
Analysis of SARS-CoV-2 Genomes from West Java, Indonesia

 ,
,  ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Analysis of Distribution of Variants and Mutations
2.3. Analysis of Protein Stabilization
2.4. Molecular Docking
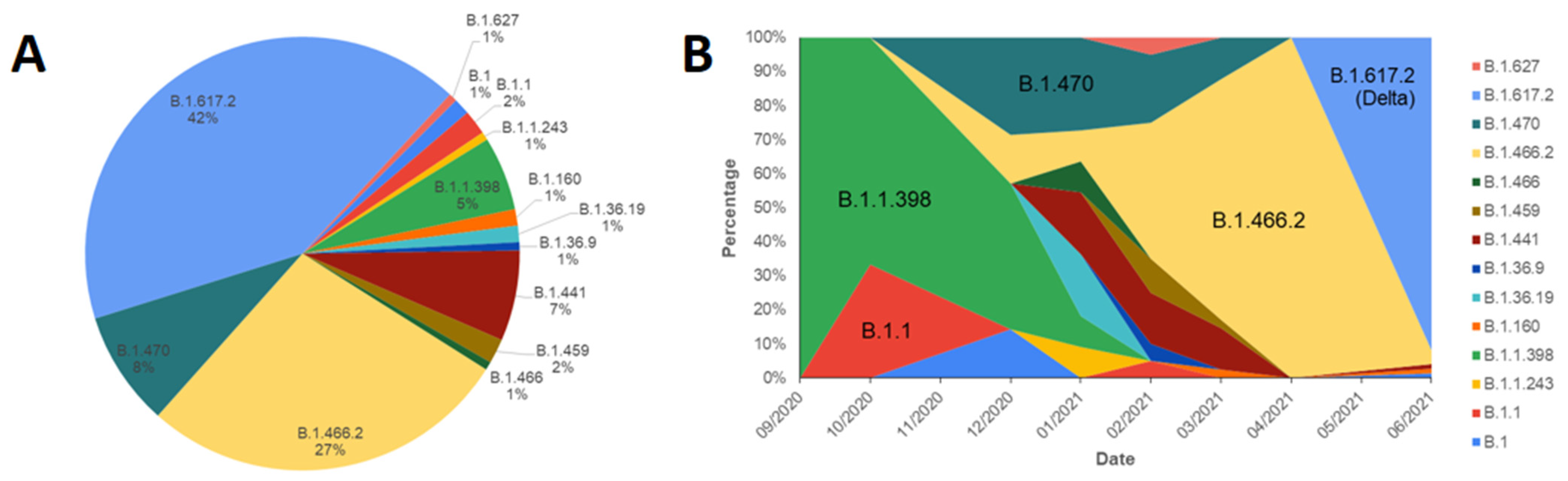
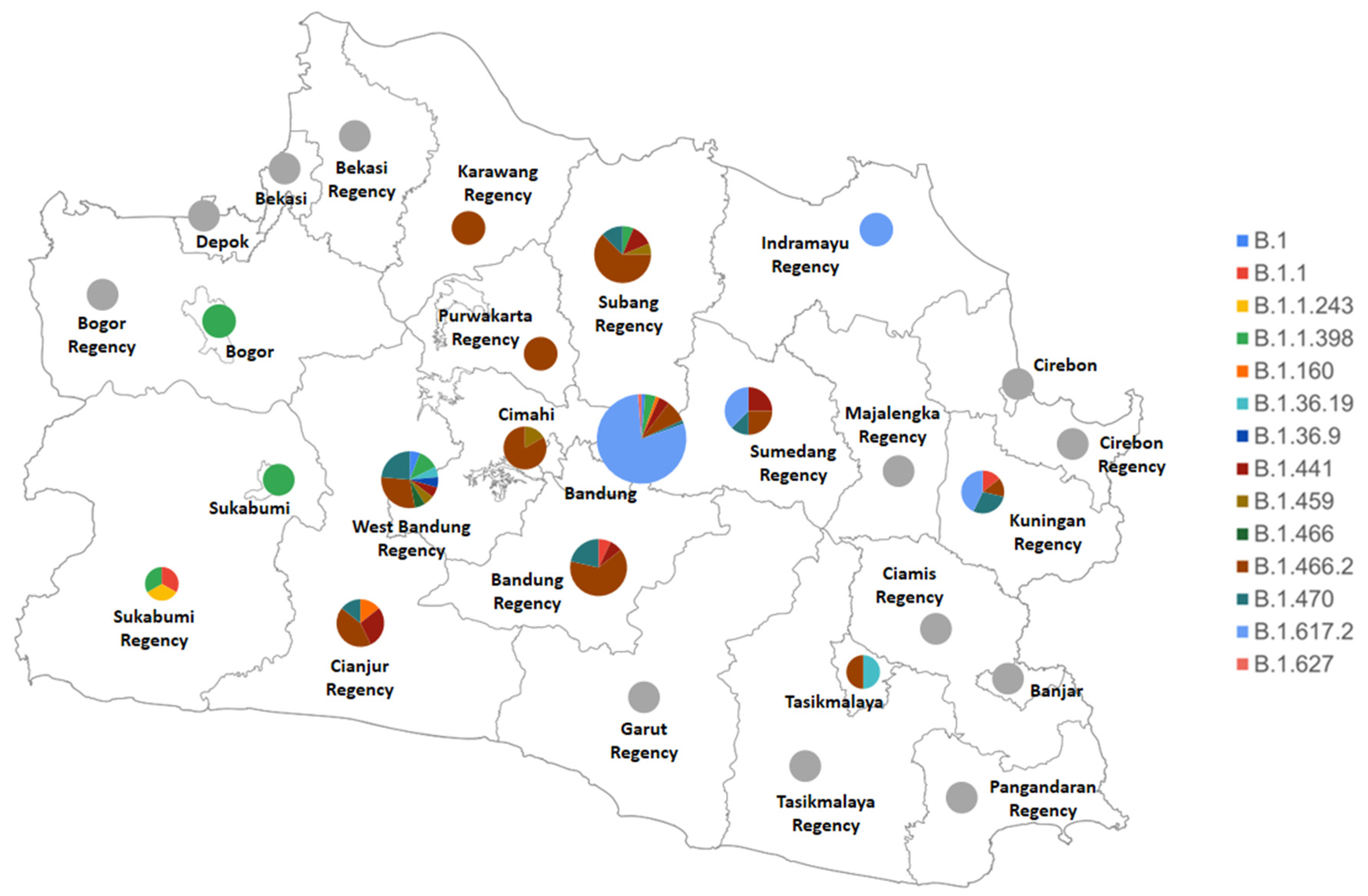
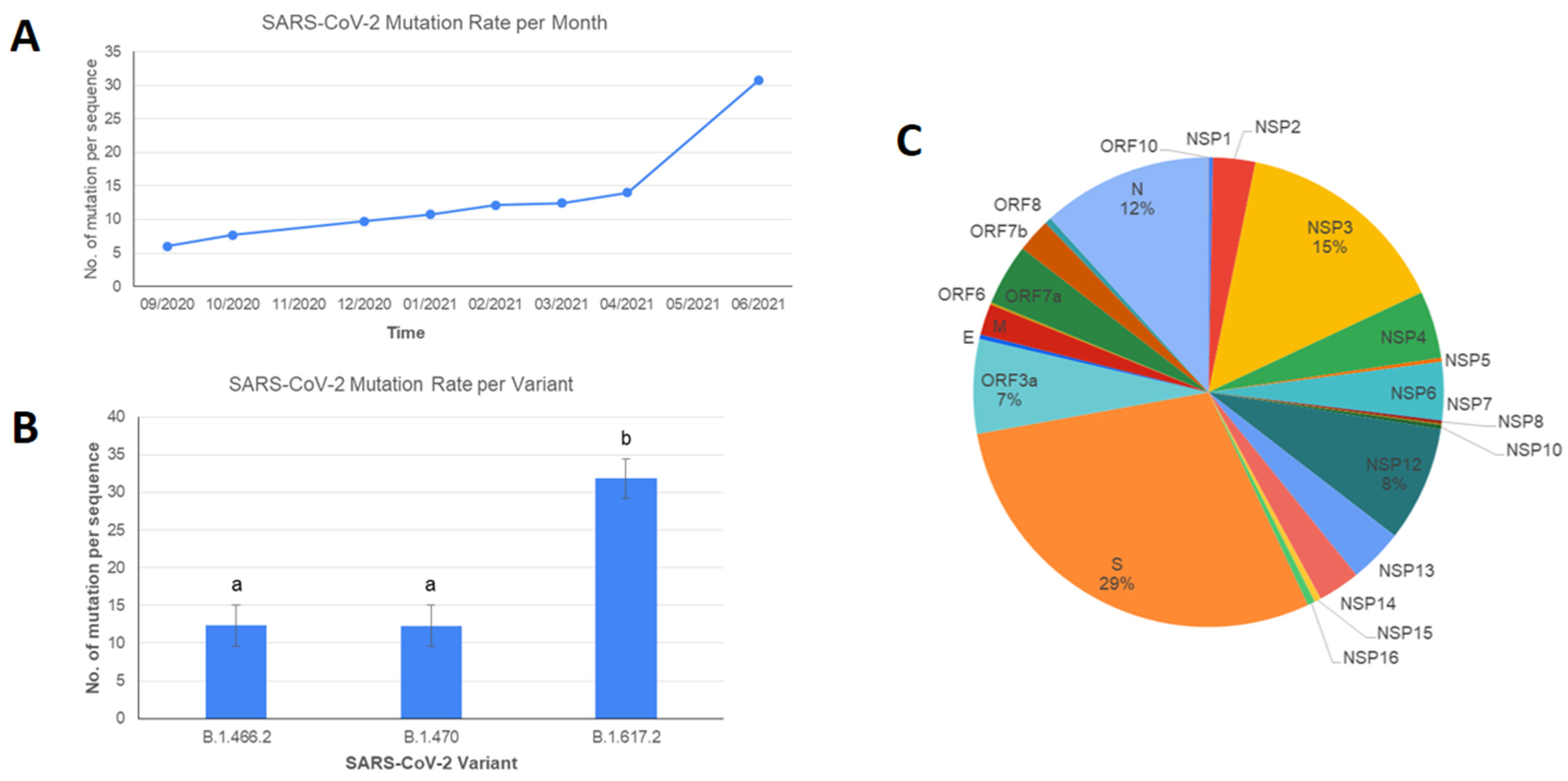
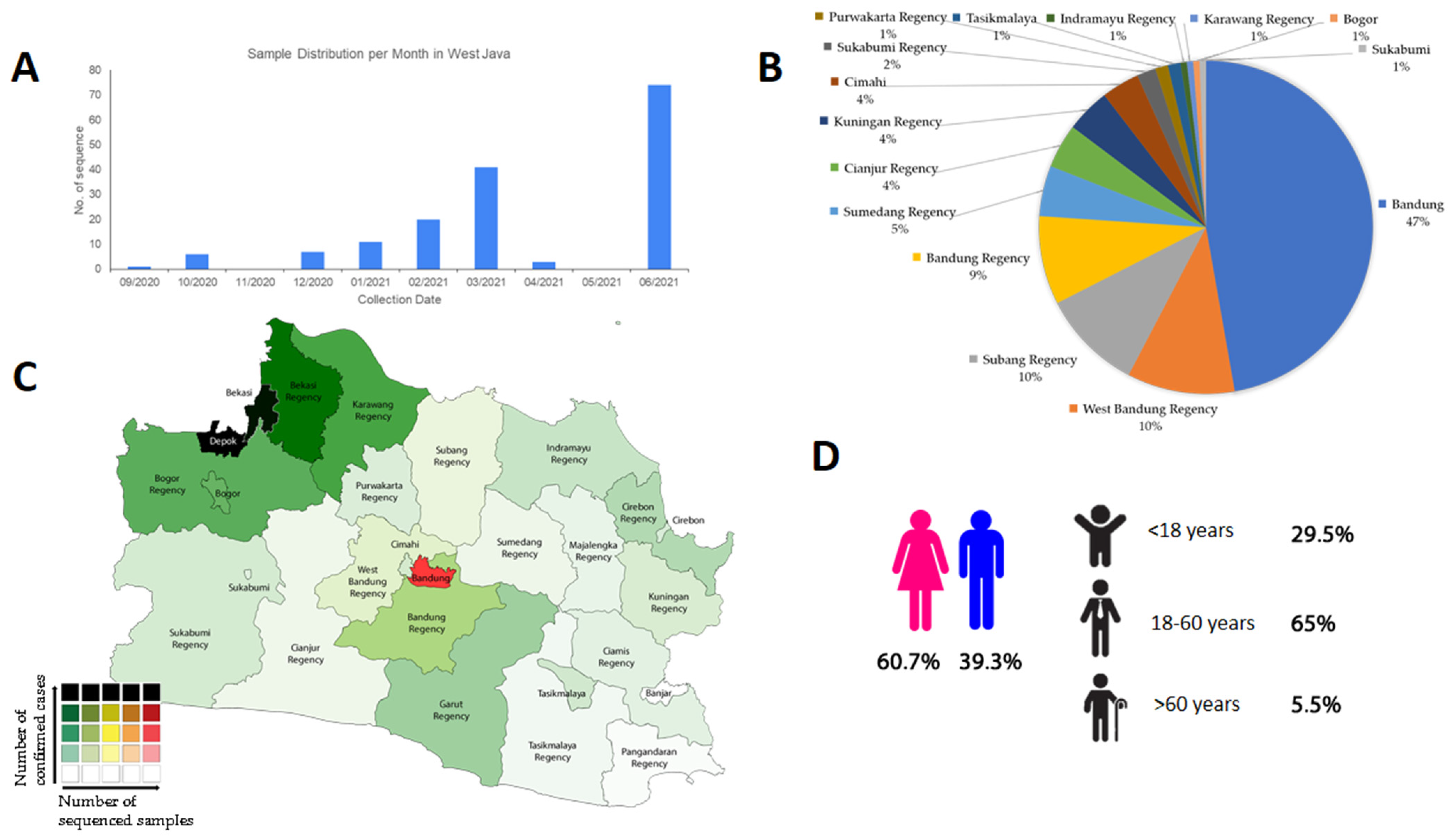
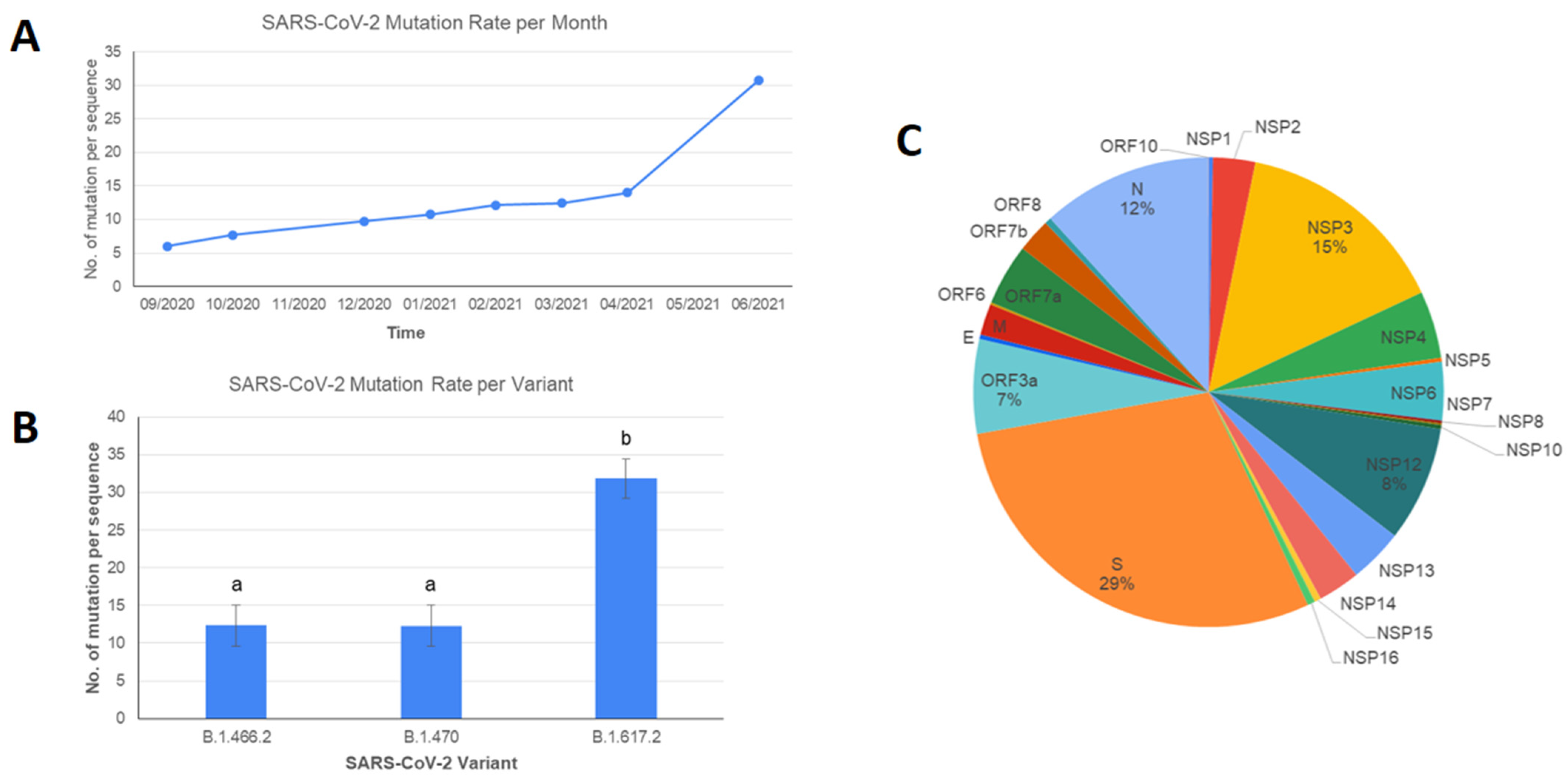
3. Results and Discussions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Worldometer. Indonesia COVID: 4,153,355 Cases and 138,116 Deaths—Worldometer. Available online: https://www.worldometers.info/coronavirus/country/indonesia/ (accessed on 1 March 2021).
- Wu, S.; Tian, C.; Liu, P.; Guo, D.; Zheng, W.; Huang, X.; Zhang, Y.; Liu, L. Effects of SARS-CoV-2 mutations on protein structures and intraviral protein–protein interactions. J. Med. Virol. 2021, 93, 2132–2140. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Urhan, A.; Abeel, T. Emergence of novel SARS-CoV-2 variants in the Netherlands. Sci. Rep. 2021, 11, 6625. [Google Scholar] [CrossRef] [PubMed]
- Alouane, T.; Laamarti, M.; Essabbar, A.; Hakmi, M.; Bouricha, E.M.; Chemao-Elfihri, M.W.; Kartti, S.; Boumajdi, N.; Bendani, H.; Laamarti, R.; et al. Genomic Diversity and Hotspot Mutations in 30,983 SARS-CoV-2 Genomes: Moving Toward a Universal Vaccine for the “Confined Virus”? Pathogens 2020, 9, 829. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Singer, J.B.; Gifford, R.J.; Cotten, M.; Robertson, D. CoV-GLUE: A Web Application for Tracking SARS-CoV-2 Genomic Variation. Preprints 2020. preprint. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. Protein Structure and Function Prediction Using I-TASSER. Curr. Protoc. Bioinform. 2015, 52, 5.8.1–5.8.15. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Durme, J.; Delgado, J.; Stricher, F.; Serrano, L.; Schymkowitz, J.; Rousseau, F. A graphical interface for the FoldX forcefield. Bioinformatics 2011, 27, 1711–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; van Dijk, M.; de Vries, S.; Bonvin, A.M. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.; DeLano, W. PyMOL. Available online: https://www.pymol.org/pymol.html? (accessed on 10 September 2021).
- Aisyah, D.N.; Mayadewi, C.A.; Diva, H.; Kozlakidis, Z.; Adisasmito, W. A spatial-temporal description of the SARS-CoV-2 infections in Indonesia during the first six months of outbreak. PLoS ONE 2020, 15, e0243703. [Google Scholar] [CrossRef]
- Cahyani, I.; Putro, E.W.; Ridwanuloh, A.M.; Wibowo, S.H.B.; Hariyatun, H.; Syahputra, G.; Akbariani, G.; Utomo, A.R.; Ilyas, M.; Loose, M.W.; et al. Genome Profiling of SARS-CoV-2 in Indonesia, ASEAN, and the Neighbouring East Asian Countries: Features, Challenges, and Achievements. bioRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Leong, R.; Lee, T.-S.J.; Chen, Z.; Zhang, C.; Xu, J. Global Temporal Patterns of Age Group and Sex Distributions of COVID-19. Infect. Dis. Rep. 2021, 13, 582–596. [Google Scholar] [CrossRef]
- PANGO. Cov-Lineages. Available online: https://cov-lineages.org/lineage_list.html (accessed on 31 August 2021).
- World Health Organization. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 31 August 2021).
- Chookajorn, T.; Kochakarn, T.; Wilasang, C.; Kotanan, N.; Modchang, C. Southeast Asia is an emerging hotspot for COVID-19. Nat. Med. 2021, 27, 1495–1496. [Google Scholar] [CrossRef] [PubMed]
- Media Nusantara. Bandung Barat Alami Lonjakan Kasus Covid-19. Available online: https://mediaindonesia.com/nusantara/410683/bandung-barat-alami-lonjakan-kasus-covid-19 (accessed on 31 August 2021).
- Karim, F. SARS-CoV-2 Sequences in Indonesia. Available online: https://varian.id/ (accessed on 31 August 2021).
- Indonesian Ministry of Health. IKHTISAR MINGGUAN COVID-19|Indonesia, 14–20 Agustus 2021. 2021. Available online: https://www.kemkes.go.id/downloads/resources/download/laporan-mingguan-covid/Laporan-Mingguan-Penanganan-Covid-19_25-Agustus.pdf (accessed on 31 August 2021).
- World Health Organization. Coronavirus Disease 2019 (COVID-19)|Situation Report—40, INDONESIA. 2021. Available online: https://cdn.who.int/media/docs/default-source/searo/indonesia/covid19/external-situation-report-40_27-january-2021.pdf (accessed on 31 August 2021).
- World Health Organization. Coronavirus Disease 2019 (COVID-19)|Situation Report—70, INDONESIA. 2021. Available online: https://cdn.who.int/media/docs/default-source/searo/indonesia/covid19/external-situation-report-70_1-september-2021.pdf (accessed on 31 August 2021).
- World Health Organization. Coronavirus Disease 2019 (COVID-19)|Situation Report—69, INDONESIA. 2021. Available online: https://cdn.who.int/media/docs/default-source/searo/indonesia/covid19/external-situation-report-69_25-august-2021.pdf?sfvrsn=c74a9ec7_5 (accessed on 31 August 2021).
- Hagen, A. How Dangerous Is the Delta Variant (B.1.617.2)? American Society of Microbiology. 2021. Available online: https://asm.org/Articles/2021/July/How-Dangerous-is-the-Delta-Variant-B-1-617-2 (accessed on 31 August 2021).
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Wang, R.; Chen, J.; Gao, K.; Hozumi, Y.; Yin, C.; Wei, G.W. Analysis of SARS-CoV-2 mutations in the United States suggests presence of four substrains and novel variants. Commun. Biol. 2021, 4, 228. [Google Scholar] [CrossRef] [PubMed]
- Suratekar, R.; Ghosh, P.; Niesen, M.J.M.; Donadio, G.; Anand, P.; Soundararajan, V.; Venkatakrishnan, A.J. High diversity in Delta variant across countries revealed via genome-wide analysis of SARS-CoV-2 beyond the Spike protein. bioRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Kar, T.; Narsaria, U.; Basak, S.; Deb, D.; Castiglione, F.; Mueller, D.M.; Srivastava, A.P. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 2020, 10, 10895. [Google Scholar] [CrossRef]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Xia, H.; Rajsbaum, R.; Xia, X.; Wang, H.; Shi, P.Y. Ubiquitination of SARS-CoV-2 ORF7a promotes antagonism of interferon response. Cell. Mol. Immunol. 2021, 18, 746–748. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fibriani, A.; Stephanie, R.; Alfiantie, A.A.; Siregar, A.L.F.; Pradani, G.A.P.; Yamahoki, N.; Purba, W.S.; Alamanda, C.N.C.; Rahmawati, E.; Rachman, R.W.; et al. Analysis of SARS-CoV-2 Genomes from West Java, Indonesia. Viruses 2021, 13, 2097. https://doi.org/10.3390/v13102097
Fibriani A, Stephanie R, Alfiantie AA, Siregar ALF, Pradani GAP, Yamahoki N, Purba WS, Alamanda CNC, Rahmawati E, Rachman RW, et al. Analysis of SARS-CoV-2 Genomes from West Java, Indonesia. Viruses. 2021; 13(10):2097. https://doi.org/10.3390/v13102097
Chicago/Turabian StyleFibriani, Azzania, Rebecca Stephanie, Afifah Alifia Alfiantie, Agust Leo Fany Siregar, Gusti Ayu Prani Pradani, Nicholas Yamahoki, William Steflandel Purba, Cut Nur Cinthia Alamanda, Ema Rahmawati, Rifky Waluyajati Rachman, and et al. 2021. "Analysis of SARS-CoV-2 Genomes from West Java, Indonesia" Viruses 13, no. 10: 2097. https://doi.org/10.3390/v13102097
APA StyleFibriani, A., Stephanie, R., Alfiantie, A. A., Siregar, A. L. F., Pradani, G. A. P., Yamahoki, N., Purba, W. S., Alamanda, C. N. C., Rahmawati, E., Rachman, R. W., Robiani, R., & Ristandi, R. B. (2021). Analysis of SARS-CoV-2 Genomes from West Java, Indonesia. Viruses, 13(10), 2097. https://doi.org/10.3390/v13102097