4FH under hypoxic conditions or eIF4FM in response to stress or proliferative cues, respectively [55], are indisputable.
The impact of translational repression, especially on RNA viruses, is highlighted by the fact that many evolved alternative initiation strategies to translate their genome independently of eIF2α, including the use of IRES elements [56] and the possibility to switch from cap-dependent to cap-independent translation [57,58,59,60].
2.3. From Translational Suppression to SG Formation
When translation initiation is blocked under stress conditions, stalled translation preinitiation complexes accumulate. At the same time, translation elongation continues and polysomes disassemble as a consequence of ribosome run-off. Upon acute and strong inhibition of translation initiation, non-translating mRNAs condensate together with RBPs into microscopically visible, non-membranous cytosolic SGs through a liquid–liquid phase separation event [61]. Phase separation is likely initiated by the appearance of long stretches of mRNA not covered by ribosomes, which engage in RNA–RNA interactions [62] and serve as scaffolds for numerous RBPs that promote phase separation through their intrinsically disordered regions. RBPs with an essential role in nucleating SGs include RasGAP-associated endoribonuclease 1 (G3BP1), T cell internal antigen 1 (TIA1), TIA1-related protein (TIAR), Caprin1, and fragile X mental retardation protein (FMRP) [63]. Recently, ubiquitin-associated protein 2-like (UBAP2L) [64,65,66,67], cold shock domain containing E1 (CSDE1), and proline-rich coiled-coil 2C (PRRC2C) were added to the growing list of SG-nucleating proteins [64]. Overexpression of single proteins such as TIA1 or G3BP1 can drive SG formation, even in the absence of stress [68,69], indicating that a shift in the equilibrium between solubilizing and aggregation-prone proteins is sufficient to induce phase separation of the cytosol.
Interestingly, SGs were found to consist of a more stable inner core, stabilized by direct protein–protein and RNA–protein interactions, and a dynamic shell-like outer layer that is characterized by multiple, multivalent low-affinity interactions between proteins and RNAs [70]. SGs are very dynamic structures, which rapidly assembly under stress conditions and disassemble within minutes when cells recover from stress [71,72]. Depending on the type of stress, SGs vary in size and number, and differ with respect to some of their mRNA and protein constituents. During oxidative stress, for example, SGs move in a microtubule-dependent manner and grow in size by fusion [71,73].
Given their dynamic nature, it is not surprising that proteins and mRNAs shuttle in and out of SGs in the seconds to minute range [71,74,75], showing that SG components are in constant exchange with the cytosol. SGs also exchange proteins and mRNAs with processing bodies (PBs), a different type of cytosolic RNA granule that contains RNA degrading enzymes and functions in mRNA silencing [76]. SGs and PBs often exist in close proximity and may represent different stages of an “mRNP cycle” [13].
A peculiar type of SG dynamic was discovered by our laboratory upon chronic hepatitis C virus (HCV) infection, which leads to recurring cycles of SG assembly and disassembly, following an oscillation of translational on- and off-states. The stochastic nature of SG assembly and disassembly is controlled by the antagonistic action of PKR and GADD34, which repeatedly phosphorylate and dephosphorylate eIF2α. This oscillating stress response is widely observed upon infection with RNA viruses including Newcastle disease virus (NDV) and Sendai virus (SeV) [77]. Our current understanding is that oscillating SGs represent a long-term strategy by which infected cells suppress viral protein production and replication intermittently, while enabling host protein production in between SG phases. Moreover, rapid SG oscillations seem to correlate with enhanced cell survival, suggesting a role in balancing the burden of antiviral defense with cellular homeostasis.
3. Stress Kinases—Mediators of Viral Translational Inhibition
Translation inhibition is an important pillar of the antiviral response. Viruses evolved multiple strategies to target all steps of translation initiation in order to evade host-induced translational shutoff and promote the synthesis of their own proteins. Since these strategies have been covered in several excellent reviews [26,27,56], we will focus here on how stress kinases contribute to the induction of the ISR upon virus infection and how viruses directly target these kinases. While global translation suppression upon infection with RNA viruses is primarily mediated by PKR, activation of other stress kinases, alone or in combination with PKR, has been reported. They can be considered as additional antiviral barriers, especially when viruses have evolved strategies to counteract PKR.
3.1. Protein Kinase R (EIF2AK2)
Among the four eIF2α-kinases, PKR, formerly called DAI, contributes to the sensing of viral infection and is the object of extensive research [78,79,80]. PKR is an IFN-induced effector protein [81] that is activated upon binding of dsRNA molecules [82]. The binding of dsRNA to the two dsRNA-binding motifs (dsRBD) within the PKR N-terminal domain promotes a structural reorientation, which allows for PKR dimerization and subsequent activation by autophosphorylation of PKR C-terminal kinase domain. These structural rearrangements are required for binding and phosphorylation of eIF2α [82,83,84,85]. One of the earliest molecules found to activate PKR was a hairpin loop of the hepatitis delta virus genome, within the self-cleaving ribozyme region [86,87,88]. A similar hairpin structure was discovered in the human immunodeficiency virus (HIV) genome within the transactivation-response region [89]. In general terms, viral activators of PKR represent dsRNA regions longer than 30 bp [85,86], including the genome of dsRNA viruses like rotaviruses [90], dsRNA replication intermediates of positive and negative ssRNA viruses [91,92] as well as dsRNA products from antiparallel transcription of DNA viruses such as vaccinia virus (VACV) [93]. The activation of PKR by short-stem loops has been suggested to be 5′ triphosphate-dependent [94,95], though this observation is discussed controversially [96,97].
In recent years, PKR was shown to have functions, beyond the detection of viral dsRNA, in cellular processes such as mitosis and apoptosis [98,99], in metabolic as well as autoimmune diseases [100,101,102,103], and in long-term memory [104,105,106]. Cellular activators of PKR include mitochondrial dsRNAs [99], dsRNAs derived from inverted Alu repeats [98,107], non-coding small nucleolar RNAs [100], ribotoxin-induced or damaged rRNAs as well as unmodified rRNAs or tRNAs [108,109,110]. RNAs that inhibit PKR have also been identified, e.g., circular RNAs [103] and the human non-coding RNA 886 [111,112].
Viruses have evolved a multitude of strategies to counteract PKR. For instance, many RNA viruses encode proteins that bind and shield dsRNA from PKR detection, exemplified by VACV E3L [113], influenza A virus (IAV) accessory protein NS1 [114], Middle East respiratory syndrome coronavirus accessory protein 4a [115,116], reovirus sigma 3 protein [117], and Ebola virus protein VP35 [118,119]. Other viruses such as human parainfluenza virus type 3 (HPIV3) sequester viral RNA in inclusion bodies to avoid detection by PKR [120]. Herpes simplex virus (HSV) 1 and 2 encode an endoribonuclease, the virion host shutoff protein that degrades RNA to avoid PKR activation early during infection [121,122,123]. Negative ssRNA viruses such as measles virus (MV), influenza virus, and SeV avoid detection by PKR by making sure that only a small number of dsRNA replication intermediates accumulate in the cytosol [91]. This is achieved by the virally encoded accessory protein C, which attenuates the copy-back mechanism of the viral RNA polymerase during replication [124,125,126].
Other viruses suppress PKR by encoding proteins that directly inhibit the kinase function, e.g., HCV non-structural protein NS5A [127], Japanese encephalitis virus (JEV) non-structural protein NS2A [128], human cytomegalovirus protein TRS1 [129], and Kaposi’s sarcoma-associated herpesvirus lytic protein ORF57 [130]. An interesting variation of this theme is the expression of small regulatory RNAs by some DNA viruses, which antagonize PKR activation by competing with dsRNA binding. Examples include the adenovirus VAI RNAs and the Epstein–Barr virus transcripts EBER-1 and EBER-2 [131,132,133], all of which bind to but do not activate PKR.
An alternative strategy adopted by some viruses is mimicry. Baculovirus protein PK2, for example, is an inactive kinase with homology to PKR, which leads to the formation of inactive PKR heterodimers [134,135]. The VACV K3L protein is an eIF2α homologue, which competes with eIF2α for PKR binding and thereby reduces eIF2α phosphorylation [113]. Finally, PKR can be targeted to proteasomal degradation upon infection with Rift Valley fever virus by the NSs protein [136,137,138].
3.2. PKR-Like Endoplasmic Reticulum Kinase (EIF2AK3)
During their life cycle, many RNA viruses perturb or hijack ER functions by (i) remodeling ER membranes to form viral replication and assembly sites [139], (ii) utilizing and competing for the host protein glycosylation machinery [140,141], and (iii) encoding viral proteins with viroporin function that alter ER calcium homeostasis [142]. Virus-imposed interference with ER functions causes ER stress and induces the three signaling branches of the UPR through activation of IRE1-XBP1, ATF6, and PERK, whereby infected cells aim to reestablish ER homeostasis. PERK, as one player of the UPR, prevents protein production, hence alleviating the ER burden [143]. For certain viruses, PERK activation was reported to be beneficial, notably for NDV [144] and Seneca Valley virus [145]. Here, PERK-induced autophagy is essential for viral replication. However, in multiple cases, PERK has adverse effects for viral replication. Transmissible gastroenteritis virus (TGEV) but also West Nile virus (WNV) and Langat virus replication, for instance, are inhibited via PERK signaling [146,147,148].
Hence, it is not surprising that viruses counteract PERK activity. NDV, e.g., mediates PERK cleavage [149], whereas dengue virus (DENV) infection was found to inhibit PERK activation by a yet unknown mechanism [150]. The VACV K3L protein was reported to reduce PERK activity in vitro [151], and similar results were obtained for the HCV-encoded glycoprotein E2. This protein induces ER stress on the one hand [152], but acts as a pseudo-substrate and thereby inhibits PERK activation on the other hand [153]. These examples illustrate that viruses strongly interfere with ER functions but at the same time, often try to prevent the consequent activation of PERK. Interestingly, PERK can also get activated by viral proteins such as JEV-encoded protein NS4B, which induces PERK dimerization and is known to be important for the pathogenesis of encephalitis [154]. This is consistent with the fact that PERK’s implication in virus infection might differ depending on the virus type or infection stage.
3.3. General Control Nonderepressible 2 (EIF2AK4)
GCN2 is usually activated under conditions of amino acid starvation through its histidyl-tRNA synthetase-related domain that binds uncharged tRNAs [155]. This domain also binds to viral RNA genomes in vitro, notably those of sindbis virus (SINV), poliovirus, and HIV-1, and in vivo studies confirmed that infection with SINV and HIV-1 leads to activation of GCN2, eIF2α phosphorylation, and translational inhibition of viral RNA [156,157]. Furthermore, HIV-1 induces protease-mediated cleavage of GCN2, indicating that GCN2 has an antiviral function that the virus tries to suppress [157]. This notion is supported by the observation that GCN2-deficient or GCN2-mutant mice are more susceptible to infection with SINV [156], murine cytomegalovirus, and human adenovirus [158].
In the future, it will be important to further characterize the mechanism behind GCN2 activation in virus-infected cells since this might also be an indirect consequence of virus- or IFN-γ-induced amino acid deprivation.
3.4. Heme-Regulated Inhibitor (EIF2AK1)
A direct implication of HRI in virus infection was reported only recently for its fish homologues. In flounder cells (Paralichthys olivaceus), an upregulation of HRI was observed upon infection with Scophthalmus maximus rhabdovirus and poly(I:C) treatment, at both the mRNA and protein levels [159]. Furthermore, overexpression of HRI in orange-spotted grouper (Epinephelus coioides) resulted in an increase in eIF2α phosphorylation and inhibition of red-spotted-grouper nervous necrosis virus (RGNNV) replication [160]. Both studies suggested that HRI might have a similar function to PKR in fish.
Normally, HRI is activated by high intracellular levels of reactive oxygen species (ROS) and other imbalances of redox homeostasis, e.g., those induced by arsenite [161]. Several viruses interfere with mitochondrial, peroxisomal, and ER functions and can thereby lead to ROS production, as observed during infection with influenza virus [162], flaviviruses such as DENV, WNV, and JEV [163], as well as chronic viruses such as HCV and hepatitis B virus [164,165,166,167], HIV [168], and poxviruses [169]. In many cases, ROS production represents a necessary event for viral replication or other processes such as capping [170,171]. At the same time, viruses manipulate the antioxidative defense system to maintain ROS levels in a range that is optimal for their purpose, without inducing cell death [172]. However, in contrast to fish, there are no reports, to our knowledge, showing an activation and implication of HRI upon virus infections in mammalian cells.
4. Stress Kinases—Mediators of Innate Immune Signaling
Beside their function in translational control, stress kinases were reported to be involved in a multitude of immune modulatory processes. In the following section, we summarize the currently known connections between the immune and stress sensing pathways.
4.1. Innate Immune Signaling Pathways
Innate immune sensors represent the first line of defense against viral infection. They span a wide range of receptor families: RIG-I-like receptors (RLRs) including retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5), Toll-like receptors (TLRs), C-type-lectin receptors, NOD-like receptors, AIM2-like receptors, and cytosolic DNA sensors (CDSs) such as ZBP-1, GMP-AMP synthase (cGAS), DDX41, and IFI16. Due to differences in their cellular localization (plasma membrane, endosomes, cytosol) and binding preference (proteins, dsDNA, ssRNA, dsRNA), the diversity of these sensors enables the detection of a large panel of PAMPs. Upon ligand binding, receptors initiate specific intracellular signaling cascades via different signaling adapters. RLRs signal via the mitochondrial-associated adaptor protein MAVS, CDSs via the ER adaptor molecule stimulator of interferon response CGAMP interactor (STING), and endosomal TLRs via TRIF and MyD88 [173,174,175,176,177].
Notably, all these signaling pathways converge on the activation of the transcription factors IFN-regulatory factor (IRF) 3 and 7 by the TANK-binding kinase 1 (TBK1) and nuclear factor-κ-B (NF-κB) by the IκB kinase (IKK) complex [9,10,178,179]. Nuclear translocation of IRF3/7 phosphorylated forms leads to the induction of type I IFN genes. Upon secretion, IFNs act in an autocrine and paracrine manner, engage with IFN receptors and induce Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling [180], leading to the transcriptional activation of hundreds of IFN-stimulated genes (ISGs) with antiviral activity [181], including PKR. Thereby, type I IFNs establish an antiviral state that is critical for containment of viral infections. In parallel, phosphorylation of IκB by the IKK complex results in its dissociation from NF-κB and its degradation. In turn, NF-κB translocates into the nucleus where it initiates transcription of numerous pro-inflammatory cytokines and chemokines (e.g., interleukin (IL)-1, IL-6, IL-8, tumor necrosis factor α (TNF-α)), as well as anti-apoptotic proteins (e.g., Bcl-2, cFLIP, Fas) that promote cell survival [182,183].
The expression of cytokines and IFNs is further controlled at the posttranscriptional level via mitogen-activated kinases Erk1/2 and p38 MAPK signaling, through the downstream kinases Mnk1/2 and MK2. The phosphorylation of eIF4E by Mnk1/2, for instance, upregulates translation of IκB and IRF-1 [184,185]. Moreover, MK2-dependent phosphorylation of Tristetraprolin (TTP), an RBP that binds to AU-rich elements (AREs) located in the 3′ UTR of many cytokine and IFN mRNAs, prevents rapid degradation of these mRNAs and further promotes their translation [186,187,188].
4.2. Impact of Stress Kinases on Innate Immune Signaling Pathways
IFN and cytokine production is not only regulated by the abovementioned immune sensing pathways. Several lines of evidence indicate that eIF2α-kinases modulate immune signaling pathways at several levels (Figure 1).
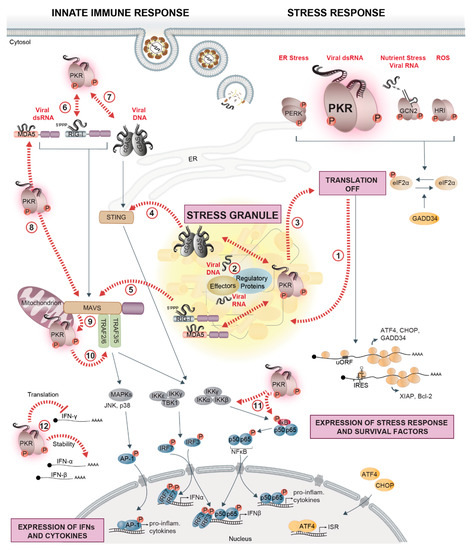
Figure 1.
PKR at the crossroads of virus sensing, innate immune signaling, and stress response pathways. Cells evolved several cytosolic sentinels that detect virus infection and initiate defense mechanisms, including innate immune and stress responses. Cytosolic nucleic acid sensors RIG-I, MDA5, and cGAS signal via the mitochondrial adapter protein MAVS and the ER-adapter protein STING, triggering downstream signaling cascades via the IKK complex (IKKγ, IKKα, IKKβ) and IKKε/IKKγ/TBK1. The IKK complex phosphorylates the NF-κB (p50, p65) inhibitor IκB, whose degradation enables the nuclear translocation of NF-κB and transcriptional activation of IFN-β and pro-inflammatory cytokines. TBK1, on the other hand, phosphorylates IRF3/7, whose nuclear translocation mediates the transcriptional activation of IFN-α/β (left side). Cytosolic dsRNA that accumulates during viral replication is sensed by the stress kinase PKR. As a consequence, phosphorylation of eIF2α strongly represses the translation of most cellular mRNAs, while the translation of factors related to the stress response (ATF4, CHOP, and GADD34) or cell survival (XIAP and Bcl-2) is selectively favored (right side). The other eIF2α-kinases, GCN2, PERK, and HRI, contribute to translation suppression by detecting virus-induced changes in cellular homeostasis such as nutrient deprivation and accumulation of reactive oxygen species (ROS) or unfolded proteins in the ER. In the cytosol, untranslated mRNAs condense together with numerous RBPs and form SGs (1). Upon infection with certain viruses, innate immune sensors, PKR, regulators of stress and immune sensors, and interferon (IFN)-induced effectors localize in stress granules (SGs) together with viral RNA or DNA, forming a signaling platform (2) that coordinates and potentiates the antiviral response (3,4,5). PKR is at the crossroads of stress and innate immune signaling pathways: PKR interacts with RIG-I, promotes its activation, and amplifies the downstream signaling cascade (6). A similar interaction exists with cGAS (7). PKR promotes MDA5 filament formation, is activated by MDA5 to enhance downstream MAVS signaling (8), and, in turn, can also be activated by MAVS (9). PKR affects pro-inflammatory responses by interacting with TRAFs to activate the NF-κB signaling and potentially the JNK/p38 MAPK pathway leading to AP-1 activation (10), or directly regulates NF-κB activity via phosphorylation of IκB and IKK (11). Finally, PKR is involved in controlling the stability and translation of IFN mRNAs (12). Black arrows indicate signaling pathways; dashed red arrows indicate crossroads between stress response pathways, innate immune signaling, and SGs.
A first mechanism is a direct consequence of translational repression by the eIF2α-kinases, which lowers the levels of key regulatory proteins, especially labile proteins such as IκB, A20, and SHIP-1, leading to elevated activity of NF-κB and IRF3 [189,190,191,192]. A second mechanism involves CHOP, a central uORF-regulated transcription factor induced during the ISR. CHOP causes transcriptional inhibition of peroxisome proliferator-activated receptor γ, which, in turn, is a negative regulator of NF-κB transcriptional activity [193]. Thereby, CHOP indirectly augments NF-κB activity under stress conditions. Furthermore, PKR-mediated translation inhibition was found to be required for full activation of the stress-activated JNK upon poly(I:C) transfection [194].
Independently of its function in translational control, PKR orchestrates a variety of immune and survival pathways, thereby influencing cell fate decisions. For instance, PKR has been implicated in NF-κB activation by directly phosphorylating the NF-κB inhibitor IκB [195]. However, later reports indicated that PKR activates NF-κB signaling indirectly, either in a translation-dependent manner as described above, or by providing a signaling platform via its dsRBD, which recruits various signaling molecules and allows PKR to function as a scaffold independently of its kinase activity. Accordingly, PKR was shown to interact with the β subunit of the IKK complex [196,197,198] as well as with TNF receptor-associated factor (TRAF) 2, TRAF5, and TRAF6 [199]. Additionally, PKR was found to directly interact with components of the RIG-I/MDA5 signaling pathway. For instance, during the very early response to HCV infection, PKR interacts with MAVS and TRAF3, thereby inducing ISG15. Subsequent ISGylation of RIG-I interferes with RIG-I activation and thus, limits IFN induction [200]. In contrast, DHX36-mediated activation of PKR in response to IAV and NDV infection promotes RIG-I activation [201]. PKR was also reported to associate with MDA5 and to stimulate IFN-β production via the MAVS-IRF3/7 signaling cascade in response to VACV infection [202]. In addition, the interaction of PKR with MAVS is suggested to promote PKR activation in response to dsRNA [202,203]. cGAS together with G3BP1 was shown to form a complex with PKR, which is important for cGAS activation and IFN production upon dsDNA exposure. Vice versa, PKR was also activated within the complex [204]. Interactions of PKR with immune-related transcription factors, including STAT1 [205] and STAT3 [206], have also been reported. Finally, PKR coordinates IKK, JNK, and p38 MAPK activity in response to pro-inflammatory stimuli such as TNF-α and IL-1 [207,208].
Another mode by which PKR affects immune signaling is through binding to cis-acting regulatory elements in cytokine mRNAs. Binding of PKR to an element in the TNF-α 3′ UTR was shown to promote TNF-α pre-mRNA splicing [209] and binding to the IFN-γ 5′ UTR appears to suppress IFN-γ translation beyond the effect of PKR on global protein synthesis [210]. PKR has also been implicated in sustaining IFN-α/β mRNA integrity in response to a subset of RNA viruses that activate MDA5 specifically, but the exact mechanism still needs to be clarified [211].
Unlike those identified for PKR, links between the other stress kinases, namely GCN2, PERK, and HRI, and innate immune signaling pathways, are less well understood and are often not studied in the context of virus infections. In vitro, HRI mediates NF-κB activation by phosphorylating IκB [212]. Further to this, HRI was shown to be important in fish for the NF-κB-mediated immune response to RGNNV [160]. Reports about GCN2 focus on its ability to negatively regulate inflammatory responses or positively influence antigen-presentation [213,214,215]. Direct interactions with immune signaling components remain to be investigated. PERK acts as an activator of the JAK1/STAT3 signaling pathway in the context of neuroinflammation [216] and was shown to be important for poly(I:C)-induced TLR inflammatory signaling [217]. On the other hand, PERK activation was reported to induce the degradation of the type I IFN receptor IFNAR1, and thereby inhibits JAK/STAT-mediated production of many ISGs. Viruses such as HCV or vesicular stomatitis virus (VSV) take advantage of that and actively trigger IFNAR1 degradation via PERK activation [218,219]. Further investigations about a possible direct implication of HRI, GCN2, and PERK in innate immune signaling are needed.
Downstream of the eIF2α-kinases, signal transducers also link the ISR to the IFN signaling. ATF4, which is translationally activated upon eIF2α phosphorylation, directly interacts with IRF7, and affects IFN-α/β induction in response to viral infection. This interaction is bidirectional, since IRF7 upregulates ATF4 expression and activity, while ATF4 inhibits IRF7 activation [220]. Moreover, GADD34 expression was proposed to be induced by the MAVS-IRF3/7 pathway in response to poly (I:C) or infection with VSV [192]. Additionally, GADD34 is involved in negative feedback upon TNF-α activation: it recruits CUE domain-containing 2 (CUEDC2) to PP1 and thereby leads to the dephosphorylation of IKKα and β and a decrease in NF-κB activity [221].
Altogether, the translational inhibition of unstable immune-regulatory proteins, as well as interactions of stress kinases or downstream signal transducers with components of the immune signaling cascade, promote the transcription of IFNs and pro-inflammatory cytokines. Hence, induction of the stress response, especially the activation of PKR, upon viral infections strongly contributes to establishing a pro-inflammatory and antiviral state. At the same time, PKR, PERK, ATF4, and GADD34, however, also initiate distinct negative feedback loops in order to fine-tune and possibly locally and temporally restrict IFN production to prevent an overactivation of the innate immune system.
An unsolved question is how antiviral proteins are synthesized when global translation is repressed. While uORFs facilitate translation of mRNAs under such conditions, as exemplified by ATF4 [222], most mRNAs encoding antiviral proteins do not appear to harbor uORFs. GADD34 is induced as a negative feedback regulator upon translational inhibition, leading to dephosphorylation of eIF2α and resumption of translation. Mathematic modeling in combination with flow cytometry experiments suggest that this negative feedback loop contributes to the stochastic expression of IFN in individual cells upon poly(I:C) treatment and VSV infection, whereby transcriptional induction of IFNs during translation-off states is followed by synthesis of IFNs during translation-on states [192,223]. Recently, two different laboratories made another important contribution to the understanding of how antiviral factors are preferentially produced in response to viral infections. They discovered that ribonuclease L (RNase L), which is activated in response to dsRNA, strongly depletes cellular mRNA pools through wide-spread mRNA degradation, but specifically leaves mRNAs encoding IFNs, cytokines, and other defense proteins intact [224,225,226]. The mechanism by which these mRNAs are excluded from RNase L-mediated cleavage remains unclear, but probably involves regulatory sequences, RNA secondary structures, and RBPs that protect individual mRNAs from degradation. Hence, it appears that the combination of increased transcription and selective mRNA stabilization and translation permits the synthesis of antiviral factors under conditions of repressed global translation.
An early link between the ISR, in particular the formation of SGs, and the temporal control of cytokine production comes from studies on the adaptive immune system. In naïve T helper cells, IL-4 mRNA was found to accumulate in SG-like foci in the cytoplasm during T cell priming, concomitant with elevated phosphorylation of eIF2α. The release of these mRNAs then allowed for the rapid production of cytokines during T cell restimulation [227], suggesting that SGs can exert storage and regulatory functions outside of classical stress conditions.
5. SGs as Immune Signaling Platforms in Antiviral Defense
While SGs assemble in response to translational shut-off, they are not necessary for translation suppression under stress conditions. Rather, SGs were proposed to function as (i) hubs for modulating local protein and mRNA concentrations; (ii) timers for the stress response, marking a “window of opportunity” during which the stress can be resolved or an apoptotic program will be initiated; (iii) joint assemblies of unfolded proteins and translation complexes that coordinate the activities of the protein synthesis and folding machineries; (iv) storage sites for pre-assembled initiation complexes, allowing for rapid resumption of protein synthesis when cells recover from stress; (v) signaling platforms that connect stress sensors with effectors of immune responses, especially in the context of viral infection [15,16,18,228,229].
Viruses deploy many different strategies to inhibit the formation of SGs, suggesting that SGs serve an antiviral function. In particular, flaviviruses have evolved numerous mechanisms to interfere with SG assembly. The core protein of JEV, for instance, interacts with Caprin1 to relocalize G3BP1 and USP10 to the perinuclear region, thereby preventing SG formation [230]. Other SG components seem to be sequestered by viral RNAs, as reported for TIA1 and TIAR, which bind to the genomic RNA 3′ end of DENV, WNV, and tick-borne encephalitis virus [231,232]. At later stages of infection, flavivirus genomes are degraded by the cellular exoribonuclease XRN1 from the 5′ end up to a compact pseudoknot structure in the 3′ UTR, leading to the cytosolic accumulation of the remaining subgenomic flaviviral RNA (sfRNA) [233,234]. G3BP1, G3BP2, Caprin1, and USP10 are sequestered by sfRNAs, which leads to inhibition of ISG mRNA translation and thus, attenuation of the antiviral response [235]. A similar sequestration strategy was observed for SeV infection, during which trailer RNAs, i.e., small abortive products of viral genome replication, bind TIAR, and inhibit SG formation [236,237,238]. Our own work showed that flaviviruses uncouple the stress response from translation control by blocking both eIF2α phosphorylation and SG assembly, while protein synthesis is still suppressed [58]. In line with this observation, other laboratories reported that the expression of single viral proteins such as Zika virus (ZIKV) capsid, NS3, NS2B-3, or NS4A is sufficient to inhibit SG assembly [239,240,241]. The precise mechanism behind this inhibition remains to be uncovered. In the following, we will focus on the role of SGs within the complex signaling network that controls cellular reprogramming towards an antiviral state (Table 1 and Figure 2).

Table 1.
Localization of proteins with antiviral functions in SGs during stress or viral infection.
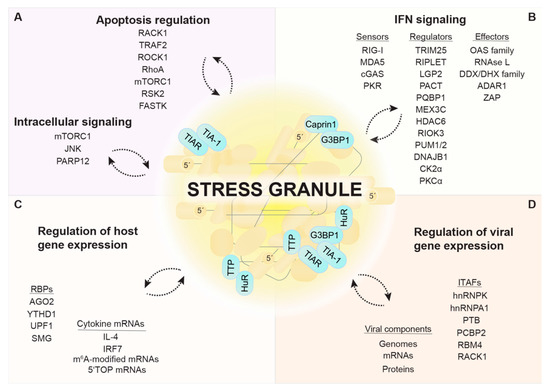
Figure 2.
SGs as immune and stress signaling platforms. SGs function as immune and signaling platforms by phase-separating and concentrating regulatory proteins involved in apoptosis induction and other intracellular signaling pathways (A) and IFN signaling. (B). Certain RBPs and cellular mRNAs (e.g., cytokine mRNAs) preferentially localize in SGs (C). Viral components and ITAFs that control viral gene expression are also detected in SGs (D). SG composition is highly stress- and cell type-specific. Notably, many components have been detected in SGs under metabolic or environmental stress conditions, while an investigation in the context of viral infection is still missing. The localization and function of some SG components is dependent on the interaction with specific SG core proteins, indicated in turquoise.
5.1. G3BP1 at the Interface between SGs and the IFN Response
G3BP1 turns out to be a target of particular importance for viruses, which aim to interfere with its function as a SG nucleator and regulator of immune responses. Moreover, G3BP1 appears to facilitate replication of many viruses. Several viral proteins were shown to directly recruit G3BP1 to sites of viral replication, e.g., HCV polymerase NS5B [271,296,297,298], Chikungunya virus nsP3 [299,300], Semliki Forest virus and SINV Nsp3 [301,302], and Junín virus N protein [303]. Murine norovirus inhibits the formation of canonical SGs not only by redistributing G3BP1 together with the NS3 protein to sites of viral replication, but also by modifying the interactome of G3BP1 [304]. Notably, G3BP1 is directly involved in translation of the norovirus genome by associating with the VPg viral cap complex to help ribosome recruitment [305].
In recent years, G3BP1 was also found to promote both the inflammatory and the IFN responses. Overexpression of G3BP1 was shown to induce the formation of SGs to which innate immune factors such as PKR, oligoadenylate synthetase (OAS) 2, and RNase L are recruited, which, in turn, promote the production of certain cytokines such as IL-17, MIP-3a/b, and MCP-5 via activation of the NF-κB and JNK pathways [256]. Independently of its role in SG assembly, G3BP1 was also shown to interact with the RNA sensor RIG-I and positively regulates its downstream signaling [306,307], as well as with the DNA sensor cGAS, thereby enhancing IFN-β production [308]. This might be the reason why several members of the Picornaviridae and related viruses have evolved G3BP1-cleaving proteases, as reported for the Leader protein of foot-and-mouth disease virus, Theiler’s murine encephalomyelitis virus (TMEV) and mengovirus [280,309,310], the proteinase 3C of poliovirus, coxsackievirus B3 and encephalomyocarditis virus (EMCV) [245,311,312,313], as well as the 3C-like proteinase NS6 of feline calicivirus [314]. Hence, G3BP1 and SGs seem to be at the nexus of the stress response and innate immune signaling. The role of SGs as a signaling platform of the antiviral response will be described in the paragraph below.
5.2. SGs, a Platform to Initiate IFN Signaling?
In cells infected with IAV lacking the NS1 protein (IAVΔNS1) and thus, unable to counteract PKR, viral RNA was found to co-localize in SGs together with the immune sensors RIG-I and MDA5 as well as other antiviral effectors such as OAS, RNase L, and PKR. A similar redistribution of RIG-I was also observed upon infection with EMCV, adenovirus, and SINV. In these infection models, the inhibition of SG assembly by depletion of essential SG components resulted in a strong attenuation of IFN production and an increase in viral replication. Based on these results, Onomoto et al. proposed the term “antiviral SGs” (avSGs) to indicate that SGs may serve as a platform for the activation of innate immune sensors by viral RNA and the subsequent initiation of the IFN response [242]. While most RNA viruses are sensed by RIG-I alone or by RIG-I and MDA5 together [315], MDA5 is essential for IFN-α/β induction in response to Picornaviridae infection [316,317]. In agreement with the idea of avSGs, MDA5 and dsRNA were found to locate in EMCV-induced SGs at early times post infection. At later stages of EMCV infection, cleavage of G3BP1 by the EMCV protease 3C resulted in SG dissolution concomitant with a reduced IFN-β and cytokine response, which could be rescued expressing a cleavage-resistant G3BP1 mutant [245]. The characterization of avSGs formed upon infection with SeV, again revealed the presence of many antiviral components and SG formation was shown to be important for IFN-β production and viral restriction [263]. Furthermore, late during NDV infection, uncapped viral RNA(+) derived from read-through transcription was found to accumulate together with RIG-I in SGs and trigger RIG-I activation. Consistent with the idea of avSGs, IFN-β induction could be reduced by the disruption of SGs in this model [243].
In contrast to these findings, some studies suggest that SG formation may not be necessary for IFN production upon virus infection. During infection with a mutant mengovirus lacking the Leader protein, for instance, localization of MDA5 to SGs was not a prerequisite for IFN induction. However, it may be important to note that viral dsRNA could not be detected within SGs in this infection model, which may indicate that SGs adopt an antiviral function as platforms for RIG-I or MDA5 activation only when they accumulate viral RNA [95]. Another study showed that disruption of SGs in IAVΔNS1- and SeV-infected cells did not reduce but actually increased RIG-I-mediated IFN-β production [202], which is in direct contradiction with previous observations [242,263]. It is therefore still unclear whether the changes observed in the IFN response ensue from the disruption of SGs as a coordinating platform or are due to the immunomodulatory function of single SG components. Additional experiments based on genetic ablation of other SG components, interference with SG assembly by cycloheximide or ISRIB treatments, or the use of eIF2α S51A mutant cells might be necessary to answer this question
Finally, MDA5 and RIG-I were also detected in SGs formed under other stress conditions such as heat shock and arsenite treatment [95,242], which raises the possibility that MDA5/RIG-I can also assemble with endogenous (e.g., damaged) RNAs in SGs, or that these sensors passively phase separate in SGs.
Whether SGs contribute to the antiviral response upon infection with DNA viruses is even less clear. Upon DNA virus infection, viral DNA is sensed by specialized DNA sensors, among those cGAS. In addition, viral transcripts, cytosolic noncoding RNAs transcribed by the RNA polymerase III, as well as Alu-derived host RNAs can activate MDA5 or RIG-I [318]. In the case of VACVΔE3L, MDA5-mediated IFN-β production was unchanged upon SG dissolution, and rather dependent on the immunomodulatory function of PKR [202]. HSV-1 infection induces formation of SGs, in which dsRNA (whose origin is unclear) was detected. In this infection model, IFN production was induced by the DNA sensing cGAS-STING pathway rather than by MDA5/RIG-I, and preceded PKR activation and SG formation, indicating that SGs are dispensable for cGAS-mediated IFN production upon HSV-1 infection [121]. Interestingly, cGAS was reported to associate with dsDNA, G3BP1, and PKR in a complex and one study claimed the presence in cytoplasmic foci upon dsDNA transfection [204,308]. While it is not clear if these foci represent canonical SGs, they were proposed to be essential for cGAS activation and downstream IFN-β production [204]. Of note, polyglutamine binding protein 1 (PQBP1), identified as a co-sensor of cGAS in the context of HIV infection [319], was shown to localize to SGs [70,255]. Hence, SGs might support cGAS-mediated IFN production under specific conditions or for particular virus types. However, this needs to be further investigated.
Given these discrepancies, the link between SGs and innate immune signaling appears to be complex. Besides the co-localization of innate immune sensors with viral RNAs or DNAs within SGs, the type of virus and the presence or absence of regulatory factors are likely to affect the potential of SGs to serve as platforms initiating the IFN response. It should further be noted that studies so far have mainly focused on the impact of SGs on the production of IFN-β or cytokines such as IL-6, RANTES, and CXCL10. Hence, it would be interesting to investigate how SGs might influence the expression of other IFN types and subtypes, and how this could affect virus replication.
5.3. Regulators of the Innate Immune Sensors
RLR activity is tightly controlled by multiple posttranslational modifications and protein interactions. Tripartite motif protein 25 (TRIM25) and Riplet are two examples of E3 ubiquitin ligases, which, according to a current model, mediate consecutive K63-linked ubiquitination of RIG-I. The first ubiquitination event mediated by Riplet leads to the exposure of the RIG-I CARD domains and their subsequent ubiquitination by TRIM25. Thereby RIG-I gets activated, oligomerizes, and initiates downstream MAVS signaling [320]. Both E3 ubiquitin ligases were found to be recruited together with RIG-I to SGs when cells were exposed to poly(I:C) [249]. It is not clear, however, whether co-localization of these proteins within SGs is important for the sequential ubiquitination of RIG-I and downstream signaling events. Using a bimolecular fluorescence complementation assay and super-resolution microscopy in SeV-infected cells, RIG-I was identified in two distinct complexes—RIG-I/TRIM25 and RIG-I/MAVS—with different cellular localization [248]. While TRIM25/RIG-I complexes localized in SGs, RIG-I/MAVS complexes remained attached to mitochondrial membranes. An interaction between MAVS and TRIM25 was barely observed [248], contrasting previous studies in which TRIM25 was proposed to act as an ubiquitin ligase of MAVS [321,322]. RIG-I/TRIM25 complexes further appeared to be in close proximity with mitochondria upon virus stimulation [248], consistent with previous reports about physical contacts between avSGs and MAVS [242,243]. These findings suggest that RIG-I first needs to interact with TRIM25 within SGs to become ubiquitinated and activated before it is released from the complex to activate MAVS at mitochondria. It should be noted that TRIM25 also has a second function: when bound to FAT10, it stabilizes the protein, which acts as a negative regulator of RIG-I and prevents the formation of avSGs under conditions of inflammation [323]. Recent in vivo studies suggest that Riplet alone is sufficient for RIG-I activation [324,325], and confirmed a role of Riplet in modulating the kinetics of RIG-I recruitment into SGs. However, the ability of Riplet to activate RIG-I signaling was not dependent on SG formation [324]. Apart from Riplet and TRIM25, other E3 ubiquitin ligases can mediate RIG-I ubiquitination including MEX3C, an RNA-binding E3 ligase that was found to localize in NDV-induced avSGs and plays an essential role in RIG-I-mediated IFN signaling [244]. Moreover, RIG-I activity is regulated by the activity of several kinases, phosphatases, and acetyl transferases, some of which localize to SGs such as histone deacetylase 6 (HDAC6) [252], PKC-α [251], and the α-subunits of CKII [250].
MDA5 activity is similarly regulated by posttranslational modifications [326]. RIO kinase 3 (RIOK3) is involved in maintaining MDA5 inactive under normal conditions by phosphorylating its C-terminal domain, thereby interfering with MDA5 filament formation on dsRNA and downstream IFN signaling. Upon poly(I:C) exposure, RIOK3 was shown to be partially recruited to SGs, together with MDA5 [246]. DNAJ heat shock protein family (Hsp40) member B1 (DNAJB1), a member of the Hsp40 family, was identified as an interactor of MDA5 that negatively regulates MDA5-MAVS signaling. Upon poly(I:C) treatment, DNAJB1 in conjunction with Hsp70 was found to co-localize with SG markers and the mitochondrial membrane, negatively affecting MDA5 oligomerization and MAVS aggregation [247]. If and how the co-localization of MDA5 together with the DNAJB1-Hsp70 complex or RIOK3 within SGs affects MDA5-MAVS signaling still needs to be investigated.
Laboratory of genetics and physiology 2 (LGP2, DHX58) belongs to the RLR family and acts as a modulatory co-receptor of RIG-I and MDA5 [315]. LGP2 has been shown to localize in SGs upon IAVΔNS1 infection [242]. Moreover, the positive activator of PKR (PACT, PRKRA) was identified as an LGP2 interactor essential for RLR regulation [327], and also shown to localize in SGs upon treatment with hippuristanol [257], a steroid compound that interferes with cap-dependent translation [270]. Interestingly, PACT was also found in a complex with RIG-I and MDA5, positively regulating their downstream signaling [328,329]. In addition, two members of the pumilio RBP family, PUM1 and PUM2, localized to SGs and were found to associate with LGP2 upon NDV infection, enhancing its dsRNA binding ability through conformational changes and thereby increasing the IFN response [253]. Whether co-localization of LGP2 with PACT, PUM1, and PUM2 in SGs is necessary for the regulatory effects on RIG-I and MDA5 activity is currently not clear.
5.4. Stress Kinase PKR and Its Regulators
Localization of the dsRNA-sensor PKR in SGs is a prime example of their functional relevance [121,256,330]. PKR is recruited to SGs through its direct interaction with the NTF2 and PXXP domains of G3BP1 [330]. By complex formation with G3BP1 and Caprin1, PKR can be activated in a dsRNA-independent manner. The high local concentrations of all three components in SGs are likely what drive formation of the tripartite G3BP1/Caprin-1/PKR complex. Activated PKR is subsequently released from SGs into the cytosol, where it phosphorylates eIF2α and thereby promotes both translation repression and SG persistence [330]. Hence, SGs are part of a feed-forward loop that amplifies PKR activity and restricts viral replication. This amplification mechanism might explain why so many viruses simultaneously target PKR and SG assembly. One may speculate that the ubiquitin-specific peptidase USP10, which binds to G3BP1 and inhibits SG formation [331], could affect PKR activation and other antiviral signaling events in SGs.
In contrast, activation of PKR by MAVS was reported to occur outside of SGs prior to its recruitment into SGs [203]. This finding corroborates the idea that PKR is activated in sequential steps and that SGs help to maintain PKR activity at later stages of the infection. Interestingly, PKR was also reported to localize both inside and at the outer membrane of mitochondria [99]. Since SGs and mitochondria are often found in close proximity [248], it is tempting to speculate that PKR shuttles between the two compartments. Accordingly, recruitment of PKR to SGs could affect not only the activation status of PKR within protein complexes inside SGs, but also within protein complexes that specifically form inside or at mitochondria. This scenario is well in line with the tight collaboration between SGs and mitochondria in the RIG-I/MAVS activation pathway described above (Figure 3).
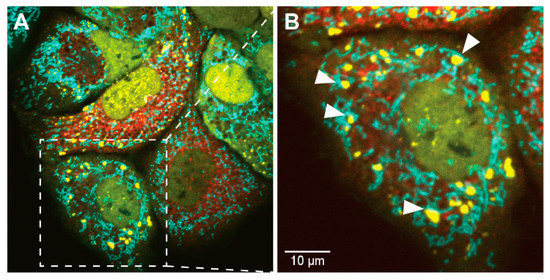
Figure 3.
SG localization in close proximity to mitochondria. Shown are live-cell microscopy images of HCV-induced dynamic assembly and disassembly of SGs. (A) human hepatocarcinoma Huh7 cells, stably expressing YFP-TIA1 (in yellow) and mTurquoise2-mito [339], a mitochondrial targeting peptide (in cyan), were infected with an HCV-mCherry reporter virus (in red). (B) Cropped section. White arrows indicate examples of SGs that are localized in close proximity to mitochondria (scale bar 10 µM).
Besides G3BP1 and Caprin1, PACT, transactivation response element RNA-binding protein (TRBP), and P58IPK modulate PKR activity by directly targeting its kinase domain. It is generally thought that PACT stimulates PKR whereas TRBP and P58IPK inhibit PKR activity, although exceptions have been reported and may be cell-context specific [332,333,334,335,336]. The interaction between PKR, PACT, and TRBP is especially important for cellular physiology during viral infections and has implications in stress recovery and the RNA-induced silencing pathway [335,337,338]. However, it is currently unclear whether the recruitment of PKR into SGs affects its interaction with PACT, TRBP, and P58IPK. Upon hippuristanol treatment, PACT localizes in SGs where it was proposed to form a complex with argonaute 2 (AGO2) [257]. Furthermore, the SG proteins TIA1 and TIAR were found to suppress PKR activity, although they seem to exert this effect by controlling PACT pre-mRNA splicing, independently of their role in SG assembly [336]. In addition to PACT [257], several modulators of PKR activity, including NFAR1/2 [258,259], adenosine deaminase RNA-specific 1 (ADAR1) [77,262] and Staufen [260], were found to localize in SGs or modulate SGs formation. Further studies are needed to assess whether these PKR regulators relocalize into, or are specifically excluded from, SGs upon virus infection. The differential activation of PKR within different complexes and cellular compartments is likely to dictate the spectrum of PKR targets and thereby decides whether PKR has pro-apoptotic, anti-apoptotic, inflammatory, or translational effects.
5.5. Oligoadenylate Synthase and RNase L
Proteins of the OAS family are IFN-induced effectors that are activated by dsRNA binding [340]. OAS proteins polymerize ATPs into 2′-5′-linked oligoadenylates (2-5A), whose lengths vary from dimers to 30-mers [341]. Trimeric or longer 2-5A molecules serve as unique secondary messengers to activate RNase L, which induces RNA degradation [342]. Studies indicate the presence of several proteins of the OAS family within SGs, including OAS1, OAS2, and OAS-like protein (OASL), as well as its mouse homolog OASL1 [159,242,256,267]. OASL, in contrast to the other members of the OAS family, lacks 2-5A synthetase activity but instead, contains ubiquitin-like (UBL) domains [343,344]. OASL was found to co-localize with RIG-I in SGs upon SeV infection and enhances RIG-I-signaling via its UBL domains, mimicking K63-linked ubiquitination. A similar function was suggested for one of the mouse homologues, OASL2 [266]. Interestingly, in the case of DNA virus infections, OASL/OASL2 had the opposite effect, negatively regulating cGAS signaling [345]. OASL1, the second mouse OASL homologue, was similarly shown to be recruited to IAV-induced SGs and to interact with many SG components, including PKR and MDA5 [267]. OASL1 was proposed to act both as a positive and negative regulator of immune signaling. By binding to viral RNA, OASL1 supports MDA5-mediated IFN production early in infection, while its binding to IRF7 mRNA prevents IRF7 synthesis and thereby dampens transcriptional activation of IFN at later time points. However, while SGs are supposed to be important for the activation of the MDA5-MAVS signaling, translational control of IRF7 was not affected by the absence of SGs [267]. Whether the localization of the active 2-5A synthetases OAS1 and OAS2 within SGs has an impact on their function still needs to be clarified.
RNase L activation upon binding of 2-5A results in the cleavage of viral and cellular ssRNAs into small fragments with 5′-hydroxyl and 3′-monophosphate ends [346]. These fragments, which tend to form duplex structures, can activate RIG-I/MDA5-MAVS signaling pathways and enhance the response to virus infection [347,348]. Recently, RNase L was reported to localize in SGs upon transfection of 2-5A. The resulting RNase L cleavage products were found to activate PKR, hence triggering SG formation and subsequent induction of IRF3-mediated IFN response [263]. Contradictory to these findings, earlier reports indicate that RNase L activity reduces PKR protein levels and eIF2α phosphorylation via destabilizing PKR mRNA [349]. In addition, RNase L was shown to inhibit SG assembly or reduce the size of SGs, likely through the global decay of cellular mRNAs, while antiviral mRNAs (e.g., IFN-β mRNA) escape degradation [224,225,226]. Hence, depending on the cell type and the stage of infection, RNase L can apparently enhance or restrict SG formation and the SG-associated antiviral response. In turn, SGs may potentially serve as platforms for interactions between RNase L and viral RNAs, thus enhancing the antiviral activity of RNase L.
5.6. Editing of dsRNA by Adenosine Deaminase
ADAR1 is an IFN-induced RNA editing enzyme that catalyzes the C6 deamination of adenosine (A) to inosine (I) within dsRNAs [350]. In cellular RNAs, A-to-I editing weakens RNA duplex structures [351], thereby avoiding the activation of PKR, RIG-I, and MDA5 by self-dsRNAs, most importantly Alu-derived dsRNAs [352]. ADAR1 is recruited to SGs in response to different stresses, IFN treatment, poly(I:C) transfection, and MV and HCV infection [77,245,262,353,354]. Considerable evidence supports the notion that ADAR1 acts as an immune suppressor by promoting the survival and replication of RNA viruses. Wild type MV infection causes induction of IFN-β and subsequent activation of ADAR1. Infection with C-deficient MV, which is not able to counteract PKR, induces SGs in ADAR1 knockout but not in ADAR1-sufficient cells, indicating that ADAR1 antagonizes SG formation [353,354,355]. Thus, ADAR1 sustains viral replication through inhibition of both SG formation and IFN production. However, this proviral effect of ADAR1 activity appears to be virus-specific, since ADAR1 was also found to work as an antiviral immune modulator against HCV infection [356]. Therefore, ADAR1 seems to be a regulator for both SG assembly and antiviral immune responses, yet it remains to be determined whether the recruitment of ADAR1 to SGs influences the decision of whether ADAR1 acts in a pro- or antiviral manner.
5.7. Zinc-Finger Antiviral Protein
Zinc-finger antiviral protein (ZAP or PARP13) is an IFN-induced effector protein, which is expressed in two isoforms (ZAP-S and ZAP-L), both lacking poly(ADP-ribose) polymerase (PARP) activity [357]. ZAP is recruited to SGs and participates together with other family members in maintaining SG integrity [264]. In addition, it has a range of different antiviral functions, depending on the factors it interacts with. Acting as an RNA sensor, ZAP was shown to bind to murine leukemia virus (MLV) transcripts, mediating their degradation through recruitment of the RNA exosome machinery. Interestingly, exosome components such as EXOSC5 were found to localize within RNA granules (SGs and PBs) in a ZAP-dependent manner. Similarly, MLV transcripts were recruited [358]. However, it is not clear if the co-localization of these factors into granules is important for viral restriction. Another study found that ZAP was similarly recruited to SINV-induced SGs together with viral RNA. ZAP mutants revealed that recruitment to SGs correlates with ZAP antiviral activity. However, this study also indicated that ZAP interaction partners are important for antiviral function [265]. TRIM25, as an example, was shown to be an essential co-factor for the ability of ZAP to block SINV translation [359]. Beside the exosome, ZAP was further shown to interact with RNA-induced silencing complex (RISC) components such as AGO2, repressing the miRNA ability against antiviral factors, hence mediating an increase in many ISGs [360]. In addition, ZAP-S was reported to interact with RIG-I to strengthen IFN signaling upon IAV and NDV infections [361]. Contradictory to this study, upon knockdown of ZAP-S and exposure with poly(U/UC) RNA, others reported an increase in IFN-β, IFN-λ2, and IFN-λ3 mRNA levels, indicating that ZAP-S has a function in the degradation of those mRNAs, therefore rather in the resolution of the antiviral response. In addition, they claimed that the different cellular localization of both isoforms (with ZAP-S excluded from sites of viral replication) has an impact on their binding preference for host or viral RNA [362]. It would be interesting to investigate how the co-localization of ZAP-S/ZAP-L target RNAs and interactors within SGs shapes ZAP-dependent antiviral functions.
5.8. Other DEAD/H-Box Proteins
DEAD/H-box proteins (DDX, DHX) are a large family of helicases involved in multiple cellular processes, which cover nearly all aspects of RNA metabolism [363,364] and include the RNA sensors RIG-I (DDX58) and MDA5 (RH116). DEAD/H-box proteins can be engaged in cytosolic viral RNA sensing, promote or inhibit IFN and cytokine signaling, function as co-factors for other immune sensors and regulators, and control viral replication by protein/protein and protein/RNA interactions. Whether these proteins promote or inhibit viral infections seems to be virus-specific and depend on the cellular context. The helicases DDX1, DDX3, and DDX21, for instance, are frequently required for efficient cytokine and IFN production, whereas DDX19 has been described as a negative regulator of type I IFN production in the context of viral infections [20]. DHX36 (RHAU) was shown to interact with PKR in an RNA-dependent manner, leading to activation of PKR, subsequent SG assembly, and enhanced RIG-I signaling upon IAVΔNS1 and NDV infection [201]. DHX30 was shown to exert an antiviral role by directly interacting with and stimulating ZAP activity against MLV [365]. Proteome analyses of purified SGs induced by oxidative stress confirmed the presence of multiple DEAD/H-box proteins inside SGs, including DDX1, DDX2 (eIF4A), DDX3, DDX6 (Rck), DDX19 (Dbp5), DDX21, DDX47, and DDX50 as well as DHX30 and DHX36 [70]. Localization in SGs could also be confirmed by immunofluorescence microscopy for DDX1, DDX2, DDX3, DDX6, DDX19, and DHX36 [201,268,269,270,271,272,273,274,275,276,277]. Currently, it is not well understood if the immune regulatory functions of these proteins are affected by their localization in SGs, and localization in SGs during viral infections has so far only been addressed for DDX3, DDX6, and DHX36 [201,271,274,366].
For the RNA helicase DDX3, the connection to viral infections and SG assembly has been studied extensively [367]. Furthermore, HDAC6-mediated deacetylation of DDX3 is needed for the maturation of SGs upon stress, a process by which SGs fuse and grow in size [368]. The ability of DDX3 to induce SG assembly was demonstrated to depend on DDX3′s interaction with eIF4E. A DDX3 mutant with impaired eIF4E binding instead did not only show reduced SG assembly but also reduced cell survival upon stress [272]. In the context of HCV infection, recruitment of DDX3 into SGs has a proviral effect. By recruiting DDX3 into SGs, HCV can prevent DDX3 from exerting its positive effect on the translation of PACT mRNA [366]. Moreover, DDX3 was shown to recruit IKKα into SGs concomitant with activation of IKKα. This activation, however, did not induce pro-inflammatory NF-κB signaling, but enhanced proviral lipogenic gene expression [271]. Future studies will have to show how DDX3 mediates IKKα activation in SGs, if this mechanism operates also upon infection with other viruses, and whether this mode of IKKα activation can also exert antiviral effects via the canonical NF-κB signaling.
Finally, DDX6 was shown to co-localize with RIG-I in SGs upon infection with an NS1-deficient Influenza B virus, and promote IFN-β induction [274]. However, the effect of DDX6 on IFN induction seems to be independent of SGs since it was also observed in cells infected with wild type influenza B virus, which inhibits SG formation [274]. Taken together, experimental data for modulation of DEAD/H-box protein activities via recruitment to SGs, especially in the context of viral infections, are scarce and await further investigation.
7. Conclusions
Knowing whether RBPs and cellular or viral RNAs are actively “recruited”, “sequestered”, or just passively “phase separated” into SGs is much more than a matter of semantics—it is key to a mechanistic understanding of how SGs shape host–virus interactions and coordinate signaling events. Evidence for the presence of a specific RNA or protein in SGs largely relies on microscopy analyses, which have recently been complemented by the development of labeling methods, purification protocols, and the use of high-resolution imaging. However, most SG studies are performed in cells exposed to oxidative stress or other harsh conditions, and many aspects of SG biology still need to be addressed in virus-infected cells.
The co-evolution of virus and host can be described as an arms race that needs to be carefully balanced with the consequences of collateral damage. The dynamic nature of SGs gives testimony to this contest: while some viruses effectively suppress SG assembly, others face an initial wave of SGs that they can only curb during later stages of infection. The appearance of oscillating SGs during chronic virus infections is maybe the most stunning strategy by which cells contain “the enemy within” without suffocating from the antiviral response.
It is tempting to speculate that the condensation state, composition, and function of SGs may change over time as viral infections progress. Further studies building on the recent technical advances will be required to mechanistically dissect the antiviral role of SGs and provide a time-resolved understanding of how stress response pathways and innate immune signaling intersect in the context of viral infections. This may eventually allow us to harness the SGs’ “dance with the devil” for antiviral therapeutic approaches against important human pathogens.
Author Contributions
Writing—original draft preparation, N.E., K.H. and Z.S. Writing—review and editing, G.S. and A.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) project number 278001972-TRR 186 to A.R. and G.S., project number 240245660-SFB 1129 to A.R., and project number 201348542-SFB 1036 to G.S.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Mir, M.A.; Panganiban, A.T. A protein that replaces the entire cellular eIF4F complex. EMBO J. 2008, 27, 3129–3139. [Google Scholar] [CrossRef]
- Schulz, F.; Yutin, N.; Ivanova, N.N.; Ortega, D.R.; Lee, T.K.; Vierheilig, J.; Daims, H.; Horn, M.; Wagner, M.; Jensen, G.J.; et al. Giant viruses with an expanded complement of translation system components. Science 2017, 356, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; De Lamballerie, X.; Yutin, N.; Asgari, S.; Bigot, Y.; Bideshi, D.K.; Cheng, X.W.; Federici, B.A.; Van Etten, J.L.; Koonin, E.V.; et al. “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch. Virol. 2013, 158, 2517–2521. [Google Scholar] [CrossRef]
- Sullivan, M.B.; Huang, K.H.; Ignacio-Espinoza, J.C.; Berlin, A.M.; Kelly, L.; Weigele, P.R.; DeFrancesco, A.S.; Kern, S.E.; Thompson, L.R.; Young, S.; et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ. Microbiol. 2010, 12, 3035–3056. [Google Scholar] [CrossRef] [PubMed]
- Sencilo, A.; Jacobs-Sera, D.; Russell, D.A.; Ko, C.C.; Bowman, C.A.; Atanasova, N.S.; Osterlund, E.; Oksanen, H.M.; Bamford, D.H.; Hatfull, G.F.; et al. Snapshot of haloarchaeal tailed virus genomes. RNA Biol. 2013, 10, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Abergel, C.; Rudinger-Thirion, J.; Giege, R.; Claverie, J.M. Virus-encoded aminoacyl-tRNA synthetases: Structural and functional characterization of mimivirus TyrRS and MetRS. J. Virol. 2007, 81, 12406–12417. [Google Scholar] [CrossRef]
- Mizuno, C.M.; Guyomar, C.; Roux, S.; Lavigne, R.; Rodriguez-Valera, F.; Sullivan, M.B.; Gillet, R.; Forterre, P.; Krupovic, M. Numerous cultivated and uncultivated viruses encode ribosomal proteins. Nat. Commun. 2019, 10, 752. [Google Scholar] [CrossRef]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef]
- Jensen, S.; Thomsen, A.R. Sensing of RNA viruses: A review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef]
- Ma, Z.; Ni, G.; Damania, B. Innate Sensing of DNA Virus Genomes. Annu. Rev. Virol. 2018, 5, 341–362. [Google Scholar] [CrossRef]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2alpha kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30, 963–969. [Google Scholar] [CrossRef]
- Kedersha, N.; Ivanov, P.; Anderson, P. Stress granules and cell signaling: More than just a passing phase? Trends Biochem. Sci. 2013, 38, 494–506. [Google Scholar] [CrossRef]
- Mahboubi, H.; Stochaj, U. Cytoplasmic stress granules: Dynamic modulators of cell signaling and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 884–895. [Google Scholar] [CrossRef]
- Onomoto, K.; Yoneyama, M.; Fung, G.; Kato, H.; Fujita, T. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014, 35, 420–428. [Google Scholar] [CrossRef]
- McCormick, C.; Khaperskyy, D.A. Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 2017, 17, 647–660. [Google Scholar] [CrossRef]
- Poblete-Duran, N.; Prades-Perez, Y.; Vera-Otarola, J.; Soto-Rifo, R.; Valiente-Echeverria, F. Who Regulates Whom? An Overview of RNA Granules and Viral Infections. Viruses 2016, 8, 180. [Google Scholar] [CrossRef]
- Zhang, Q.; Sharma, N.R.; Zheng, Z.M.; Chen, M. Viral Regulation of RNA Granules in Infected Cells. Virol. Sin. 2019, 34, 175–191. [Google Scholar] [CrossRef]
- Montero, H.; Trujillo-Alonso, V. Stress granules in the viral replication cycle. Viruses 2011, 3, 2328–2338. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Lloyd, R.E. Cytoplasmic RNA Granules and Viral Infection. Annu. Rev. Virol. 2014, 1, 147–170. [Google Scholar] [CrossRef] [PubMed]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef]
- Spriggs, K.A.; Bushell, M.; Willis, A.E. Translational regulation of gene expression during conditions of cell stress. Mol. Cell 2010, 40, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Qian, S.B. Translational reprogramming in cellular stress response. Wiley Interdiscip. Rev. RNA 2014, 5, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Mathews, M.B.; Mohr, I. Tinkering with translation: Protein synthesis in virus-infected cells. Cold Spring Harb Perspect. Biol. 2013, 5, a012351. [Google Scholar] [CrossRef]
- Stern-Ginossar, N.; Thompson, S.R.; Mathews, M.B.; Mohr, I. Translational Control in Virus-Infected Cells. Cold Spring Harb Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef]
- Hinnebusch, A.G. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 2011, 75, 434–467. [Google Scholar] [CrossRef]
- Harvey, R.F.; Smith, T.S.; Mulroney, T.; Queiroz, R.M.L.; Pizzinga, M.; Dezi, V.; Villenueva, E.; Ramakrishna, M.; Lilley, K.S.; Willis, A.E. Trans-acting translational regulatory RNA binding proteins. Wiley Interdiscip. Rev. RNA 2018, 9, e1465. [Google Scholar] [CrossRef]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef]
- Galloway, A.; Cowling, V.H. mRNA cap regulation in mammalian cell function and fate. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.L.; Diamond, M.S. Innate immune restriction and antagonism of viral RNA lacking 2-O methylation. Virology 2015, 479-480, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Aitken, C.E.; Lorsch, J.R. A mechanistic overview of translation initiation in eukaryotes. Nat. Struct. Mol. Biol. 2012, 19, 568–576. [Google Scholar] [CrossRef]
- Pavitt, G.D. Regulation of translation initiation factor eIF2B at the hub of the integrated stress response. Wiley Interdiscip. Rev. RNA 2018, 9, e1491. [Google Scholar] [CrossRef]
- Jousse, C.; Oyadomari, S.; Novoa, I.; Lu, P.; Zhang, Y.; Harding, H.P.; Ron, D. Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. J. Cell Biol. 2003, 163, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Kastan, J.P.; Dobrikova, E.Y.; Bryant, J.D.; Gromeier, M. CReP mediates selective translation initiation at the endoplasmic reticulum. Sci. Adv. 2020, 6, eaba0745. [Google Scholar] [CrossRef]
- Connor, J.H.; Weiser, D.C.; Li, S.; Hallenbeck, J.M.; Shenolikar, S. Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol. Cell. Biol. 2001, 21, 6841–6850. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef]
- Kojima, E.; Takeuchi, A.; Haneda, M.; Yagi, A.; Hasegawa, T.; Yamaki, K.; Takeda, K.; Akira, S.; Shimokata, K.; Isobe, K. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: Elucidation by GADD34-deficient mice. FASEB J. 2003, 17, 1573–1575. [Google Scholar] [CrossRef]
- Novoa, I.; Zhang, Y.; Zeng, H.; Jungreis, R.; Harding, H.P.; Ron, D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003, 22, 1180–1187. [Google Scholar] [CrossRef]
- Haghighat, A.; Mader, S.; Pause, A.; Sonenberg, N. Repression of cap-dependent translation by 4E-binding protein 1: Competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 1995, 14, 5701–5709. [Google Scholar] [CrossRef] [PubMed]
- Young, S.K.; Wek, R.C. Upstream Open Reading Frames Differentially Regulate Gene-specific Translation in the Integrated Stress Response. J. Biol. Chem. 2016, 291, 16927–16935. [Google Scholar] [CrossRef] [PubMed]
- Weingarten-Gabbay, S.; Elias-Kirma, S.; Nir, R.; Gritsenko, A.A.; Stern-Ginossar, N.; Yakhini, Z.; Weinberger, A.; Segal, E. Comparative genetics. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science 2016, 351. [Google Scholar] [CrossRef] [PubMed]
- Hellen, C.U.; Sarnow, P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 2001, 15, 1593–1612. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A.; Hatzoglou, M. Cellular IRES-mediated translation: The war of ITAFs in pathophysiological states. Cell Cycle 2011, 10, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.K.; Krausslich, H.G.; Nicklin, M.J.; Duke, G.M.; Palmenberg, A.C.; Wimmer, E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol. 1988, 62, 2636–2643. [Google Scholar] [CrossRef]
- Pelletier, J.; Sonenberg, N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 1988, 334, 320–325. [Google Scholar] [CrossRef]
- Holcik, M.; Lefebvre, C.; Yeh, C.; Chow, T.; Korneluk, R.G. A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat. Cell Biol. 1999, 1, 190–192. [Google Scholar] [CrossRef]
- Sherrill, K.W.; Byrd, M.P.; Van Eden, M.E.; Lloyd, R.E. BCL-2 translation is mediated via internal ribosome entry during cell stress. J. Biol. Chem. 2004, 279, 29066–29074. [Google Scholar] [CrossRef]
- Marques-Ramos, A.; Candeias, M.M.; Menezes, J.; Lacerda, R.; Willcocks, M.; Teixeira, A.; Locker, N.; Romao, L. Cap-independent translation ensures mTOR expression and function upon protein synthesis inhibition. RNA 2017, 23, 1712–1728. [Google Scholar] [CrossRef]
- Kwon, O.S.; An, S.; Kim, E.; Yu, J.; Hong, K.Y.; Lee, J.S.; Jang, S.K. An mRNA-specific tRNAi carrier eIF2A plays a pivotal role in cell proliferation under stress conditions: Stress-resistant translation of c-Src mRNA is mediated by eIF2A. Nucleic Acids Res. 2017, 45, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.B.; Jaffrey, S.R. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Coots, R.A.; Liu, X.M.; Mao, Y.; Dong, L.; Zhou, J.; Wan, J.; Zhang, X.; Qian, S.B. m(6)A Facilitates eIF4F-Independent mRNA Translation. Mol. Cell 2017, 68, 504–514.e7. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.J.D.; Lee, S. A Cap for Every Occasion: Alternative eIF4F Complexes. Trends Biochem. Sci. 2016, 41, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Jaafar, Z.A.; Kieft, J.S. Viral RNA structure-based strategies to manipulate translation. Nat. Rev. Microbiol. 2019, 17, 110–123. [Google Scholar] [CrossRef]
- Edgil, D.; Polacek, C.; Harris, E. Dengue virus utilizes a novel strategy for translation initiation when cap-dependent translation is inhibited. J. Virol. 2006, 80, 2976–2986. [Google Scholar] [CrossRef]
- Roth, H.; Magg, V.; Uch, F.; Mutz, P.; Klein, P.; Haneke, K.; Lohmann, V.; Bartenschlager, R.; Fackler, O.T.; Locker, N.; et al. Flavivirus infection uncouples translation suppression from cellular stress responses. mBio 2017, 8. [Google Scholar] [CrossRef]
- Song, Y.; Mugavero, J.; Stauft, C.B.; Wimmer, E. Dengue and Zika Virus 5′ Untranslated Regions Harbor Internal Ribosomal Entry Site Functions. mBio 2019, 10. [Google Scholar] [CrossRef]
- Wang, Q.S.; Jan, E. Switch from cap- to factorless IRES-dependent 0 and +1 frame translation during cellular stress and dicistrovirus infection. PLoS ONE 2014, 9, e103601. [Google Scholar] [CrossRef]
- Panas, M.D.; Ivanov, P.; Anderson, P. Mechanistic insights into mammalian stress granule dynamics. J. Cell Biol. 2016, 215, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Van Treeck, B.; Protter, D.S.W.; Matheny, T.; Khong, A.; Link, C.D.; Parker, R. RNA self-assembly contributes to stress granule formation and defining the stress granule transcriptome. Proc. Natl. Acad. Sci. USA 2018, 115, 2734–2739. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, P.; Kedersha, N.; Anderson, P. Stress Granules and Processing Bodies in Translational Control. Cold Spring Harb Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.Y.; Dunham, W.H.; Hong, S.J.; Knight, J.D.R.; Bashkurov, M.; Chen, G.I.; Bagci, H.; Rathod, B.; MacLeod, G.; Eng, S.W.M.; et al. High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol. Cell 2018, 69, 517–532.e11. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, L.; Cieren, A.; Barbieri, S.; Khong, A.; Schwager, F.; Parker, R.; Gotta, M. UBAP2L Forms Distinct Cores that Act in Nucleating Stress Granules Upstream of G3BP1. Curr. Biol. 2020, 30, 698–707.e6. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kedersha, N.; Lee, D.S.W.; Strom, A.R.; Drake, V.; Riback, J.A.; Bracha, D.; Eeftens, J.M.; Iwanicki, A.; Wang, A.; et al. Competing Protein-RNA Interaction Networks Control Multiphase Intracellular Organization. Cell 2020, 181, 306–324.e28. [Google Scholar] [CrossRef]
- Huang, C.; Chen, Y.; Dai, H.; Zhang, H.; Xie, M.; Zhang, H.; Chen, F.; Kang, X.; Bai, X.; Chen, Z. UBAP2L arginine methylation by PRMT1 modulates stress granule assembly. Cell Death Differ. 2020, 27, 227–241. [Google Scholar] [CrossRef]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef]
- Tourriere, H.; Chebli, K.; Zekri, L.; Courselaud, B.; Blanchard, J.M.; Bertrand, E.; Tazi, J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 2003, 160, 823–831. [Google Scholar] [CrossRef]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Cherkasova, V.; Bankhead, P.; Bukau, B.; Stoecklin, G. Translation suppression promotes stress granule formation and cell survival in response to cold shock. Mol. Biol. Cell 2012, 23, 3786–3800. [Google Scholar] [CrossRef] [PubMed]
- Chernov, K.G.; Barbet, A.; Hamon, L.; Ovchinnikov, L.P.; Curmi, P.A.; Pastre, D. Role of microtubules in stress granule assembly: Microtubule dynamical instability favors the formation of micrometric stress granules in cells. J. Biol. Chem. 2009, 284, 36569–36580. [Google Scholar] [CrossRef] [PubMed]
- Mollet, S.; Cougot, N.; Wilczynska, A.; Dautry, F.; Kress, M.; Bertrand, E.; Weil, D. Translationally repressed mRNA transiently cycles through stress granules during stress. Mol. Biol. Cell 2008, 19, 4469–4479. [Google Scholar] [CrossRef]
- Zhang, J.; Okabe, K.; Tani, T.; Funatsu, T. Dynamic association-dissociation and harboring of endogenous mRNAs in stress granules. J. Cell Sci. 2011, 124, 4087–4095. [Google Scholar] [CrossRef]
- Hubstenberger, A.; Courel, M.; Benard, M.; Souquere, S.; Ernoult-Lange, M.; Chouaib, R.; Yi, Z.; Morlot, J.B.; Munier, A.; Fradet, M.; et al. P-Body Purification Reveals the Condensation of Repressed mRNA Regulons. Mol. Cell 2017, 68, 144–157 e145. [Google Scholar] [CrossRef]
- Ruggieri, A.; Dazert, E.; Metz, P.; Hofmann, S.; Bergeest, J.P.; Mazur, J.; Bankhead, P.; Hiet, M.S.; Kallis, S.; Alvisi, G.; et al. Dynamic Oscillation of Translation and Stress Granule Formation Mark the Cellular Response to Virus Infection. Cell Host Microbe 2012. [Google Scholar] [CrossRef]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef]
- Bou-Nader, C.; Gordon, J.M.; Henderson, F.E.; Zhang, J. The search for a PKR code-differential regulation of protein kinase R activity by diverse RNA and protein regulators. RNA 2019, 25, 539–556. [Google Scholar] [CrossRef]
- Pindel, A.; Sadler, A. The role of protein kinase R in the interferon response. J. Interferon Cytokine Res. 2011, 31, 59–70. [Google Scholar] [CrossRef]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.S.; Kerr, I.M.; Williams, B.R.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Cole, J.L. Activation of PKR: An open and shut case? Trends Biochem. Sci. 2007, 32, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.C.; Dever, T.E.; Sicheri, F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell 2005, 122, 887–900. [Google Scholar] [CrossRef]
- Dey, M.; Cao, C.; Dar, A.C.; Tamura, T.; Ozato, K.; Sicheri, F.; Dever, T.E. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell 2005, 122, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, P.A.; Anderson, E.; Lary, J.; Cole, J.L. Mechanism of PKR Activation by dsRNA. J. Mol. Biol. 2008, 381, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.D.; Mathews, M.B. The regulation of the protein kinase PKR by RNA. Biochimie 1996, 78, 909–914. [Google Scholar] [CrossRef]
- Circle, D.A.; Neel, O.D.; Robertson, H.D.; Clarke, P.A.; Mathews, M.B. Surprising specificity of PKR binding to delta agent genomic RNA. RNA 1997, 3, 438–448. [Google Scholar]
- Heinicke, L.A.; Bevilacqua, P.C. Activation of PKR by RNA misfolding: HDV ribozyme dimers activate PKR. RNA 2012, 18, 2157–2165. [Google Scholar] [CrossRef]
- Heinicke, L.A.; Wong, C.J.; Lary, J.; Nallagatla, S.R.; Diegelman-Parente, A.; Zheng, X.; Cole, J.L.; Bevilacqua, P.C. RNA dimerization promotes PKR dimerization and activation. J. Mol. Biol. 2009, 390, 319–338. [Google Scholar] [CrossRef]
- Rojas, M.; Arias, C.F.; Lopez, S. Protein kinase R is responsible for the phosphorylation of eIF2alpha in rotavirus infection. J. Virol. 2010, 84, 10457–10466. [Google Scholar] [CrossRef]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [PubMed]
- Son, K.N.; Liang, Z.; Lipton, H.L. Double-Stranded RNA Is Detected by Immunofluorescence Analysis in RNA and DNA Virus Infections, Including Those by Negative-Stranded RNA Viruses. J. Virol. 2015, 89, 9383–9392. [Google Scholar] [CrossRef] [PubMed]
- Willis, K.L.; Langland, J.O.; Shisler, J.L. Viral double-stranded RNAs from vaccinia virus early or intermediate gene transcripts possess PKR activating function, resulting in NF-kappaB activation, when the K1 protein is absent or mutated. J. Biol. Chem. 2011, 286, 7765–7778. [Google Scholar] [CrossRef] [PubMed]
- Nallagatla, S.R.; Hwang, J.; Toroney, R.; Zheng, X.; Cameron, C.E.; Bevilacqua, P.C. 5′-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science 2007, 318, 1455–1458. [Google Scholar] [CrossRef] [PubMed]
- Langereis, M.A.; Feng, Q.; van Kuppeveld, F.J. MDA5 localizes to stress granules, but this localization is not required for the induction of type I interferon. J. Virol. 2013, 87, 6314–6325. [Google Scholar] [CrossRef] [PubMed]
- Mayo, C.B.; Wong, C.J.; Lopez, P.E.; Lary, J.W.; Cole, J.L. Activation of PKR by short stem-loop RNAs containing single-stranded arms. RNA 2016, 22, 1065–1075. [Google Scholar] [CrossRef]
- Safran, S.A.; Eckert, D.M.; Leslie, E.A.; Bass, B.L. PKR activation by noncanonical ligands: A 5′-triphosphate requirement versus antisense contamination. RNA 2019, 25, 1192–1201. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, J.H.; Park, J.E.; Cho, J.; Yi, H.; Kim, V.N. PKR is activated by cellular dsRNAs during mitosis and acts as a mitotic regulator. Genes Dev. 2014, 28, 1310–1322. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Kim, S.; Kim, M.; Kang, M.G.; Kwak, C.; Kang, M.; Kim, B.; Rhee, H.W.; Kim, V.N. PKR Senses Nuclear and Mitochondrial Signals by Interacting with Endogenous Double-Stranded RNAs. Mol. Cell 2018, 71, 1051–1063 e1056. [Google Scholar] [CrossRef]
- Youssef, O.A.; Safran, S.A.; Nakamura, T.; Nix, D.A.; Hotamisligil, G.S.; Bass, B.L. Potential role for snoRNAs in PKR activation during metabolic stress. Proc. Natl. Acad. Sci. USA 2015, 112, 5023–5028. [Google Scholar] [CrossRef]
- Nakamura, T.; Furuhashi, M.; Li, P.; Cao, H.; Tuncman, G.; Sonenberg, N.; Gorgun, C.Z.; Hotamisligil, G.S. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 2010, 140, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Kunz, R.C.; Zhang, C.; Kimura, T.; Yuan, C.L.; Baccaro, B.; Namiki, Y.; Gygi, S.P.; Hotamisligil, G.S. A critical role for PKR complexes with TRBP in Immunometabolic regulation and eIF2alpha phosphorylation in obesity. Cell Rep. 2015, 11, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.X.; Li, X.; Nan, F.; Jiang, S.; Gao, X.; Guo, S.K.; Xue, W.; Cui, Y.; Dong, K.; Ding, H.; et al. Structure and Degradation of Circular RNAs Regulate PKR Activation in Innate Immunity. Cell 2019, 177, 865–880.e21. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.D.; Bak, M.S.; Kim, S.J.; Rhee, S.; Lee, Y.S. Restoring synaptic plasticity and memory in mouse models of Alzheimer’s disease by PKR inhibition. Mol. Brain 2017, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.J.; Huang, W.; Kalikulov, D.; Yoo, J.W.; Placzek, A.N.; Stoica, L.; Zhou, H.; Bell, J.C.; Friedlander, M.J.; Krnjevic, K.; et al. Suppression of PKR promotes network excitability and enhanced cognition by interferon-gamma-mediated disinhibition. Cell 2011, 147, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2018, 11, 480. [Google Scholar] [CrossRef]
- Chu, W.M.; Ballard, R.; Carpick, B.W.; Williams, B.R.; Schmid, C.W. Potential Alu function: Regulation of the activity of double-stranded RNA-activated kinase PKR. Mol. Cell. Biol. 1998, 18, 58–68. [Google Scholar] [CrossRef]
- Zhou, H.R.; He, K.; Landgraf, J.; Pan, X.; Pestka, J.J. Direct activation of ribosome-associated double-stranded RNA-dependent protein kinase (PKR) by deoxynivalenol, anisomycin and ricin: A new model for ribotoxic stress response induction. Toxins 2014, 6, 3406–3425. [Google Scholar] [CrossRef]
- Yuen, K.C.; Xu, B.; Krantz, I.D.; Gerton, J.L. NIPBL Controls RNA Biogenesis to Prevent Activation of the Stress Kinase PKR. Cell Rep. 2016, 14, 93–102. [Google Scholar] [CrossRef]
- Nallagatla, S.R.; Jones, C.N.; Ghosh, S.K.; Sharma, S.D.; Cameron, C.E.; Spremulli, L.L.; Bevilacqua, P.C. Native tertiary structure and nucleoside modifications suppress tRNA’s intrinsic ability to activate the innate immune sensor PKR. PLoS ONE 2013, 8, e57905. [Google Scholar] [CrossRef]
- Lee, K.; Kunkeaw, N.; Jeon, S.H.; Lee, I.; Johnson, B.H.; Kang, G.Y.; Bang, J.Y.; Park, H.S.; Leelayuwat, C.; Lee, Y.S. Precursor miR-886, a novel noncoding RNA repressed in cancer, associates with PKR and modulates its activity. RNA 2011, 17, 1076–1089. [Google Scholar] [CrossRef] [PubMed]
- Calderon, B.M.; Conn, G.L. Human noncoding RNA 886 (nc886) adopts two structurally distinct conformers that are functionally opposing regulators of PKR. RNA 2017, 23, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.V.; Chang, H.W.; Jacobs, B.L.; Kaufman, R.J. The E3L and K3L vaccinia virus gene products stimulate translation through inhibition of the double-stranded RNA-dependent protein kinase by different mechanisms. J. Virol. 1993, 67, 1688–1692. [Google Scholar] [CrossRef] [PubMed]
- Khaperskyy, D.A.; Hatchette, T.F.; McCormick, C. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J. 2012, 26, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Narayanan, K.; Wada, M.; Makino, S. Inhibition of Stress Granule Formation by Middle East Respiratory Syndrome Coronavirus 4a Accessory Protein Facilitates Viral Translation, Leading to Efficient Virus Replication. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Rabouw, H.H.; Langereis, M.A.; Knaap, R.C.; Dalebout, T.J.; Canton, J.; Sola, I.; Enjuanes, L.; Bredenbeek, P.J.; Kikkert, M.; de Groot, R.J.; et al. Middle East Respiratory Coronavirus Accessory Protein 4a Inhibits PKR-Mediated Antiviral Stress Responses. PLoS Pathog. 2016, 12, e1005982. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Shatkin, A.J. Double-stranded RNA-dependent protein kinase (PKR) is regulated by reovirus structural proteins. Virology 1997, 234, 364–371. [Google Scholar] [CrossRef]
- Nelson, E.V.; Schmidt, K.M.; Deflube, L.R.; Doganay, S.; Banadyga, L.; Olejnik, J.; Hume, A.J.; Ryabchikova, E.; Ebihara, H.; Kedersha, N.; et al. Ebola Virus Does Not Induce Stress Granule Formation during Infection and Sequesters Stress Granule Proteins within Viral Inclusions. J. Virol. 2016, 90, 7268–7284. [Google Scholar] [CrossRef]
- Le Sage, V.; Cinti, A.; McCarthy, S.; Amorim, R.; Rao, S.; Daino, G.L.; Tramontano, E.; Branch, D.R.; Mouland, A.J. Ebola virus VP35 blocks stress granule assembly. Virology 2017, 502, 73–83. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, Y.; Tang, Q.; Yang, X.; Qin, Y.; Chen, M. Inclusion bodies of human parainfluenza virus type 3 inhibit antiviral stress granule formation by shielding viral RNAs. PLoS Pathog. 2018, 14, e1006948. [Google Scholar] [CrossRef]
- Burgess, H.M.; Mohr, I. Defining the Role of Stress Granules in Innate Immune Suppression by the Herpes Simplex Virus 1 Endoribonuclease VHS. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Dauber, B.; Poon, D.; Dos Santos, T.; Duguay, B.A.; Mehta, N.; Saffran, H.A.; Smiley, J.R. The Herpes Simplex Virus Virion Host Shutoff Protein Enhances Translation of Viral True Late mRNAs Independently of Suppressing Protein Kinase R and Stress Granule Formation. J. Virol. 2016, 90, 6049–6057. [Google Scholar] [CrossRef] [PubMed]
- Finnen, R.L.; Zhu, M.; Li, J.; Romo, D.; Banfield, B.W. Herpes Simplex Virus 2 Virion Host Shutoff Endoribonuclease Activity Is Required To Disrupt Stress Granule Formation. J. Virol. 2016, 90, 7943–7955. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Komatsu, T.; Kitagawa, Y.; Sada, K.; Gotoh, B. Sendai virus C protein plays a role in restricting PKR activation by limiting the generation of intracellular double-stranded RNA. J. Virol. 2008, 82, 10102–10110. [Google Scholar] [CrossRef]
- Boonyaratanakornkit, J.; Bartlett, E.; Schomacker, H.; Surman, S.; Akira, S.; Bae, Y.S.; Collins, P.; Murphy, B.; Schmidt, A. The C proteins of human parainfluenza virus type 1 limit double-stranded RNA accumulation that would otherwise trigger activation of MDA5 and protein kinase R. J. Virol. 2011, 85, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, C.K.; Radeke, M.J.; Cattaneo, R.; Samuel, C.E. Measles virus C protein impairs production of defective copyback double-stranded viral RNA and activation of protein kinase R. J. Virol. 2014, 88, 456–468. [Google Scholar] [CrossRef]
- Toroney, R.; Nallagatla, S.R.; Boyer, J.A.; Cameron, C.E.; Bevilacqua, P.C. Regulation of PKR by HCV IRES RNA: Importance of domain II and NS5A. J. Mol. Biol. 2010, 400, 393–412. [Google Scholar] [CrossRef]
- Tu, Y.C.; Yu, C.Y.; Liang, J.J.; Lin, E.; Liao, C.L.; Lin, Y.L. Blocking double-stranded RNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J. Virol. 2012, 86, 10347–10358. [Google Scholar] [CrossRef]
- Ziehr, B.; Vincent, H.A.; Moorman, N.J. Human Cytomegalovirus pTRS1 and pIRS1 Antagonize Protein Kinase R To Facilitate Virus Replication. J. Virol. 2016, 90, 3839–3848. [Google Scholar] [CrossRef]
- Sharma, N.R.; Majerciak, V.; Kruhlak, M.J.; Zheng, Z.M. KSHV inhibits stress granule formation by viral ORF57 blocking PKR activation. PLoS Pathog. 2017, 13, e1006677. [Google Scholar] [CrossRef]
- Kitajewski, J.; Schneider, R.J.; Safer, B.; Munemitsu, S.M.; Samuel, C.E.; Thimmappaya, B.; Shenk, T. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 1986, 45, 195–200. [Google Scholar] [CrossRef]
- Clarke, P.A.; Schwemmle, M.; Schickinger, J.; Hilse, K.; Clemens, M.J. Binding of Epstein-Barr virus small RNA EBER-1 to the double-stranded RNA-activated protein kinase DAI. Nucleic Acids Res. 1991, 19, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Sharp, T.V.; Schwemmle, M.; Jeffrey, I.; Laing, K.; Mellor, H.; Proud, C.G.; Hilse, K.; Clemens, M.J. Comparative analysis of the regulation of the interferon-inducible protein kinase PKR by Epstein-Barr virus RNAs EBER-1 and EBER-2 and adenovirus VAI RNA. Nucleic Acids Res. 1993, 21, 4483–4490. [Google Scholar] [CrossRef] [PubMed]
- Dever, T.E.; Sripriya, R.; McLachlin, J.R.; Lu, J.; Fabian, J.R.; Kimball, S.R.; Miller, L.K. Disruption of cellular translational control by a viral truncated eukaryotic translation initiation factor 2alpha kinase homolog. Proc. Natl. Acad. Sci. USA 1998, 95, 4164–4169. [Google Scholar] [CrossRef]
- Li, J.J.; Cao, C.; Fixsen, S.M.; Young, J.M.; Ono, C.; Bando, H.; Elde, N.C.; Katsuma, S.; Dever, T.E.; Sicheri, F. Baculovirus protein PK2 subverts eIF2alpha kinase function by mimicry of its kinase domain C-lobe. Proc. Natl. Acad. Sci. USA 2015, 112, E4364–E4373. [Google Scholar] [CrossRef]
- Ikegami, T.; Narayanan, K.; Won, S.; Kamitani, W.; Peters, C.J.; Makino, S. Rift Valley fever virus NSs protein promotes post-transcriptional downregulation of protein kinase PKR and inhibits eIF2alpha phosphorylation. PLoS Pathog. 2009, 5, e1000287. [Google Scholar] [CrossRef]
- Mudhasani, R.; Tran, J.P.; Retterer, C.; Kota, K.P.; Whitehouse, C.A.; Bavari, S. Protein Kinase R Degradation Is Essential for Rift Valley Fever Virus Infection and Is Regulated by SKP1-CUL1-F-box (SCF)FBXW11-NSs E3 Ligase. PLoS Pathog. 2016, 12, e1005437. [Google Scholar] [CrossRef]
- Habjan, M.; Pichlmair, A.; Elliott, R.M.; Overby, A.K.; Glatter, T.; Gstaiger, M.; Superti-Furga, G.; Unger, H.; Weber, F. NSs protein of rift valley fever virus induces the specific degradation of the double-stranded RNA-dependent protein kinase. J. Virol. 2009, 83, 4365–4375. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Bartenschlager, R. Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 2016, 8, 160. [Google Scholar] [CrossRef]
- Tatu, U.; Hammond, C.; Helenius, A. Folding and oligomerization of influenza hemagglutinin in the ER and the intermediate compartment. EMBO J. 1995, 14, 1340–1348. [Google Scholar] [CrossRef]
- Dubuisson, J.; Rice, C.M. Hepatitis C virus glycoprotein folding: Disulfide bond formation and association with calnexin. J. Virol. 1996, 70, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.; Griffin, S. Viroporins: Structure, function and potential as antiviral targets. J. Gen. Virol. 2015, 96, 2000–2027. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.H.; Sun, Y.J.; Zhang, F.Q.; Zhang, X.R.; Qiu, X.S.; Yu, L.P.; Wu, Y.T.; Ding, C. Newcastle disease virus NP and P proteins induce autophagy via the endoplasmic reticulum stress-related unfolded protein response. Sci. Rep. 2016, 6, 24721. [Google Scholar] [CrossRef]
- Hou, L.; Dong, J.; Zhu, S.; Yuan, F.; Wei, L.; Wang, J.; Quan, R.; Chu, J.; Wang, D.; Jiang, H.; et al. Seneca valley virus activates autophagy through the PERK and ATF6 UPR pathways. Virology 2019, 537, 254–263. [Google Scholar] [CrossRef]
- Ambrose, R.L.; Mackenzie, J.M. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J. Virol. 2011, 85, 2723–2732. [Google Scholar] [CrossRef]
- Lewy, T.G.; Offerdahl, D.K.; Grabowski, J.M.; Kellman, E.; Mlera, L.; Chiramel, A.; Bloom, M.E. PERK-Mediated Unfolded Protein Response Signaling Restricts Replication of the Tick-Borne Flavivirus Langat Virus. Viruses 2020, 12, 328. [Google Scholar] [CrossRef]
- Xue, M.; Fu, F.; Ma, Y.; Zhang, X.; Li, L.; Feng, L.; Liu, P. The PERK Arm of the Unfolded Protein Response Negatively Regulates Transmissible Gastroenteritis Virus Replication by Suppressing Protein Translation and Promoting Type I Interferon Production. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Liao, Y.; Gu, F.; Mao, X.; Niu, Q.; Wang, H.; Sun, Y.; Song, C.; Qiu, X.; Tan, L.; Ding, C. Regulation of de novo translation of host cells by manipulation of PERK/PKR and GADD34-PP1 activity during Newcastle disease virus infection. J. Gen. Virol. 2016, 97, 867–879. [Google Scholar] [CrossRef]
- Pena, J.; Harris, E. Dengue virus modulates the unfolded protein response in a time-dependent manner. J. Biol. Chem. 2011, 286, 14226–14236. [Google Scholar] [CrossRef]
- Sood, R.; Porter, A.C.; Ma, K.; Quilliam, L.A.; Wek, R.C. Pancreatic eukaryotic initiation factor-2alpha kinase (PEK) homologues in humans, Drosophila melanogaster and Caenorhabditis elegans that mediate translational control in response to endoplasmic reticulum stress. Biochem. J. 2000, 346 Pt. 2, 281–293. [Google Scholar] [CrossRef]
- Liberman, E.; Fong, Y.L.; Selby, M.J.; Choo, Q.L.; Cousens, L.; Houghton, M.; Yen, T.S. Activation of the grp78 and grp94 promoters by hepatitis C virus E2 envelope protein. J. Virol. 1999, 73, 3718–3722. [Google Scholar] [CrossRef] [PubMed]
- Pavio, N.; Romano, P.R.; Graczyk, T.M.; Feinstone, S.M.; Taylor, D.R. Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2alpha kinase PERK. J. Virol. 2003, 77, 3578–3585. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xin, X.; Wang, T.; Wan, J.; Ou, Y.; Yang, Z.; Yu, Q.; Zhu, L.; Guo, Y.; Wu, Y.; et al. Japanese Encephalitis Virus Induces Apoptosis and Encephalitis by Activating the PERK Pathway. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C.; Jackson, B.M.; Hinnebusch, A.G. Juxtaposition of domains homologous to protein kinases and histidyl-tRNA synthetases in GCN2 protein suggests a mechanism for coupling GCN4 expression to amino acid availability. Proc. Natl. Acad. Sci. USA 1989, 86, 4579–4583. [Google Scholar] [CrossRef] [PubMed]
- Berlanga, J.J.; Ventoso, I.; Harding, H.P.; Deng, J.; Ron, D.; Sonenberg, N.; Carrasco, L.; de Haro, C. Antiviral effect of the mammalian translation initiation factor 2alpha kinase GCN2 against RNA viruses. EMBO J. 2006, 25, 1730–1740. [Google Scholar] [CrossRef]
- Del Pino, J.; Jimenez, J.L.; Ventoso, I.; Castello, A.; Munoz-Fernandez, M.A.; de Haro, C.; Berlanga, J.J. GCN2 has inhibitory effect on human immunodeficiency virus-1 protein synthesis and is cleaved upon viral infection. PLoS ONE 2012, 7, e47272. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Eidenschenk, C.; Arnold, C.N.; Siggs, O.M.; Sun, L.; Brandl, K.; Mullen, T.M.; Nemerow, G.R.; Moresco, E.M.; Beutler, B. Increased susceptibility to DNA virus infection in mice with a GCN2 mutation. J. Virol. 2012, 86, 1802–1808. [Google Scholar] [CrossRef]
- Zhu, R.; Zhang, Y.B.; Chen, Y.D.; Dong, C.W.; Zhang, F.T.; Zhang, Q.Y.; Gui, J.F. Molecular cloning and stress-induced expression of paralichthys olivaceus heme-regulated initiation factor 2alpha kinase. Dev. Comp. Immunol. 2006, 30, 1047–1059. [Google Scholar] [CrossRef]
- Zang, S.; Zhang, X.; Li, C.; Wang, L.; Wei, J.; Qin, Q. HRI of Epinephelus coioides is a critical factor in the grouper immune response to RGNNV infection. Fish Shellfish Immunol. 2019, 87, 659–668. [Google Scholar] [CrossRef]
- Lu, L.; Han, A.P.; Chen, J.J. Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol. Cell. Biol. 2001, 21, 7971–7980. [Google Scholar] [CrossRef] [PubMed]
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392. [Google Scholar] [CrossRef] [PubMed]
- Valadao, A.L.; Aguiar, R.S.; de Arruda, L.B. Interplay between Inflammation and Cellular Stress Triggered by Flaviviridae Viruses. Front. Microbiol. 2016, 7, 1233. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Fujita, N.; Sugimoto, R.; Urawa, N.; Horiike, S.; Kobayashi, Y.; Iwasa, M.; Ma, N.; Kawanishi, S.; Watanabe, S.; et al. Hepatic oxidative DNA damage is associated with increased risk for hepatocellular carcinoma in chronic hepatitis C. Br. J. Cancer 2008, 98, 580–586. [Google Scholar] [CrossRef]
- Vasallo, C.; Gastaminza, P. Cellular stress responses in hepatitis C virus infection: Mastering a two-edged sword. Virus Res. 2015, 209, 100–117. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Siddiqui, A. Regulatory mechanisms of viral hepatitis B and C. J. Biosci. 2003, 28, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell. Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef]
- Liem, J.; Liu, J. Stress Beyond Translation: Poxviruses and More. Viruses 2016, 8, 169. [Google Scholar] [CrossRef]
- Gullberg, R.C.; Jordan Steel, J.; Moon, S.L.; Soltani, E.; Geiss, B.J. Oxidative stress influences positive strand RNA virus genome synthesis and capping. Virology 2015, 475, 219–229. [Google Scholar] [CrossRef]
- Camini, F.C.; da Silva Caetano, C.C.; Almeida, L.T.; de Brito Magalhaes, C.L. Implications of oxidative stress on viral pathogenesis. Arch. Virol. 2017, 162, 907–917. [Google Scholar] [CrossRef]
- Lee, C. Therapeutic Modulation of Virus-Induced Oxidative Stress via the Nrf2-Dependent Antioxidative Pathway. Oxid. Med. Cell. Longev. 2018, 2018, 6208067. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, N.; Burnaevskiy, N.; Chen, D.; Gonugunta, V.K.; Alto, N.M.; Yan, N. STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe 2015, 18, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Majzoub, K.; Wrensch, F.; Baumert, T.F. The Innate Antiviral Response in Animals: An Evolutionary Perspective from Flagellates to Humans. Viruses 2019, 11, 758. [Google Scholar] [CrossRef]
- Tan, X.; Sun, L.; Chen, J.; Chen, Z.J. Detection of Microbial Infections Through Innate Immune Sensing of Nucleic Acids. Annu Rev. Microbiol. 2018, 72, 447–478. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Platanias, L.C. Mnk Kinases in Cytokine Signaling and Regulation of Cytokine Responses. Biomol. Concepts 2012, 3, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Herdy, B.; Jaramillo, M.; Svitkin, Y.V.; Rosenfeld, A.B.; Kobayashi, M.; Walsh, D.; Alain, T.; Sean, P.; Robichaud, N.; Topisirovic, I.; et al. Translational control of the activation of transcription factor NF-kappaB and production of type I interferon by phosphorylation of the translation factor eIF4E. Nat. Immunol. 2012, 13, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Stoecklin, G.; Stubbs, T.; Kedersha, N.; Wax, S.; Rigby, W.F.; Blackwell, T.K.; Anderson, P. MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 2004, 23, 1313–1324. [Google Scholar] [CrossRef]
- Schott, J.; Stoecklin, G. Networks controlling mRNA decay in the immune system. Wiley Interdiscip. Rev. RNA 2010, 1, 432–456. [Google Scholar] [CrossRef]
- Tiedje, C.; Ronkina, N.; Tehrani, M.; Dhamija, S.; Laass, K.; Holtmann, H.; Kotlyarov, A.; Gaestel, M. The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet. 2012, 8, e1002977. [Google Scholar] [CrossRef]
- Deng, J.; Lu, P.D.; Zhang, Y.; Scheuner, D.; Kaufman, R.J.; Sonenberg, N.; Harding, H.P.; Ron, D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 2004, 24, 10161–10168. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Wek, R.C. GCN2 phosphorylation of eIF2alpha activates NF-kappaB in response to UV irradiation. Biochem. J. 2005, 385, 371–380. [Google Scholar] [CrossRef]
- McAllister, C.S.; Taghavi, N.; Samuel, C.E. Protein kinase PKR amplification of interferon beta induction occurs through initiation factor eIF-2alpha-mediated translational control. J. Biol. Chem. 2012, 287, 36384–36392. [Google Scholar] [CrossRef] [PubMed]
- Dalet, A.; Arguello, R.J.; Combes, A.; Spinelli, L.; Jaeger, S.; Fallet, M.; Vu Manh, T.P.; Mendes, A.; Perego, J.; Reverendo, M.; et al. Protein synthesis inhibition and GADD34 control IFN-beta heterogeneous expression in response to dsRNA. EMBO J. 2017, 36, 761–782. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Choi, H.J.; Yang, H.; Do, K.H.; Kim, J.; Moon, Y. Repression of peroxisome proliferator-activated receptor gamma by mucosal ribotoxic insult-activated CCAAT/enhancer-binding protein homologous protein. J. Immunol. 2010, 185, 5522–5530. [Google Scholar] [CrossRef]
- Iordanov, M.S.; Paranjape, J.M.; Zhou, A.; Wong, J.; Williams, B.R.; Meurs, E.F.; Silverman, R.H.; Magun, B.E. Activation of p38 mitogen-activated protein kinase and c-Jun NH(2)-terminal kinase by double-stranded RNA and encephalomyocarditis virus: Involvement of RNase L, protein kinase R, and alternative pathways. Mol. Cell. Biol. 2000, 20, 617–627. [Google Scholar] [CrossRef]
- Kumar, A.; Haque, J.; Lacoste, J.; Hiscott, J.; Williams, B.R. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc. Natl. Acad. Sci. USA 1994, 91, 6288–6292. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, M.C.; Weil, R.; Dam, E.; Hovanessian, A.G.; Meurs, E.F. PKR stimulates NF-kappaB irrespective of its kinase function by interacting with the IkappaB kinase complex. Mol. Cell. Biol. 2000, 20, 4532–4542. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, M.C.; Daurat, C.; Ottone, C.; Meurs, E.F. The N-terminus of PKR is responsible for the activation of the NF-kappaB signaling pathway by interacting with the IKK complex. Cell Signal. 2006, 18, 1865–1875. [Google Scholar] [CrossRef]
- Zamanian-Daryoush, M.; Mogensen, T.H.; DiDonato, J.A.; Williams, B.R. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol. Cell. Biol. 2000, 20, 1278–1290. [Google Scholar] [CrossRef]
- Gil, J.; Garcia, M.A.; Gomez-Puertas, P.; Guerra, S.; Rullas, J.; Nakano, H.; Alcami, J.; Esteban, M. TRAF family proteins link PKR with NF-kappa B activation. Mol. Cell. Biol. 2004, 24, 4502–4512. [Google Scholar] [CrossRef]
- Arnaud, N.; Dabo, S.; Akazawa, D.; Fukasawa, M.; Shinkai-Ouchi, F.; Hugon, J.; Wakita, T.; Meurs, E.F. Hepatitis C virus reveals a novel early control in acute immune response. PLoS Pathog. 2011, 7, e1002289. [Google Scholar] [CrossRef]
- Yoo, J.S.; Takahasi, K.; Ng, C.S.; Ouda, R.; Onomoto, K.; Yoneyama, M.; Lai, J.C.; Lattmann, S.; Nagamine, Y.; Matsui, T.; et al. DHX36 Enhances RIG-I Signaling by Facilitating PKR-Mediated Antiviral Stress Granule Formation. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.M.; Santa Maria, F.G.; Lahiri, T.; Friedman, E.; Marie, I.J.; Levy, D.E. PKR Transduces MDA5-Dependent Signals for Type I IFN Induction. PLoS Pathog. 2016, 12, e1005489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, Y.; Xia, J.; He, J.; Pu, J.; Xie, J.; Wu, S.; Feng, L.; Huang, X.; Zhang, P. IPS-1 plays an essential role in dsRNA-induced stress granule formation by interacting with PKR and promoting its activation. J. Cell Sci. 2014, 127, 2471–2482. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Sun, H.; Yin, L.; Li, J.; Mei, S.; Xu, F.; Wu, C.; Liu, X.; Zhao, F.; Zhang, D.; et al. PKR-dependent cytosolic cGAS foci are necessary for intracellular DNA sensing. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.H.; Tam, N.W.; Yang, Y.L.; Cuddihy, A.R.; Li, S.; Kirchhoff, S.; Hauser, H.; Decker, T.; Koromilas, A.E. Physical association between STAT1 and the interferon-inducible protein kinase PKR and implications for interferon and double-stranded RNA signaling pathways. EMBO J. 1997, 16, 1291–1304. [Google Scholar] [CrossRef]
- Deb, A.; Zamanian-Daryoush, M.; Xu, Z.; Kadereit, S.; Williams, B.R. Protein kinase PKR is required for platelet-derived growth factor signaling of c-fos gene expression via Erks and Stat3. EMBO J. 2001, 20, 2487–2496. [Google Scholar] [CrossRef]
- Takada, Y.; Ichikawa, H.; Pataer, A.; Swisher, S.; Aggarwal, B.B. Genetic deletion of PKR abrogates TNF-induced activation of IkappaBalpha kinase, JNK, Akt and cell proliferation but potentiates p44/p42 MAPK and p38 MAPK activation. Oncogene 2007, 26, 1201–1212. [Google Scholar] [CrossRef]
- Williams, B.R. Signal integration via PKR. Sci. STKE 2001, 2001, re2. [Google Scholar] [CrossRef]
- Osman, F.; Jarrous, N.; Ben-Asouli, Y.; Kaempfer, R. A cis-acting element in the 3′-untranslated region of human TNF-alpha mRNA renders splicing dependent on the activation of protein kinase PKR. Genes Dev. 1999, 13, 3280–3293. [Google Scholar] [CrossRef]
- Ben-Asouli, Y.; Banai, Y.; Pel-Or, Y.; Shir, A.; Kaempfer, R. Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell 2002, 108, 221–232. [Google Scholar] [CrossRef]
- Schulz, O.; Pichlmair, A.; Rehwinkel, J.; Rogers, N.C.; Scheuner, D.; Kato, H.; Takeuchi, O.; Akira, S.; Kaufman, R.J.; Reis e Sousa, C. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 2010, 7, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Baltimore, D. Activation in vitro of NF-kappa B by phosphorylation of its inhibitor I kappa B. Nature 1990, 344, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Fougeray, S.; Mami, I.; Bertho, G.; Beaune, P.; Thervet, E.; Pallet, N. Tryptophan depletion and the kinase GCN2 mediate IFN-gamma-induced autophagy. J. Immunol. 2012, 189, 2954–2964. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, R.; Loebbermann, J.; Nakaya, H.I.; Khan, N.; Ma, H.; Gama, L.; Machiah, D.K.; Lawson, B.; Hakimpour, P.; Wang, Y.C.; et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature 2016, 531, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, R.; Khan, N.; Nakaya, H.I.; Li, S.; Loebbermann, J.; Maddur, M.S.; Park, Y.; Jones, D.P.; Chappert, P.; Davoust, J.; et al. Vaccine activation of the nutrient sensor GCN2 in dendritic cells enhances antigen presentation. Science 2014, 343, 313–317. [Google Scholar] [CrossRef]
- Meares, G.P.; Liu, Y.; Rajbhandari, R.; Qin, H.; Nozell, S.E.; Mobley, J.A.; Corbett, J.A.; Benveniste, E.N. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol. Cell. Biol. 2014, 34, 3911–3925. [Google Scholar] [CrossRef]
- Mijosek, V.; Lasitschka, F.; Warth, A.; Zabeck, H.; Dalpke, A.H.; Weitnauer, M. Endoplasmic Reticulum Stress Is a Danger Signal Promoting Innate Inflammatory Responses in Bronchial Epithelial Cells. J. Innate Immun. 2016, 8, 464–478. [Google Scholar] [CrossRef]
- Liu, J.; HuangFu, W.C.; Kumar, K.G.; Qian, J.; Casey, J.P.; Hamanaka, R.B.; Grigoriadou, C.; Aldabe, R.; Diehl, J.A.; Fuchs, S.Y. Virus-induced unfolded protein response attenuates antiviral defenses via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe 2009, 5, 72–83. [Google Scholar] [CrossRef]
- Chandra, P.K.; Bao, L.; Song, K.; Aboulnasr, F.M.; Baker, D.P.; Shores, N.; Wimley, W.C.; Liu, S.; Hagedorn, C.H.; Fuchs, S.Y.; et al. HCV infection selectively impairs type I but not type III IFN signaling. Am. J. Pathol. 2014, 184, 214–229. [Google Scholar] [CrossRef]
- Liang, Q.; Deng, H.; Sun, C.W.; Townes, T.M.; Zhu, F. Negative regulation of IRF7 activation by activating transcription factor 4 suggests a cross-regulation between the IFN responses and the cellular integrated stress responses. J. Immunol. 2011, 186, 1001–1010. [Google Scholar] [CrossRef]
- Li, H.Y.; Liu, H.; Wang, C.H.; Zhang, J.Y.; Man, J.H.; Gao, Y.F.; Zhang, P.J.; Li, W.H.; Zhao, J.; Pan, X.; et al. Deactivation of the kinase IKK by CUEDC2 through recruitment of the phosphatase PP1. Nat. Immunol. 2008, 9, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [PubMed]
- Dalet, A.; Gatti, E.; Pierre, P. Integration of PKR-dependent translation inhibition with innate immunity is required for a coordinated anti-viral response. FEBS Lett. 2015, 589, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Chitrakar, A.; Rath, S.; Donovan, J.; Demarest, K.; Li, Y.; Sridhar, R.R.; Weiss, S.R.; Kotenko, S.V.; Wingreen, N.S.; Korennykh, A. Real-time 2-5A kinetics suggest that interferons beta and lambda evade global arrest of translation by RNase L. Proc. Natl. Acad. Sci. USA 2019, 116, 2103–2111. [Google Scholar] [CrossRef]
- Rath, S.; Prangley, E.; Donovan, J.; Demarest, K.; Wingreen, N.S.; Meir, Y.; Korennykh, A. Concerted 2-5A-Mediated mRNA Decay and Transcription Reprogram Protein Synthesis in the dsRNA Response. Mol. Cell 2019, 75, 1218–1228 e1216. [Google Scholar] [CrossRef]
- Burke, J.M.; Moon, S.L.; Matheny, T.; Parker, R. RNase L Reprograms Translation by Widespread mRNA Turnover Escaped by Antiviral mRNAs. Mol. Cell 2019, 75, 1203–1217.e5. [Google Scholar] [CrossRef]
- Scheu, S.; Stetson, D.B.; Reinhardt, R.L.; Leber, J.H.; Mohrs, M.; Locksley, R.M. Activation of the integrated stress response during T helper cell differentiation. Nat. Immunol. 2006, 7, 644–651. [Google Scholar] [CrossRef]
- Cherkasov, V.; Hofmann, S.; Druffel-Augustin, S.; Mogk, A.; Tyedmers, J.; Stoecklin, G.; Bukau, B. Coordination of translational control and protein homeostasis during severe heat stress. Curr. Biol. 2013, 23, 2452–2462. [Google Scholar] [CrossRef]
- Yoneyama, M.; Jogi, M.; Onomoto, K. Regulation of antiviral innate immune signaling by stress-induced RNA granules. J. Biochem. 2016, 159, 279–286. [Google Scholar] [CrossRef]
- Katoh, H.; Okamoto, T.; Fukuhara, T.; Kambara, H.; Morita, E.; Mori, Y.; Kamitani, W.; Matsuura, Y. Japanese Encephalitis Virus Core Protein Inhibits Stress Granule Formation through an Interaction with Caprin-1 and Facilitates Viral Propagation. J. Virol. 2013, 87, 489–502. [Google Scholar] [CrossRef]
- Emara, M.M.; Brinton, M.A. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 9041–9046. [Google Scholar] [CrossRef] [PubMed]
- Albornoz, A.; Carletti, T.; Corazza, G.; Marcello, A. The Stress Granule Component TIA-1 Binds Tick-Borne Encephalitis Virus RNA and Is Recruited to Perinuclear Sites of Viral Replication To Inhibit Viral Translation. J. Virol. 2014, 88, 6611–6622. [Google Scholar] [CrossRef]
- Urosevic, N.; van Maanen, M.; Mansfield, J.P.; Mackenzie, J.S.; Shellam, G.R. Molecular characterization of virus-specific RNA produced in the brains of flavivirus-susceptible and -resistant mice after challenge with Murray Valley encephalitis virus. J. Gen. Virol. 1997, 78 Pt 1, 23–29. [Google Scholar] [CrossRef]
- Pijlman, G.P.; Funk, A.; Kondratieva, N.; Leung, J.; Torres, S.; van der Aa, L.; Liu, W.J.; Palmenberg, A.C.; Shi, P.Y.; Hall, R.A.; et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Bidet, K.; Dadlani, D.; Garcia-Blanco, M.A. G3BP1, G3BP2 and CAPRIN1 Are Required for Translation of Interferon Stimulated mRNAs and Are Targeted by a Dengue Virus Non-coding RNA. PLoS Pathog. 2014, 10, e1004242. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Y.; Kedersha, N.; Anderson, P.; Emara, M.; Swiderek, K.M.; Moreno, G.T.; Brinton, M.A. Cell Proteins TIA-1 and TIAR Interact with the 3′ Stem-Loop of the West Nile Virus Complementary Minus-Strand RNA and Facilitate Virus Replication. J. Virol. 2002, 76, 11989–12000. [Google Scholar] [CrossRef]
- Ward, A.M.; Bidet, K.; Yinglin, A.; Ler, S.G.; Hogue, K.; Blackstock, W.; Gunaratne, J.; Garcia-Blanco, M.A. Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biol. 2011, 8, 1173–1186. [Google Scholar] [CrossRef]
- Iseni, F.; Garcin, D.; Nishio, M.; Kedersha, N.; Anderson, P.; Kolakofsky, D. Sendai virus trailer RNA binds TIAR, a cellular protein involved in virus-induced apoptosis. EMBO J. 2002, 21, 5141–5150. [Google Scholar] [CrossRef]
- Amorim, R.; Temzi, A.; Griffin, B.D.; Mouland, A.J. Zika virus inhibits eIF2α-dependent stress granule assembly. PLoS Negl. Trop. Dis. 2017, 11. [Google Scholar] [CrossRef]
- Bonenfant, G.; Williams, N.; Netzband, R.; Schwarz, M.C.; Evans, M.J.; Pager, C.T. Zika Virus Subverts Stress Granules To Promote and Restrict Viral Gene Expression. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Hou, S.; Kumar, A.; Xu, Z.; Airo, A.M.; Stryapunina, I.; Wong, C.P.; Branton, W.; Tchesnokov, E.; Götte, M.; Power, C.; et al. Zika Virus Hijacks Stress Granule Proteins and Modulates the Host Stress Response. J. Virol. 2017. [Google Scholar] [CrossRef]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS ONE 2012, 7, e43031. [Google Scholar] [CrossRef]
- Oh, S.W.; Onomoto, K.; Wakimoto, M.; Onoguchi, K.; Ishidate, F.; Fujiwara, T.; Yoneyama, M.; Kato, H.; Fujita, T. Leader-Containing Uncapped Viral Transcript Activates RIG-I in Antiviral Stress Granules. PLoS Pathog. 2016, 12, e1005444. [Google Scholar] [CrossRef] [PubMed]
- Kuniyoshi, K.; Takeuchi, O.; Pandey, S.; Satoh, T.; Iwasaki, H.; Akira, S.; Kawai, T. Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I-mediated antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2014, 111, 5646–5651. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.S.; Jogi, M.; Yoo, J.S.; Onomoto, K.; Koike, S.; Iwasaki, T.; Yoneyama, M.; Kato, H.; Fujita, T. Encephalomyocarditis Virus Disrupts Stress Granules, the Critical Platform for Triggering Antiviral Innate Immune Responses. J. Virol. 2013, 87, 9511–9522. [Google Scholar] [CrossRef] [PubMed]
- Takashima, K.; Oshiumi, H.; Takaki, H.; Matsumoto, M.; Seya, T. RIOK3-mediated phosphorylation of MDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep. 2015, 11, 192–200. [Google Scholar] [CrossRef]
- Takashima, K.; Oshiumi, H.; Matsumoto, M.; Seya, T. DNAJB1/HSP40 Suppresses Melanoma Differentiation-Associated Gene 5-Mitochondrial Antiviral Signaling Protein Function in Conjunction with HSP70. J. Innate Immun. 2018, 10, 44–55. [Google Scholar] [CrossRef]
- Sánchez-Aparicio, M.T.; Ayllón, J.; Leo-Macias, A.; Wolff, T.; García-Sastre, A. Subcellular Localizations of RIG-I, TRIM25, and MAVS Complexes. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Oshiumi, H.; Miyashita, M.; Matsumoto, M.; Seya, T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 2013, 9, e1003533. [Google Scholar] [CrossRef]
- Reineke, L.C.; Tsai, W.C.; Jain, A.; Kaelber, J.T.; Jung, S.Y.; Lloyd, R.E. Casein Kinase 2 Is Linked to Stress Granule Dynamics through Phosphorylation of the Stress Granule Nucleating Protein G3BP1. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef]
- Kobayashi, T.; Winslow, S.; Sunesson, L.; Hellman, U.; Larsson, C. PKCalpha binds G3BP2 and regulates stress granule formation following cellular stress. PLoS ONE 2012, 7, e35820. [Google Scholar] [CrossRef]
- Kwon, S.; Zhang, Y.; Matthias, P. The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response. Genes Dev. 2007, 21, 3381–3394. [Google Scholar] [CrossRef]
- Narita, R.; Takahasi, K.; Murakami, E.; Hirano, E.; Yamamoto, S.P.; Yoneyama, M.; Kato, H.; Fujita, T. A Novel Function of Human Pumilio Proteins in Cytoplasmic Sensing of Viral Infection. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Vessey, J.P.; Vaccani, A.; Xie, Y.; Dahm, R.; Karra, D.; Kiebler, M.A.; Macchi, P. Dendritic localization of the translational repressor Pumilio 2 and its contribution to dendritic stress granules. J. Neurosci. 2006, 26, 6496–6508. [Google Scholar] [CrossRef] [PubMed]
- Kunde, S.A.; Musante, L.; Grimme, A.; Fischer, U.; Müller, E.; Wanker, E.E.; Kalscheuer, V.M. The X-chromosome-linked intellectual disability protein PQBP1 is a component of neuronal RNA granules and regulates the appearance of stress granules. Hum. Mol. Genet. 2011, 20, 4916–4931. [Google Scholar] [CrossRef] [PubMed]
- Reineke, L.C.; Lloyd, R.E. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J. Virol. 2015, 89, 2575–2589. [Google Scholar] [CrossRef]
- Pare, J.M.; Tahbaz, N.; Lopez-Orozco, J.; LaPointe, P.; Lasko, P.; Hobman, T.C. Hsp90 regulates the function of argonaute 2 and its recruitment to stress granules and P-bodies. Mol. Biol. Cell 2009, 20, 3273–3284. [Google Scholar] [CrossRef]
- Shiina, N.; Nakayama, K. RNA granule assembly and disassembly modulated by nuclear factor associated with double-stranded RNA 2 and nuclear factor 45. J. Biol. Chem. 2014, 289, 21163–21180. [Google Scholar] [CrossRef]
- Wen, X.; Huang, X.; Mok, B.W.; Chen, Y.; Zheng, M.; Lau, S.Y.; Wang, P.; Song, W.; Jin, D.Y.; Yuen, K.Y.; et al. NF90 exerts antiviral activity through regulation of PKR phosphorylation and stress granules in infected cells. J. Immunol. 2014, 192, 3753–3764. [Google Scholar] [CrossRef]
- Thomas, M.G.; Martinez Tosar, L.J.; Desbats, M.A.; Leishman, C.C.; Boccaccio, G.L. Mammalian Staufen 1 is recruited to stress granules and impairs their assembly. J. Cell Sci. 2009, 122, 563–573. [Google Scholar] [CrossRef]
- Kit Ng, S.; Weissbach, R.; Ronson, G.E.; J Scadden, A.D. Proteins that contain a functional Z-DNA-binding domain localize to cytoplasmic stress granules. Nucleic Acids Res. 2013, 41, 9786–9799. [Google Scholar] [CrossRef]
- Weissbach, R.; Scadden, A.D.J. Tudor-SN and ADAR1 are components of cytoplasmic stress granules. RNA 2012, 18, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Manivannan, P.; Siddiqui, M.A.; Malathi, K. RNase L amplifies interferon signaling by inducing PKR-mediated antiviral stress granules. J. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.; Vyas, S.; Rood, J.E.; Bhutkar, A.; Sharp, P.A.; Chang, P. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol. Cell 2011, 42, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Law, L.M.J.; Razooky, B.S.; Li, M.M.H.; You, S.; Jurado, A.; Rice, C.M.; Macdonald, M.R. ZAP’s stress granule localization is correlated with its antiviral activity and induced by virus replication. PLoS Pathog. 2019, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, Y.; Ghosh, A.; Cuevas, R.A.; Forero, A.; Dhar, J.; Ibsen, M.S.; Schmid-Burgk, J.L.; Schmidt, T.; Ganapathiraju, M.K.; et al. Antiviral activity of human oligoadenylate synthetases-like (OASL) is mediated by enhancing retinoic acid-inducible gene I (RIG-I) signaling. Immunity 2014, 40, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Hwang, Y.S.; Kim, L.K.; Lee, S.; Lee, W.B.; Kim-Ha, J.; Kim, Y.J. OASL1 traps viral RNAs in stress granules to promote antiviral responses. Mol. Cells 2018, 41, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.; Kino, Y.; Morita, T.; Futai, E.; Sasagawa, N.; Ishiura, S. MBNL1 associates with YB-1 in cytoplasmic stress granules. J. Neurosci. Res. 2008, 86, 1994–2002. [Google Scholar] [CrossRef]
- Ozeki, K.; Sugiyama, M.; Akter, K.A.; Nishiwaki, K.; Asano-Inami, E.; Senga, T. FAM98A is localized to stress granules and associates with multiple stress granule-localized proteins. Mol. Cell. Biochem. 2019, 451, 107–115. [Google Scholar] [CrossRef]
- Mazroui, R.; Sukarieh, R.; Bordeleau, M.E.; Kaufman, R.J.; Northcote, P.; Tanaka, J.; Gallouzi, I.; Pelletier, J. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2alpha phosphorylation. Mol. Biol. Cell 2006, 17, 4212–4219. [Google Scholar] [CrossRef]
- Pène, V.; Li, Q.; Sodroski, C.; Hsu, C.-S.; Liang, T.J. Dynamic Interaction of Stress Granules, DDX3X, and IKK-α Mediates Multiple Functions in Hepatitis C Virus Infection. J. Virol. 2015, 89, 5462–5477. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.W.; Wang, W.T.; Tsai, T.Y.; Kuo, C.Y.; Li, H.K.; Wu Lee, Y.H. Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem. J. 2012, 441, 119–129. [Google Scholar] [CrossRef]
- Lai, M.C.; Lee, Y.H.; Tarn, W.Y. The DEAD-box RNA helicase DDX3 associates with export messenger ribonucleoproteins as well as tip-associated protein and participates in translational control. Mol. Biol. Cell 2008, 19, 3847–3858. [Google Scholar] [CrossRef] [PubMed]
- Núñez, R.D.; Budt, M.; Saenger, S.; Paki, K.; Arnold, U.; Sadewasser, A.; Wolff, T. The RNA helicase DDX6 associates with RIG-I to augment induction of antiviral signaling. Int. J. Mol. Sci. 2018, 19, 1877. [Google Scholar] [CrossRef] [PubMed]
- Wilczynska, A.; Aigueperse, C.; Kress, M.; Dautry, F.; Weil, D. The translational regulator CPEB1 provides a link between dcp1 bodies and stress granules. J. Cell Sci. 2005, 118, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Hochberg-Laufer, H.; Schwed-Gross, A.; Neugebauer, K.M.; Shav-Tal, Y. Uncoupling of nucleo-cytoplasmic RNA export and localization during stress. Nucleic Acids Res. 2019, 47, 4778–4797. [Google Scholar] [CrossRef]
- Chalupnikova, K.; Lattmann, S.; Selak, N.; Iwamoto, F.; Fujiki, Y.; Nagamine, Y. Recruitment of the RNA helicase RHAU to stress granules via a unique RNA-binding domain. J. Biol. Chem. 2008, 283, 35186–35198. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Long, J.C.; Caceres, J.F. hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell. Biol. 2006, 26, 5744–5758. [Google Scholar] [CrossRef]
- Fujimura, K.; Kano, F.; Murata, M. Identification of PCBP2, a facilitator of IRES-mediated translation, as a novel constituent of stress granules and processing bodies. RNA 2008, 14, 425–431. [Google Scholar] [CrossRef]
- Borghese, F.; Michiels, T. The Leader Protein of Cardioviruses Inhibits Stress Granule Assembly. J. Virol. 2011, 85, 9614–9622. [Google Scholar] [CrossRef]
- Sola, I.; Galan, C.; Mateos-Gomez, P.A.; Palacio, L.; Zuniga, S.; Cruz, J.L.; Almazan, F.; Enjuanes, L. The polypyrimidine tract-binding protein affects coronavirus RNA accumulation levels and relocalizes viral RNAs to novel cytoplasmic domains different from replication-transcription sites. J. Virol. 2011, 85, 5136–5149. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Hsu, M.; Tarn, W.Y. Cell stress modulates the function of splicing regulatory protein RBM4 in translation control. Proc. Natl. Acad. Sci. USA 2007, 104, 2235–2240. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Naiki, T.; Saito, M.; Irie, K. hnRNP K interacts with RNA binding motif protein 42 and functions in the maintenance of cellular ATP level during stress conditions. Genes Cells 2009, 14, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Detzer, A.; Engel, C.; Wunsche, W.; Sczakiel, G. Cell stress is related to re-localization of Argonaute 2 and to decreased RNA interference in human cells. Nucleic Acids Res. 2011, 39, 2727–2741. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Roberts, T.L.; Richards, R.; Woods, R.; Birrell, G.; Lim, Y.C.; Ohno, S.; Yamashita, A.; Abraham, R.T.; Gueven, N.; et al. A novel role for hSMG-1 in stress granule formation. Mol. Cell. Biol. 2011, 31, 4417–4429. [Google Scholar] [CrossRef]
- Fu, Y.; Zhuang, X. m(6)A-binding YTHDF proteins promote stress granule formation. Nat. Chem. Biol. 2020. [Google Scholar] [CrossRef]
- Catara, G.; Grimaldi, G.; Schembri, L.; Spano, D.; Turacchio, G.; Lo Monte, M.; Beccari, A.R.; Valente, C.; Corda, D. PARP1-produced poly-ADP-ribose causes the PARP12 translocation to stress granules and impairment of Golgi complex functions. Sci. Rep. 2017, 7, 14035. [Google Scholar] [CrossRef]
- Takahara, T.; Maeda, T. Transient sequestration of TORC1 into stress granules during heat stress. Mol. Cell 2012, 47, 242–252. [Google Scholar] [CrossRef]
- Thedieck, K.; Holzwarth, B.; Prentzell, M.T.; Boehlke, C.; Klasener, K.; Ruf, S.; Sonntag, A.G.; Maerz, L.; Grellscheid, S.N.; Kremmer, E.; et al. Inhibition of mTORC1 by astrin and stress granules prevents apoptosis in cancer cells. Cell 2013, 154, 859–874. [Google Scholar] [CrossRef]
- Wippich, F.; Bodenmiller, B.; Trajkovska, M.G.; Wanka, S.; Aebersold, R.; Pelkmans, L. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell 2013, 152, 791–805. [Google Scholar] [CrossRef]
- Wasserman, T.; Katsenelson, K.; Daniliuc, S.; Hasin, T.; Choder, M.; Aronheim, A. A novel c-Jun N-terminal kinase (JNK)-binding protein WDR62 is recruited to stress granules and mediates a nonclassical JNK activation. Mol. Biol. Cell 2010, 21, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, K.; Fukuda, H.; Imajoh-Ohmi, S.; Saito, H.; Takekawa, M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 2008, 10, 1324–1332. [Google Scholar] [CrossRef]
- Kim, W.J.; Back, S.H.; Kim, V.; Ryu, I.; Jang, S.K. Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol. Cell. Biol. 2005, 25, 2450–2462. [Google Scholar] [CrossRef] [PubMed]
- Eisinger-Mathason, T.S.; Andrade, J.; Groehler, A.L.; Clark, D.E.; Muratore-Schroeder, T.L.; Pasic, L.; Smith, J.A.; Shabanowitz, J.; Hunt, D.F.; Macara, I.G.; et al. Codependent functions of RSK2 and the apoptosis-promoting factor TIA-1 in stress granule assembly and cell survival. Mol. Cell 2008, 31, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Tsai, N.P.; Wei, L.N. RhoA/ROCK1 signaling regulates stress granule formation and apoptosis. Cell Signal. 2010, 22, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Pan, T.; Wu, X.; Song, W.; Wang, S.; Xu, Y.; Rice, C.M.; Macdonald, M.R.; Yuan, Z. Hepatitis C virus co-opts Ras-GTPase-activating protein-binding protein 1 for its genome replication. J. Virol. 2011, 85, 6996–7004. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y.; Kuroki, M.; Kushima, Y.; Osugi, K.; Hijikata, M.; Maki, M.; Ikeda, M.; Kato, N. Hepatitis C Virus Hijacks P-Body and Stress Granule Components around Lipid Droplets. J. Virol. 2011, 85, 6882–6892. [Google Scholar] [CrossRef]
- Garaigorta, U.; Heim, M.H.; Boyd, B.; Wieland, S.; Chisari, F.V. Hepatitis C Virus (HCV) Induces Formation of Stress Granules Whose Proteins Regulate HCV RNA Replication and Virus Assembly and Egress. J. Virol. 2012, 86, 11043–11056. [Google Scholar] [CrossRef]
- Fros, J.J.; Domeradzka, N.E.; Baggen, J.; Geertsema, C.; Flipse, J.; Vlak, J.M.; Pijlman, G.P. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J. Virol. 2012, 86, 10873–10879. [Google Scholar] [CrossRef]
- Scholte, F.E.; Tas, A.; Albulescu, I.C.; Zusinaite, E.; Merits, A.; Snijder, E.J.; van Hemert, M.J. Stress granule components G3BP1 and G3BP2 play a proviral role early in Chikungunya virus replication. J. Virol. 2015, 89, 4457–4469. [Google Scholar] [CrossRef]
- Panas, M.D.; Varjak, M.; Lulla, A.; Eng, K.E.; Merits, A.; Karlsson Hedestam, G.B.; McInerney, G.M. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol. Biol. Cell 2012, 23, 4701–4712. [Google Scholar] [CrossRef] [PubMed]
- Frolova, E.; Gorchakov, R.; Garmashova, N.; Atasheva, S.; Vergara, L.A.; Frolov, I. Formation of nsP3-specific protein complexes during Sindbis virus replication. J. Virol. 2006, 80, 4122–4134. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.L.; York, J.; Nunberg, J.H. Arenavirus infection induces discrete cytosolic structures for RNA replication. J. Virol. 2012, 86, 11301–11310. [Google Scholar] [CrossRef] [PubMed]
- Brocard, M.; Iadevaia, V.; Klein, P.; Hall, B.; Lewis, G.; Lu, J.; Burke, J.; Willcocks, M.M.; Parker, R.; Goodfellow, I.G.; et al. Norovirus infection results in eIF2α independent host translation shut-off and remodels the G3BP1 interactome evading stress granule formation. PLoS Pathog. 2020, 16. [Google Scholar] [CrossRef]
- Hosmillo, M.; Lu, J.; McAllaster, M.R.; Eaglesham, J.B.; Wang, X.; Emmott, E.; Domingues, P.; Chaudhry, Y.; Fitzmaurice, T.J.; Tung, M.K.; et al. Noroviruses subvert the core stress granule component G3BP1 to promote viral VPg-dependent translation. eLife 2019, 8. [Google Scholar] [CrossRef]
- Kim, S.S.; Sze, L.; Liu, C.; Lam, K.P. The stress granule protein G3BP1 binds viral dsRNA and RIG-I to enhance interferon-beta response. J. Biol. Chem. 2019, 294, 6430–6438. [Google Scholar] [CrossRef]
- Yang, W.; Ru, Y.; Ren, J.; Bai, J.; Wei, J.; Fu, S.; Liu, X.; Li, D.; Zheng, H. G3BP1 inhibits RNA virus replication by positively regulating RIG-I-mediated cellular antiviral response. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Liu, Z.S.; Cai, H.; Xue, W.; Wang, M.; Xia, T.; Li, W.J.; Xing, J.Q.; Zhao, M.; Huang, Y.J.; Chen, S.; et al. G3BP1 promotes DNA binding and activation of cGAS. Nat. Immunol. 2019, 20, 18–28. [Google Scholar] [CrossRef]
- Visser, L.J.; Medina, G.N.; Rabouw, H.H.; de Groot, R.J.; Langereis, M.A.; de los Santos, T.; van Kuppeveld, F.J.M. Foot-and-Mouth Disease Virus Leader Protease Cleaves G3BP1 and G3BP2 and Inhibits Stress Granule Formation. J. Virol. 2018, 93. [Google Scholar] [CrossRef]
- Ye, X.; Pan, T.; Wang, D.; Fang, L.; Ma, J.; Zhu, X.; Shi, Y.; Zhang, K.; Zheng, H.; Chen, H.; et al. Foot-and-mouth disease virus counteracts on internal ribosome entry site suppression by G3BP1 and inhibits G3BP1-mediated stress granule assembly via post-translational mechanisms. Front. Immunol. 2018, 9, 1142. [Google Scholar] [CrossRef]
- White, J.P.; Cardenas, A.M.; Marissen, W.E.; Lloyd, R.E. Inhibition of Cytoplasmic mRNA Stress Granule Formation by a Viral Proteinase. Cell Host Microbe 2007, 2, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Fung, G.; Ng, C.S.; Zhang, J.; Shi, J.; Wong, J.; Piesik, P.; Han, L.; Chu, F.; Jagdeo, J.; Jan, E.; et al. Production of a dominant-negative fragment due to G3BP1 cleavage contributes to the disruption of mitochondria-associated protective stress granules during CVB3 infection. PLoS ONE 2013, 8, e79546. [Google Scholar] [CrossRef]
- Dougherty, J.D.; Tsai, W.C.; Lloyd, R.E. Multiple poliovirus proteins repress cytoplasmic RNA granules. Viruses 2015, 7, 6127–6140. [Google Scholar] [CrossRef]
- Humoud, M.N.; Doyle, N.; Royall, E.; Willcocks, M.M.; Sorgeloos, F.; van Kuppeveld, F.; Roberts, L.O.; Goodfellow, I.G.; Langereis, M.A.; Locker, N. Feline Calicivirus Infection Disrupts Assembly of Cytoplasmic Stress Granules and Induces G3BP1 Cleavage. J. Virol. 2016, 90, 6489–6501. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Feng, Q.; Hato, S.V.; Langereis, M.A.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Karijolich, J. Know Thyself: RIG-I-Like Receptor Sensing of DNA Virus Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Yoh, S.M.; Schneider, M.; Seifried, J.; Soonthornvacharin, S.; Akleh, R.E.; Olivieri, K.C.; De Jesus, P.D.; Ruan, C.; De Castro, E.; Ruiz, P.A.; et al. PQBP1 is a proximal sensor of the cGAS-dependent innate response to HIV-1. Cell 2015, 161, 1293–1305. [Google Scholar] [CrossRef]
- Okamoto, M.; Kouwaki, T.; Fukushima, Y.; Oshiumi, H. Regulation of RIG-I Activation by K63-Linked Polyubiquitination. Front. Immunol. 2017, 8, 1942. [Google Scholar] [CrossRef]
- Castanier, C.; Zemirli, N.; Portier, A.; Garcin, D.; Bidère, N.; Vazquez, A.; Arnoult, D. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, J.; Zheng, W.; Shang, Y.; Zhao, Z.; Wang, S.; Bi, Y.; Zhang, S.; Xu, C.; Duan, Z.; et al. Cyclophilin A-regulated ubiquitination is critical for RIG-I-mediated antiviral immune responses. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Now, H.; Kim, W.J.; Kim, N.; Yoo, J.Y. Ubiquitin-like modifier FAT10 attenuates RIG-I mediated antiviral signaling by segregating activated RIG-I from its signaling platform. Sci. Rep. 2016, 6, 23377. [Google Scholar] [CrossRef] [PubMed]
- Cadena, C.; Ahmad, S.; Xavier, A.; Hou, F.; Binder, M.; Hur, S. Ubiquitin-Dependent and-Independent Roles of E3 Ligase RIPLET in Innate Immunity The E3 ligase RIPLET activates RIG-I via dual ubiquitin-dependent and-independent mechanisms that together work to discriminate the length of dsRNA sensed by RIG-I. Cell 2019, 177, 1187–1200.e16. [Google Scholar] [CrossRef] [PubMed]
- Hayman, T.J.; Hsu, A.C.; Kolesnik, T.B.; Dagley, L.F.; Willemsen, J.; Tate, M.D.; Baker, P.J.; Kershaw, N.J.; Kedzierski, L.; Webb, A.I.; et al. RIPLET, and not TRIM25, is required for endogenous RIG-I-dependent antiviral responses. Immunol. Cell Biol. 2019, 97, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef]
- Sanchez David, R.Y.; Combredet, C.; Najburg, V.; Millot, G.A.; Beauclair, G.; Schwikowski, B.; Leger, T.; Camadro, J.M.; Jacob, Y.; Bellalou, J.; et al. LGP2 binds to PACT to regulate RIG-I- and MDA5-mediated antiviral responses. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Kok, K.H.; Lui, P.Y.; Ng, M.H.; Siu, K.L.; Au, S.W.; Jin, D.Y. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 2011, 9, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Lui, P.Y.; Wong, L.R.; Ho, T.H.; Au, S.W.N.; Chan, C.P.; Kok, K.H.; Jin, D.Y. PACT Facilitates RNA-Induced Activation of MDA5 by Promoting MDA5 Oligomerization. J. Immunol. 2017, 199, 1846–1855. [Google Scholar] [CrossRef]
- Reineke, L.C.; Kedersha, N.; Langereis, M.A.; van Kuppeveld, F.J.; Lloyd, R.E. Stress granules regulate double-stranded RNA-dependent protein kinase activation through a complex containing G3BP1 and Caprin1. mBio 2015, 6, e02486. [Google Scholar] [CrossRef]
- Kedersha, N.; Panas, M.D.; Achorn, C.A.; Lyons, S.; Tisdale, S.; Hickman, T.; Thomas, M.; Lieberman, J.; McInerney, G.M.; Ivanov, P.; et al. G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol. 2016, 212, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.C.; Sen, G.C. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998, 17, 4379–4390. [Google Scholar] [CrossRef] [PubMed]
- Benkirane, M.; Neuveut, C.; Chun, R.F.; Smith, S.M.; Samuel, C.E.; Gatignol, A.; Jeang, K.T. Oncogenic potential of TAR RNA binding protein TRBP and its regulatory interaction with RNA-dependent protein kinase PKR. EMBO J. 1997, 16, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.G.; Tang, N.; Thompson, S.; Miller, J.; Katze, M.G. The 58,000-dalton cellular inhibitor of the interferon-induced double-stranded RNA-activated protein kinase (PKR) is a member of the tetratricopeptide repeat family of proteins. Mol. Cell. Biol. 1994, 14, 2331–2342. [Google Scholar] [CrossRef]
- Clerzius, G.; Shaw, E.; Daher, A.; Burugu, S.; Gelinas, J.F.; Ear, T.; Sinck, L.; Routy, J.P.; Mouland, A.J.; Patel, R.C.; et al. The PKR activator, PACT, becomes a PKR inhibitor during HIV-1 replication. Retrovirology 2013, 10, 96. [Google Scholar] [CrossRef]
- Meyer, C.; Garzia, A.; Mazzola, M.; Gerstberger, S.; Molina, H.; Tuschl, T. The TIA1 RNA-Binding Protein Family Regulates EIF2AK2-Mediated Stress Response and Cell Cycle Progression. Mol. Cell 2018, 69, 622–635 e626. [Google Scholar] [CrossRef]
- Chukwurah, E.; Patel, R.C. Stress-induced TRBP phosphorylation enhances its interaction with PKR to regulate cellular survival. Sci. Rep. 2018, 8, 1020. [Google Scholar] [CrossRef]
- Haase, A.D.; Jaskiewicz, L.; Zhang, H.; Laine, S.; Sack, R.; Gatignol, A.; Filipowicz, W. TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep. 2005, 6, 961–967. [Google Scholar] [CrossRef]
- Goedhart, J.; von Stetten, D.; Noirclerc-Savoye, M.; Lelimousin, M.; Joosen, L.; Hink, M.A.; van Weeren, L.; Gadella, T.W., Jr.; Royant, A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 2012, 3, 751. [Google Scholar] [CrossRef]
- Lohöfener, J.; Steinke, N.; Kay-Fedorov, P.; Baruch, P.; Nikulin, A.; Tishchenko, S.; Manstein, D.J.; Fedorov, R. The activation mechanism of 2′-5′-oligoadenylate synthetase gives new insights into OAS/cGAS triggers of innate immunity. Structure 2015, 23, 851–862. [Google Scholar] [CrossRef]
- Sarkar, S.N.; Bandyopadhyay, S.; Ghosh, A.; Sen, G.C. Enzymatic characteristics of recombinant medium isozyme of 2′-5′ oligoadenylate synthetase. J. Biol. Chem. 1999, 274, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Xu, L.; Zhou, A.; Hassel, B.A.; Lee, X.; Torrence, P.F.; Silverman, R.H. Intrinsic molecular activities of the interferon-induced 2-5A-dependent RNase. J. Biol. Chem. 1994, 269, 14153–14158. [Google Scholar] [PubMed]
- Hartmann, R.; Olsen, H.S.; Widder, S.; Jorgensen, R.; Justesen, J. p59OASL, a 2′-5′ oligoadenylate synthetase like protein: A novel human gene related to the 2′-5′ oligoadenylate synthetase family. Nucleic Acids Res. 1998, 26, 4121–4128. [Google Scholar] [CrossRef] [PubMed]
- Rebouillat, D.; Marie, I.; Hovanessian, A.G. Molecular cloning and characterization of two related and interferon-induced 56-kDa and 30-kDa proteins highly similar to 2′-5′ oligoadenylate synthetase. Eur. J. Biochem. 1998, 257, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Shao, L.; Sampath, P.; Zhao, B.; Patel, N.V.; Zhu, J.; Behl, B.; Parise, R.A.; Beumer, J.H.; O’Sullivan, R.J.; et al. Oligoadenylate-Synthetase-Family Protein OASL Inhibits Activity of the DNA Sensor cGAS during DNA Virus Infection to Limit Interferon Production. Immunity 2019, 50, 51–63.e5. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Jha, B.K.; Silverman, R.H. New insights into the role of RNase L in innate immunity. J. Interferon Cytokine Res. 2011, 31, 49–57. [Google Scholar] [CrossRef]
- Malathi, K.; Dong, B.; Gale, M.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef]
- Malathi, K.; Saito, T.; Crochet, N.; Barton, D.J.; Gale, M., Jr.; Silverman, R.H. RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA 2010, 16, 2108–2119. [Google Scholar] [CrossRef]
- Khabar, K.S.A.; Siddiqui, Y.M.; Al-Zoghaibi, F.; Al-Haj, L.; Dhalla, M.; Zhou, A.; Dong, B.; Whitmore, M.; Paranjape, J.; Al-Ahdal, M.N.; et al. RNase L mediates transient control of the interferon response through modulation of the double-stranded RNA-dependent protein kinase PKR. J. Biol. Chem. 2003, 278, 20124–20132. [Google Scholar] [CrossRef]
- Lamers, M.M.; van den Hoogen, B.G.; Haagmans, B.L. ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front. Immunol. 2019, 10, 1763. [Google Scholar] [CrossRef]
- Serra, M.J.; Smolter, P.E.; Westhof, E. Pronounced instability of tandem IU base pairs in RNA. Nucleic Acids Res. 2004, 32, 1824–1828. [Google Scholar] [CrossRef]
- Kim, D.D.Y.; Kim, T.T.Y.; Walsh, T.; Kobayashi, Y.; Matise, T.C.; Buyske, S.; Gabriel, A. Widespread RNA editing of embedded Alu elements in the human transcriptome. Genome Res. 2004, 14, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- John, L.; Samuel, C.E. Induction of stress granules by interferon and down-regulation by the cellular RNA adenosine deaminase ADAR1. Virology 2014, 454–455, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Okonski, K.M.; Samuel, C.E. Stress Granule Formation Induced by Measles Virus Is Protein Kinase PKR Dependent and Impaired by RNA Adenosine Deaminase ADAR1. J. Virol. 2013, 87, 756–766. [Google Scholar] [CrossRef]
- Toth, A.M.; Li, Z.; Cattaneo, R.; Samuel, C.E. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J. Biol. Chem. 2009, 284, 29350–29356. [Google Scholar] [CrossRef] [PubMed]
- Pujantell, M.; Franco, S.; Galván-Femenía, I.; Badia, R.; Castellví, M.; Garcia-Vidal, E.; Clotet, B.; de Cid, R.; Tural, C.; Martínez, M.A.; et al. ADAR1 affects HCV infection by modulating innate immune response. Antivir. Res. 2018, 156, 116–127. [Google Scholar] [CrossRef]
- Todorova, T.; Bock, F.J.; Chang, P. Poly(ADP-ribose) polymerase-13 and RNA regulation in immunity and cancer. Trends Mol. Med. 2015, 21, 373–384. [Google Scholar] [CrossRef]
- Lee, H.; Komano, J.; Saitoh, Y.; Yamaoka, S.; Kozaki, T.; Misawa, T.; Takahama, M.; Satoh, T.; Takeuchi, O.; Yamamoto, N.; et al. Zinc-finger antiviral protein mediates retinoic acid inducible gene I-like receptor-independent antiviral response to murine leukemia virus. Proc. Natl. Acad. Sci. USA 2013, 110, 12379–12384. [Google Scholar] [CrossRef]
- Li, M.M.; Lau, Z.; Cheung, P.; Aguilar, E.G.; Schneider, W.M.; Bozzacco, L.; Molina, H.; Buehler, E.; Takaoka, A.; Rice, C.M.; et al. TRIM25 Enhances the Antiviral Action of Zinc-Finger Antiviral Protein (ZAP). PLoS Pathog. 2017, 13, e1006145. [Google Scholar] [CrossRef]
- Seo, G.J.; Kincaid, R.P.; Phanaksri, T.; Burke, J.M.; Pare, J.M.; Cox, J.E.; Hsiang, T.Y.; Krug, R.M.; Sullivan, C.S. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe 2013, 14, 435–445. [Google Scholar] [CrossRef]
- Hayakawa, S.; Shiratori, S.; Yamato, H.; Kameyama, T.; Kitatsuji, C.; Kashigi, F.; Goto, S.; Kameoka, S.; Fujikura, D.; Yamada, T.; et al. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat. Immunol. 2011, 12, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Schwerk, J.; Soveg, F.W.; Ryan, A.P.; Thomas, K.R.; Hatfield, L.D.; Ozarkar, S.; Forero, A.; Kell, A.M.; Roby, J.A.; So, L.; et al. RNA-binding protein isoforms ZAP-S and ZAP-L have distinct antiviral and immune resolution functions. Nat. Immunol. 2019, 20, 1610–1620. [Google Scholar] [CrossRef]
- Jankowsky, E.; Jankowsky, A. The DExH/D protein family database. Nucleic Acids Res. 2000, 28, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Linder, P.; Jankowsky, E. From unwinding to clamping—The DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Liu, S.; Zhu, Y.; Chen, G.; Gao, G. DEXH-Box protein DHX30 is required for optimal function of the zinc-finger antiviral protein. Protein Cell 2010, 1, 956–964. [Google Scholar] [CrossRef]
- Lai, M.C.; Sun, H.S.; Wang, S.W.; Tarn, W.Y. DDX3 functions in antiviral innate immunity through translational control of PACT. FEBS J. 2016, 283, 88–101. [Google Scholar] [CrossRef]
- Valiente-Echeverria, F.; Hermoso, M.A.; Soto-Rifo, R. RNA helicase DDX3: At the crossroad of viral replication and antiviral immunity. Rev. Med. Virol. 2015, 25, 286–299. [Google Scholar] [CrossRef]
- Saito, M.; Hess, D.; Eglinger, J.; Fritsch, A.W.; Kreysing, M.; Weinert, B.T.; Choudhary, C.; Matthias, P. Acetylation of intrinsically disordered regions regulates phase separation. Nat. Chem. Biol. 2019, 15, 51–61. [Google Scholar] [CrossRef]
- Mok, B.W.; Song, W.; Wang, P.; Tai, H.; Chen, Y.; Zheng, M.; Wen, X.; Lau, S.Y.; Wu, W.L.; Matsumoto, K.; et al. The NS1 protein of influenza A virus interacts with cellular processing bodies and stress granules through RNA-associated protein 55 (RAP55) during virus infection. J. Virol. 2012, 86, 12695–12707. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Liu, Y.; Park, I.W.; Zhang, J.; Sanford, J.; He, J.J. Suppression of HIV-1 Nef translation by Sam68 mutant-induced stress granules and nef mRNA sequestration. Mol. Cell 2009, 33, 87–96. [Google Scholar] [CrossRef][Green Version]
- Simpson-Holley, M.; Kedersha, N.; Dower, K.; Rubins, K.H.; Anderson, P.; Hensley, L.E.; Connor, J.H. Formation of antiviral cytoplasmic granules during orthopoxvirus infection. J. Virol. 2011, 85, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Rozelle, D.K.; Filone, C.M.; Kedersha, N.; Connor, J.H. Activation of stress response pathways promotes formation of antiviral granules and restricts virus replication. Mol. Cell. Biol. 2014, 34, 2003–2016. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, J.; Civas, A.; Lama, Z.; Lagaudriere-Gesbert, C.; Blondel, D. Rabies Virus Infection Induces the Formation of Stress Granules Closely Connected to the Viral Factories. PLoS Pathog. 2016, 12, e1005942. [Google Scholar] [CrossRef]
- Brownsword, M.J.; Doyle, N.; Brocard, M.; Locker, N.; Maier, H.J. Infectious Bronchitis Virus Regulates Cellular Stress Granule Signaling. Viruses 2020, 12, 536. [Google Scholar] [CrossRef] [PubMed]
- Langland, J.O.; Cameron, J.M.; Heck, M.C.; Jancovich, J.K.; Jacobs, B.L. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006, 119, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, J.; Hansen, S.J.; Park, N.; Jamka, K.; Sarnow, P.; Gustin, K.E. Stable formation of compositionally unique stress granules in virus-infected cells. J. Virol. 2010, 84, 3654–3665. [Google Scholar] [CrossRef]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef]
- Krisenko, M.O.; Higgins, R.L.; Ghosh, S.; Zhou, Q.; Trybula, J.S.; Wang, W.H.; Geahlen, R.L. Syk Is Recruited to Stress Granules and Promotes Their Clearance through Autophagy. J. Biol. Chem. 2015, 290, 27803–27815. [Google Scholar] [CrossRef]
- Guo, H.; Chitiprolu, M.; Gagnon, D.; Meng, L.; Perez-Iratxeta, C.; Lagace, D.; Gibbings, D. Autophagy supports genomic stability by degrading retrotransposon RNA. Nat. Commun. 2014, 5, 5276. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhu, G.; Tang, Y.; Yan, J.; Han, S.; Yin, J.; Peng, B.; He, X.; Liu, W. HDAC6, A Novel Cargo for Autophagic Clearance of Stress Granules, Mediates the Repression of the Type I Interferon Response During Coxsackievirus A16 Infection. Front. Microbiol. 2020, 11, 78. [Google Scholar] [CrossRef]
- Frankel, L.B.; Lubas, M.; Lund, A.H. Emerging connections between RNA and autophagy. Autophagy 2017, 13, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Olasunkanmi, O.I.; Chen, S.; Mageto, J.; Zhong, Z. Virus-Induced Cytoplasmic Aggregates and Inclusions are Critical Cellular Regulatory and Antiviral Factors. Viruses 2020, 12, 399. [Google Scholar] [CrossRef]
- Wang, Y.; Duan, Y.; Han, C.; Yao, S.; Qi, X.; Gao, Y.; Maier, H.J.; Britton, P.; Chen, L.; Zhang, L.; et al. Infectious Bursal Disease Virus Subverts Autophagic Vacuoles To Promote Viral Maturation and Release. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Chen, C.J.; Shih, S.R. Regulation Mechanisms of Viral IRES-Driven Translation. Trends Microbiol. 2017, 25, 546–561. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, A.; Martinez-Salas, E. Insights into the biology of IRES elements through riboproteomic approaches. J. Biomed. Biotechnol. 2010, 2010, 458927. [Google Scholar] [CrossRef]
- Galan, A.; Lozano, G.; Pineiro, D.; Martinez-Salas, E. G3BP1 interacts directly with the FMDV IRES and negatively regulates translation. FEBS J. 2017, 284, 3202–3217. [Google Scholar] [CrossRef]
- Cammas, A.; Pileur, F.; Bonnal, S.; Lewis, S.M.; Leveque, N.; Holcik, M.; Vagner, S. Cytoplasmic relocalization of heterogeneous nuclear ribonucleoprotein A1 controls translation initiation of specific mRNAs. Mol. Biol. Cell 2007, 18, 5048–5059. [Google Scholar] [CrossRef]
- Asnani, M.; Pestova, T.V.; Hellen, C.U. PCBP2 enables the cadicivirus IRES to exploit the function of a conserved GRNA tetraloop to enhance ribosomal initiation complex formation. Nucleic Acids Res. 2016, 44, 9902–9917. [Google Scholar] [CrossRef]
- Blyn, L.B.; Towner, J.S.; Semler, B.L.; Ehrenfeld, E. Requirement of poly(rC) binding protein 2 for translation of poliovirus RNA. J. Virol. 1997, 71, 6243–6246. [Google Scholar] [CrossRef]
- Majzoub, K.; Hafirassou, M.L.; Meignin, C.; Goto, A.; Marzi, S.; Fedorova, A.; Verdier, Y.; Vinh, J.; Hoffmann, J.A.; Martin, F.; et al. RACK1 controls IRES-mediated translation of viruses. Cell 2014, 159, 1086–1095. [Google Scholar] [CrossRef]
- Dave, P.; George, B.; Sharma, D.K.; Das, S. Polypyrimidine tract-binding protein (PTB) and PTB-associated splicing factor in CVB3 infection: An ITAF for an ITAF. Nucleic Acids Res. 2017, 45, 9068–9084. [Google Scholar] [CrossRef] [PubMed]
- Gallouzi, I.E.; Brennan, C.M.; Stenberg, M.G.; Swanson, M.S.; Eversole, A.; Maizels, N.; Steitz, J.A. HuR binding to cytoplasmic mRNA is perturbed by heat shock. Proc. Natl. Acad. Sci. USA 2000, 97, 3073–3078. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Aravena, A.; Ramdohr, P.; Vallejos, M.; Valiente-Echeverria, F.; Dormoy-Raclet, V.; Rodriguez, F.; Pino, K.; Holzmann, C.; Huidobro-Toro, J.P.; Gallouzi, I.E.; et al. The Elav-like protein HuR exerts translational control of viral internal ribosome entry sites. Virology 2009, 392, 178–185. [Google Scholar] [CrossRef]
- Liu, W.; Yang, D.; Sun, C.; Wang, H.; Zhao, B.; Zhou, G.; Yu, L. hnRNP K Is a Novel Internal Ribosomal Entry Site-Transacting Factor That Negatively Regulates Foot-and-Mouth Disease Virus Translation and Replication and Is Antagonized by Viral 3C Protease. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Bevilacqua, E.; Wang, X.; Majumder, M.; Gaccioli, F.; Yuan, C.L.; Wang, C.; Zhu, X.; Jordan, L.E.; Scheuner, D.; Kaufman, R.J.; et al. eIF2alpha phosphorylation tips the balance to apoptosis during osmotic stress. J. Biol. Chem. 2010, 285, 17098–17111. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Shen, Y.; Garre, E.; Hao, X.; Krumlinde, D.; Cvijovic, M.; Arens, C.; Nystrom, T.; Liu, B.; Sunnerhagen, P. Stress granule-defective mutants deregulate stress responsive transcripts. PLoS Genet. 2014, 10, e1004763. [Google Scholar] [CrossRef] [PubMed]
- Stohr, N.; Lederer, M.; Reinke, C.; Meyer, S.; Hatzfeld, M.; Singer, R.H.; Huttelmaier, S. ZBP1 regulates mRNA stability during cellular stress. J. Cell Biol. 2006, 175, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Wilbertz, J.H.; Voigt, F.; Horvathova, I.; Roth, G.; Zhan, Y.; Chao, J.A. Single-Molecule Imaging of mRNA Localization and Regulation during the Integrated Stress Response. Mol. Cell 2019, 73, 946–958 e947. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Stress granules. Curr. Biol. 2009, 19, R397–R398. [Google Scholar] [CrossRef]
- Herdy, B.; Karonitsch, T.; Vladimer, G.I.; Tan, C.S.; Stukalov, A.; Trefzer, C.; Bigenzahn, J.W.; Theil, T.; Holinka, J.; Kiener, H.P.; et al. The RNA-binding protein HuR/ELAVL1 regulates IFN-beta mRNA abundance and the type I IFN response. Eur. J. Immunol. 2015, 45, 1500–1511. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, R.L.; Sternjohn, J.R.; Rattenbacher, B.; Vlasova, I.A.; Williams, D.A.; Hau, H.H.; Blackshear, P.J.; Bohjanen, P.R. Tristetraprolin mediates interferon-gamma mRNA decay. J. Biol. Chem. 2009, 284, 11216–11223. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Abdelmohsen, K.; Srikantan, S.; Guo, R.; Yang, X.; Martindale, J.L.; Gorospe, M. Tyrosine phosphorylation of HuR by JAK3 triggers dissociation and degradation of HuR target mRNAs. Nucleic Acids Res. 2014, 42, 1196–1208. [Google Scholar] [CrossRef]
- Contu, L.; Steiner, S.; Thiel, V.; Muhlemann, O. The Role of Stress Granules and the Nonsense-mediated mRNA Decay Pathway in Antiviral Defence. Chimia (Aarau) 2019, 73, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, N.S.; McIntyre, A.B.R.; Mattocks, M.D.; Holley, C.L.; Lazear, H.M.; Mason, C.E.; Horner, S.M. Altered m(6)A Modification of Specific Cellular Transcripts Affects Flaviviridae Infection. Mol. Cell 2020, 77, 542–555 e548. [Google Scholar] [CrossRef] [PubMed]
- Rubio, R.M.; Depledge, D.P.; Bianco, C.; Thompson, L.; Mohr, I. RNA m(6) A modification enzymes shape innate responses to DNA by regulating interferon beta. Genes Dev. 2018, 32, 1472–1484. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.; Gillis, E.; Lasman, L.; Safra, M.; Geula, S.; Soyris, C.; Nachshon, A.; Tai-Schmiedel, J.; Friedman, N.; Le-Trilling, V.T.K.; et al. m(6)A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 2019, 20, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Zhang, X.; Wang, J.; Ma, Y.; Zhang, L.; Cao, X. RNA-binding protein YTHDF3 suppresses interferon-dependent antiviral responses by promoting FOXO3 translation. Proc. Natl. Acad. Sci. USA 2019, 116, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Ohn, T.; Kedersha, N.; Hickman, T.; Tisdale, S.; Anderson, P. A functional RNAi screen links O-GlcNAc modification of ribosomal proteins to stress granule and processing body assembly. Nat. Cell Biol. 2008, 10, 1224–1231. [Google Scholar] [CrossRef]
- Shattuck, J.E.; Paul, K.R.; Cascarina, S.M.; Ross, E.D. The prion-like protein kinase Sky1 is required for efficient stress granule disassembly. Nat. Commun. 2019, 10, 3614. [Google Scholar] [CrossRef]
- Jongjitwimol, J.; Baldock, R.A.; Morley, S.J.; Watts, F.Z. Sumoylation of eIF4A2 affects stress granule formation. J. Cell Sci. 2016, 129, 2407–2415. [Google Scholar] [CrossRef]
- Jayabalan, A.K.; Sanchez, A.; Park, R.Y.; Yoon, S.P.; Kang, G.Y.; Baek, J.H.; Anderson, P.; Kee, Y.; Ohn, T. NEDDylation promotes stress granule assembly. Nat. Commun. 2016, 7, 12125. [Google Scholar] [CrossRef] [PubMed]
- Welsby, I.; Hutin, D.; Gueydan, C.; Kruys, V.; Rongvaux, A.; Leo, O. PARP12, an interferon-stimulated gene involved in the control of protein translation and inflammation. J. Biol. Chem. 2014, 289, 26642–26657. [Google Scholar] [CrossRef] [PubMed]
- Atasheva, S.; Frolova, E.I.; Frolov, I. Interferon-stimulated poly(ADP-Ribose) polymerases are potent inhibitors of cellular translation and virus replication. J. Virol. 2014, 88, 2116–2130. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, H.; Liu, P.; Li, C.; Quanquin, N.; Ji, X.; Sun, N.; Du, P.; Qin, C.F.; Lu, N.; et al. PARP12 suppresses Zika virus infection through PARP-dependent degradation of NS1 and NS3 viral proteins. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef]
- Li, W.; Simarro, M.; Kedersha, N.; Anderson, P. FAST is a survival protein that senses mitochondrial stress and modulates TIA-1-regulated changes in protein expression. Mol. Cell. Biol. 2004, 24, 10718–10732. [Google Scholar] [CrossRef]
- Samir, P.; Kesavardhana, S.; Patmore, D.M.; Gingras, S.; Malireddi, R.K.S.; Karki, R.; Guy, C.S.; Briard, B.; Place, D.E.; Bhattacharya, A.; et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 2019, 573, 590–594. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, W.; Liu, Z.; Liu, S.; Liang, X. Virus Infection and Death Receptor-Mediated Apoptosis. Viruses 2017, 9, 316. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).