
The First Sporadic Creutzfeldt–Jakob Disease Case with a Rare Molecular Subtype VV1 and 1-Octapeptide Repeat Deletion in PRNP

 ,
,  and
and
Abstract
:1. Introduction
2. Case Report
2.1. Disease Presentation
2.2. Clinical Tests
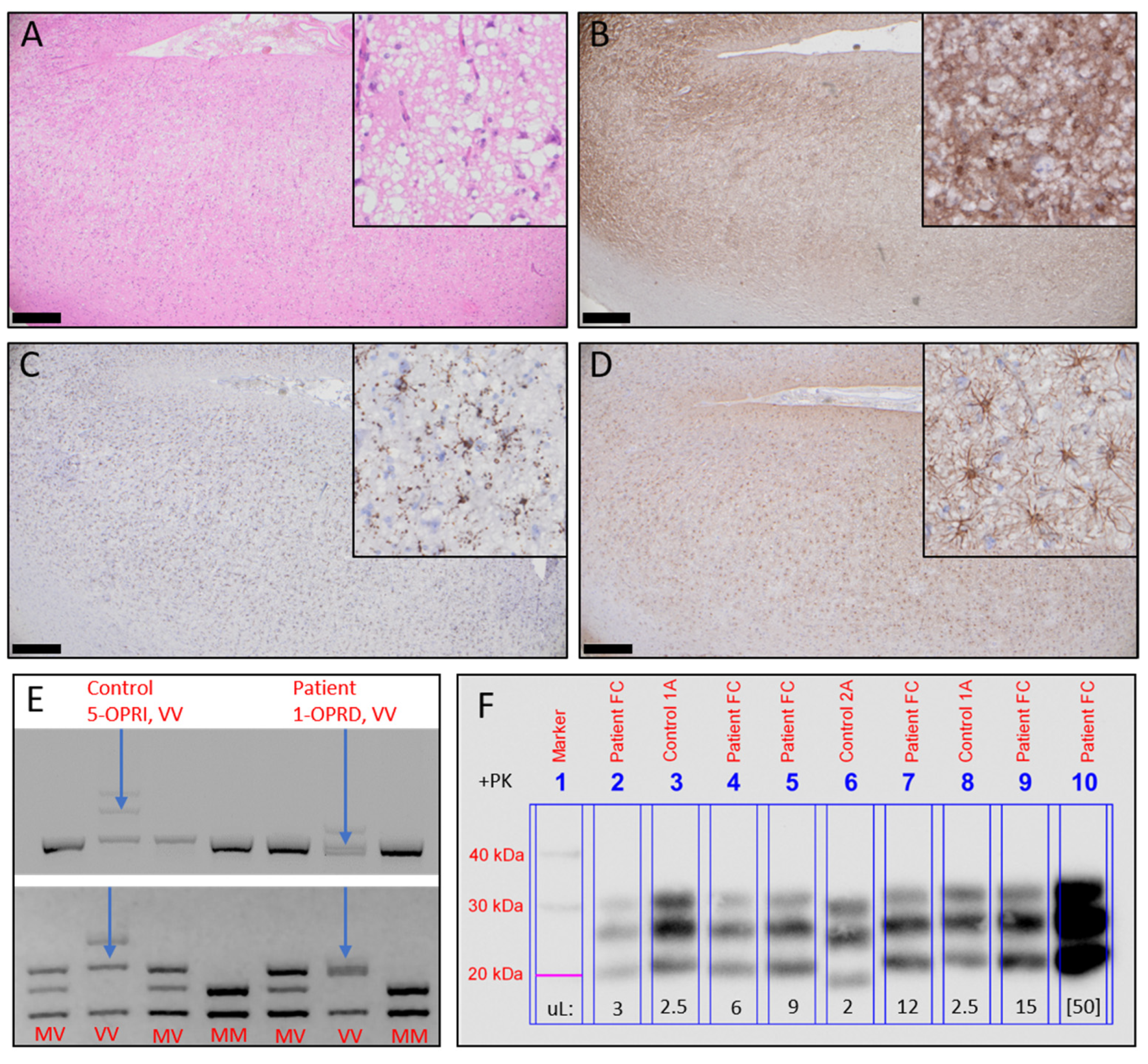
2.3. Neuropathology and Molecular Disease Subtyping
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Areškevičiūtė, A.; Broholm, H.; Melchior, L.C.; Bartoletti-Stella, A.; Parchi, P.; Capellari, S.; Scheie, D.; Lund, E.L. Molecular Characterization of the Danish Prion Diseases Cohort With Special Emphasis on Rare and Unique Cases. J. Neuropathol. Exp. Neurol. 2019, 78, 980–992. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef]
- Rossi, M.; Baiardi, S.; Parchi, P. Understanding Prion Strains: Evidence from Studies of the Disease Forms Affecting Humans. Viruses 2019, 11, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minikel, E.V.; Vallabh, S.M.; Lek, M.; Estrada, K.; Samocha, K.E.; Sathirapongsasuti, J.F.; McLean, C.Y.; Tung, J.Y.; Yu, L.P.; Gambetti, P.; et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 2016, 8, 322ra329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.H.; Koriath, C.; Jaunmuktane, Z.; Campbell, T.; Joiner, S.; Wadsworth, J.D.F.; Hosszu, L.L.P.; Brandner, S.; Parvez, A.; Truelsen, T.C.; et al. Evaluating the causality of novel sequence variants in the prion protein gene by example. Neurobiol. Aging 2018. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.O.; Takada, L.T.; Wong, K.; Forner, S.A.; Geschwind, M.D. Genetic PrP Prion Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.S.; Mahal, S.P.; Campbell, T.A.; Hill, A.F.; Sidle, K.C.L.; Laplanche, J.L.; Collinge, J. Deletions in the prion protein gene are not associated with CJD. Hum. Mol. Genet. 1993, 2, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Bishop, M.T.; Pennington, C.; Heath, C.A.; Will, R.G.; Knight, R.S. PRNP variation in UK sporadic and variant Creutzfeldt Jakob disease highlights genetic risk factors and a novel non-synonymous polymorphism. BMC Med. Genet. 2009, 10, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Areskeviciute, A.; Hogh, P.; Bartoletti-Stella, A.; Melchior, L.C.; Nielsen, P.R.; Parchi, P.; Capellari, S.; Broholm, H.; Scheie, D.; Lund, E.L. A Novel Eight Octapeptide Repeat Insertion in PRNP Causing Prion Disease in a Danish Family. J. Neuropathol. Exp. Neurol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Areskeviciute, A.; Melchior, L.C.; Broholm, H.; Krarup, L.H.; Granhoj Lindquist, S.; Johansen, P.; McKenzie, N.; Green, A.; Nielsen, J.E.; Laursen, H.; et al. Sporadic Creutzfeldt-Jakob Disease in a Woman Married Into a Gerstmann-Straussler-Scheinker Family: An Investigation of Prions Transmission via Microchimerism. J. Neuropathol. Exp. Neurol. 2018, 77, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.A.; Poulter, M.; Campbell, T.A.; Adamson, G.; Uphill, J.B.; Guerreiro, R.; Jackson, G.S.; Stevens, J.C.; Manji, H.; Collinge, J.; et al. PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum. Mutat. 2010, 31, E1551–E1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, B.; Westner, I.M.; Kallenberg, K.; Krasnianski, A.; Bartl, M.; Varges, D.; Bosenberg, C.; Kretzschmar, H.A.; Knauth, M.; Schulz-Schaeffer, W.J.; et al. Sporadic Creutzfeldt-Jakob disease—Clinical and diagnostic characteristics of the rare VV1 type. Neurology 2005, 65, 1544–1550. [Google Scholar] [CrossRef]
- Wuerz, T.P.; Bizzi, A.; Appleby, B.S. Unusual Case of Sporadic Creutzfeldt-Jakob Disease Subtype VV1. J. Neuropsychiatry Clin. Neurosci. 2015, 27, 172–173. [Google Scholar] [CrossRef] [Green Version]
- Green, A.; Sanchez-Juan, P.; Ladogana, A.; Cuadrado-Corrales, N.; Sanchez-Valle, R.; Mitrova, E.; Stoeck, K.; Sklaviadis, T.; Kulczycki, J.; Heinemann, U.; et al. CSF analysis in patients with sporadic CJD and other transmissible spongiform encephalopathies. Eur. J. Neurol. 2007, 14, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Green, A.J.E. RT-QuIC: A new test for sporadic CJD. Pract. Neurol. 2019, 19, 49–55. [Google Scholar] [CrossRef]
- Figgie, M.P., Jr.; Appleby, B.S. Clinical Use of Improved Diagnostic Testing for Detection of Prion Disease. Viruses 2021, 13, 789. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Symptom/Finding | 2019 April | 2019 August | 2019 September | 2019 October | 2019 November | 2019 December | 2020 February | 2020 May | 2020 December | 2021 January | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cognitive | Depression | - | - | Yes | Yes | Yes | - | - | - | - | - |
| Behavioral change | - | - | - | - | - | Yes | Yes | Yes | Yes | Yes | |
| Reduced intellect | - | - | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Apraxia | - | - | - | - | - | - | Yes | Yes | Yes | Yes | |
| Aphasia | - | - | - | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Memory loss | - | - | - | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Visual | Color sight change | - | - | - | - | - | - | - | - | - | - |
| Visual field loss | - | - | - | - | - | - | - | - | - | Yes | |
| Object distortion | - | - | - | - | - | - | - | - | - | - | |
| Blurred vision | - | - | - | - | - | - | - | - | - | - | |
| Cortical blindness | - | - | - | - | - | - | - | - | - | - | |
| Hallucinations | - | - | - | - | - | - | - | - | - | - | |
| Motor | Ataxia | - | - | - | - | - | - | - | - | - | - |
| Myoclonus | - | - | - | - | - | - | - | Yes | Yes | Yes | |
| Startle | - | - | - | - | - | - | - | - | Yes | Yes | |
| Mutism | - | - | - | - | - | Yes | Yes | Yes | Yes | Yes | |
| Rigidity | - | - | - | - | - | - | - | - | - | - | |
| Stupor | - | - | - | - | - | - | - | - | - | - |
| CSF Analyses | Ref. Interval | 2019 September | 2019 October | 2019 November | 2020 January |
|---|---|---|---|---|---|
| Leukocytes | <3 × 106/L | 2 | 2 | 1 | 3 (H) |
| Protein | 0.15–0.45 g/L | 0.39 | 0.41 | 0.41 | 0.356 |
| Immunglobulin-oligoclonia | - | Present | |||
| Amyloid beta-protein | >1.000 ng/L | 1.287 | 578 (L) | 1.016 | 670 (L) |
| Phosphorylated tau | <30 ng/L | 15 | 14 | 15 | 18 |
| Neurofilament light polypeptide | <890 ng/L | 1.837 (H) | 8.480 (H) | ||
| Tau protein | <300 ng/L | 881 | 1.305 (H) | 1.532 (H) | 2.310 (H) |
| RT-QuIC | Negative | ||||
| Changes in MRI | Yes | Yes | Yes | ||
| Triphasic EEG | No | No | No | ||
| Brain biopsy | PrPSc positive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Areškevičiūtė, A.; Lund, E.L.; Capellari, S.; Parchi, P.; Pinkowsky, C.T. The First Sporadic Creutzfeldt–Jakob Disease Case with a Rare Molecular Subtype VV1 and 1-Octapeptide Repeat Deletion in PRNP. Viruses 2021, 13, 2061. https://doi.org/10.3390/v13102061
Areškevičiūtė A, Lund EL, Capellari S, Parchi P, Pinkowsky CT. The First Sporadic Creutzfeldt–Jakob Disease Case with a Rare Molecular Subtype VV1 and 1-Octapeptide Repeat Deletion in PRNP. Viruses. 2021; 13(10):2061. https://doi.org/10.3390/v13102061
Chicago/Turabian StyleAreškevičiūtė, Aušrinė, Eva Løbner Lund, Sabina Capellari, Piero Parchi, and Christian Tersbøl Pinkowsky. 2021. "The First Sporadic Creutzfeldt–Jakob Disease Case with a Rare Molecular Subtype VV1 and 1-Octapeptide Repeat Deletion in PRNP" Viruses 13, no. 10: 2061. https://doi.org/10.3390/v13102061
APA StyleAreškevičiūtė, A., Lund, E. L., Capellari, S., Parchi, P., & Pinkowsky, C. T. (2021). The First Sporadic Creutzfeldt–Jakob Disease Case with a Rare Molecular Subtype VV1 and 1-Octapeptide Repeat Deletion in PRNP. Viruses, 13(10), 2061. https://doi.org/10.3390/v13102061