
Evaluating Large Spontaneous Deletions in a Bovine Cell Line Selected for Bovine Viral Diarrhea Virus Resistance

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Viruses
2.2. Whole Genome Sequence (WGS)
2.3. Annotation of RABGAP1L with Long-Read RNA Sequence Data
2.4. RNA Sequencing
2.5. Generation of MDBK Single and Triple Knockout (KO) Clones
2.5.1. gRNA Design and Production
2.5.2. Cas9 mRNA Preparation
2.5.3. Tissue Culture and Transfection for Single KO Clones
2.5.4. Single-Cell Derived Clonal Isolation and Genotyping
2.5.5. Mutation Detection
2.5.6. Selection of Clones for Downstream Testing
2.5.7. Generation of Triple Gene KO
2.6. SDS-PAGE and Immunoblotting
2.7. Multistep Virus Growth Curves
3. Results
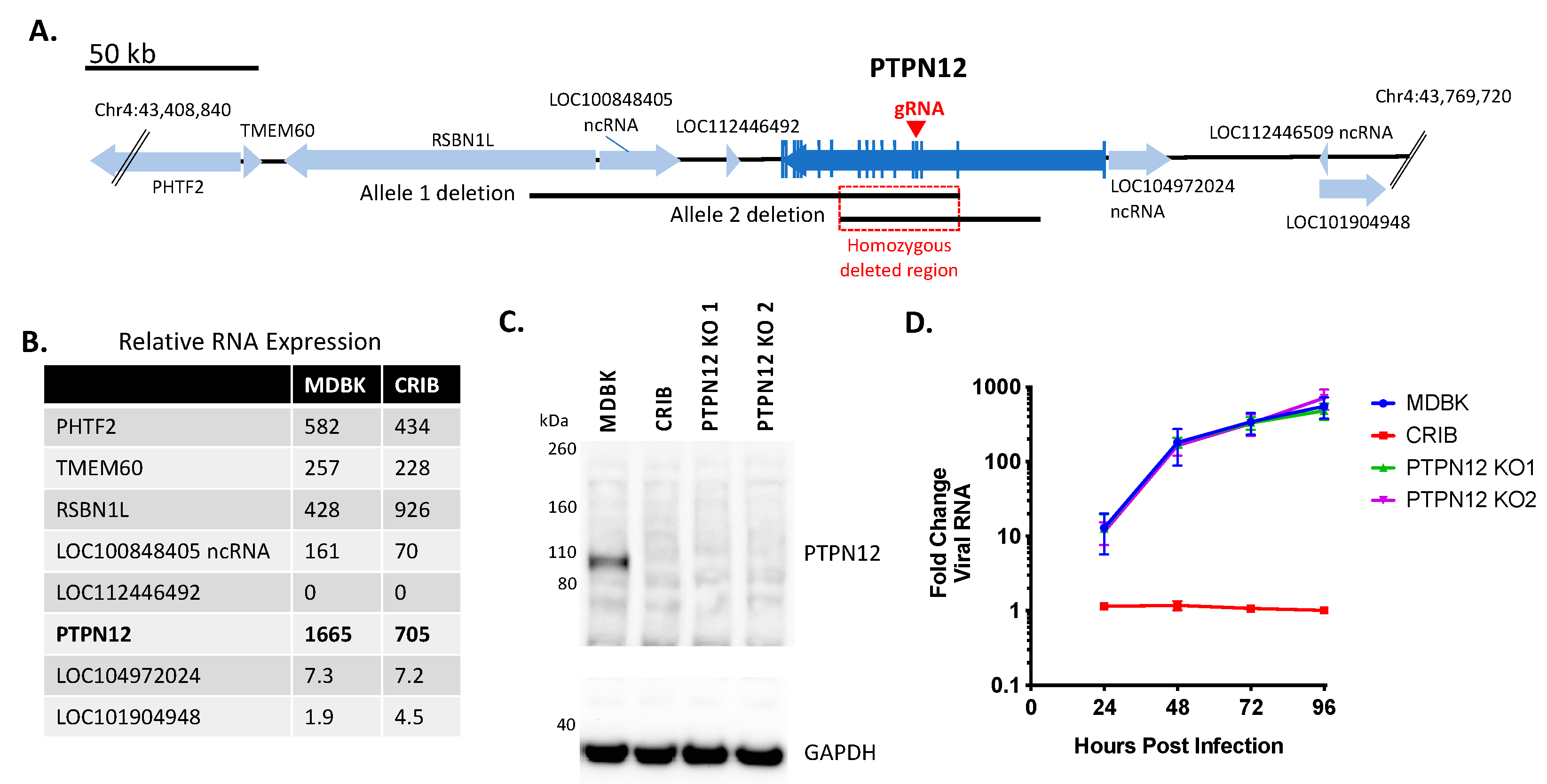
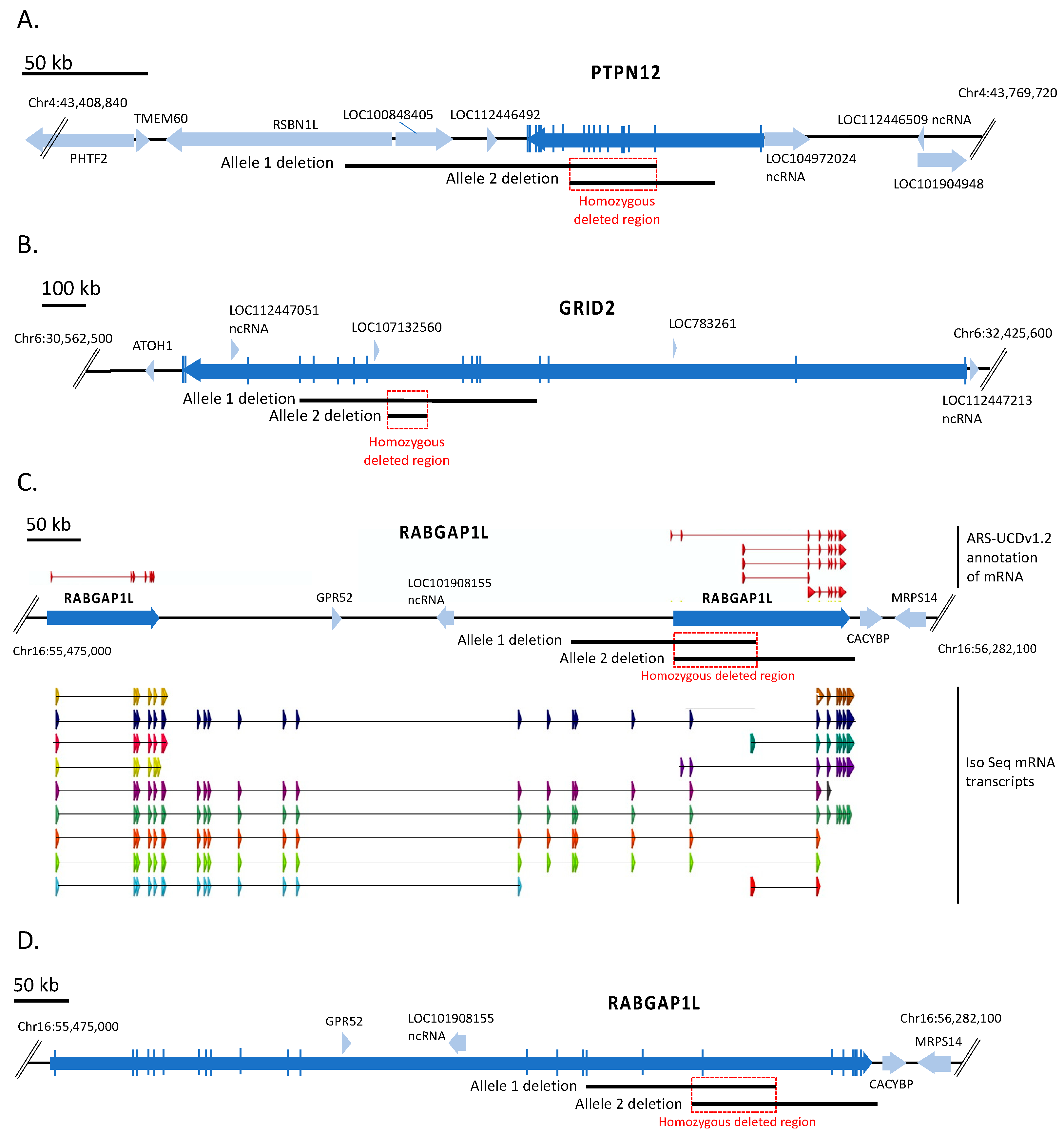
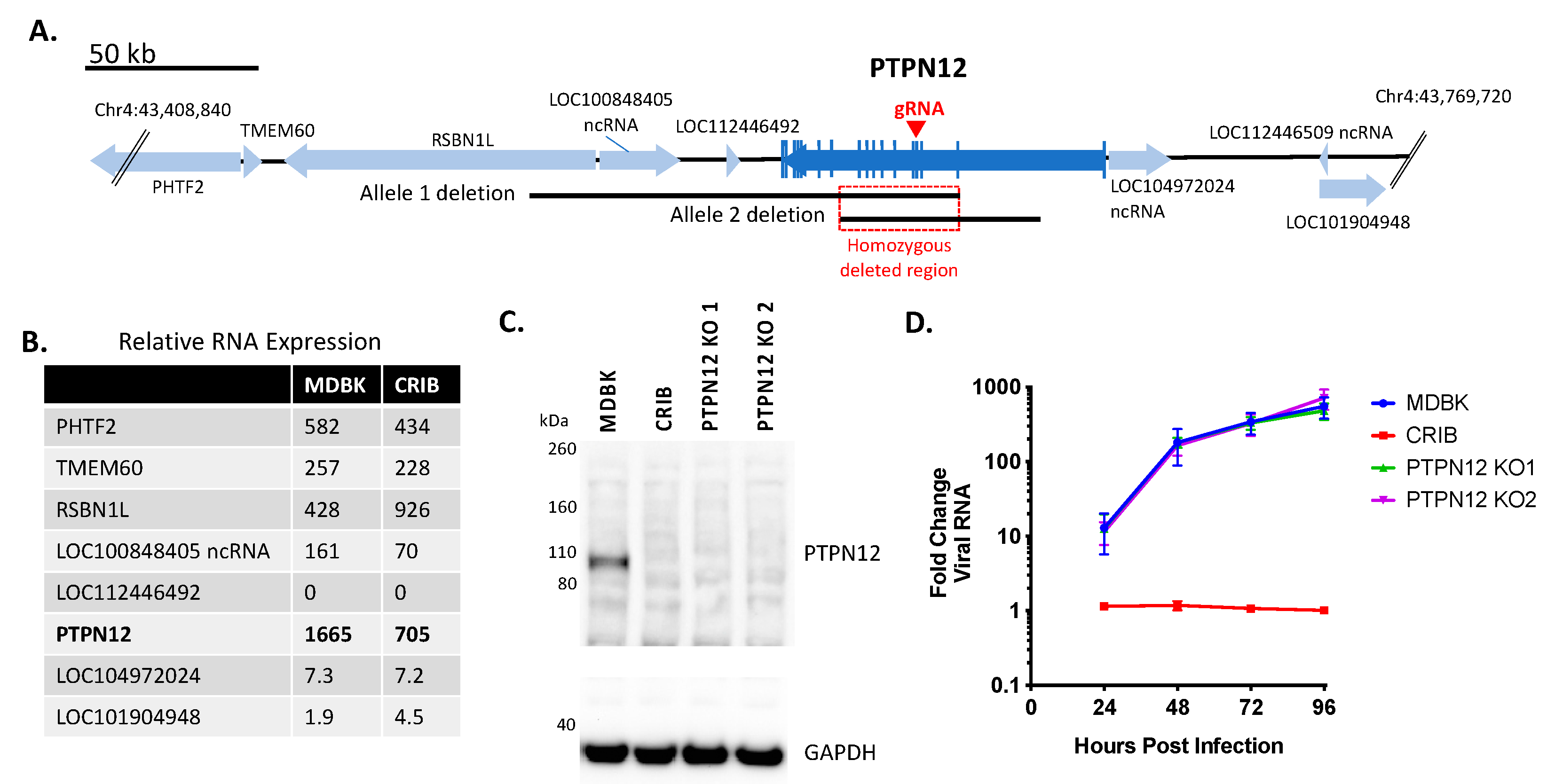
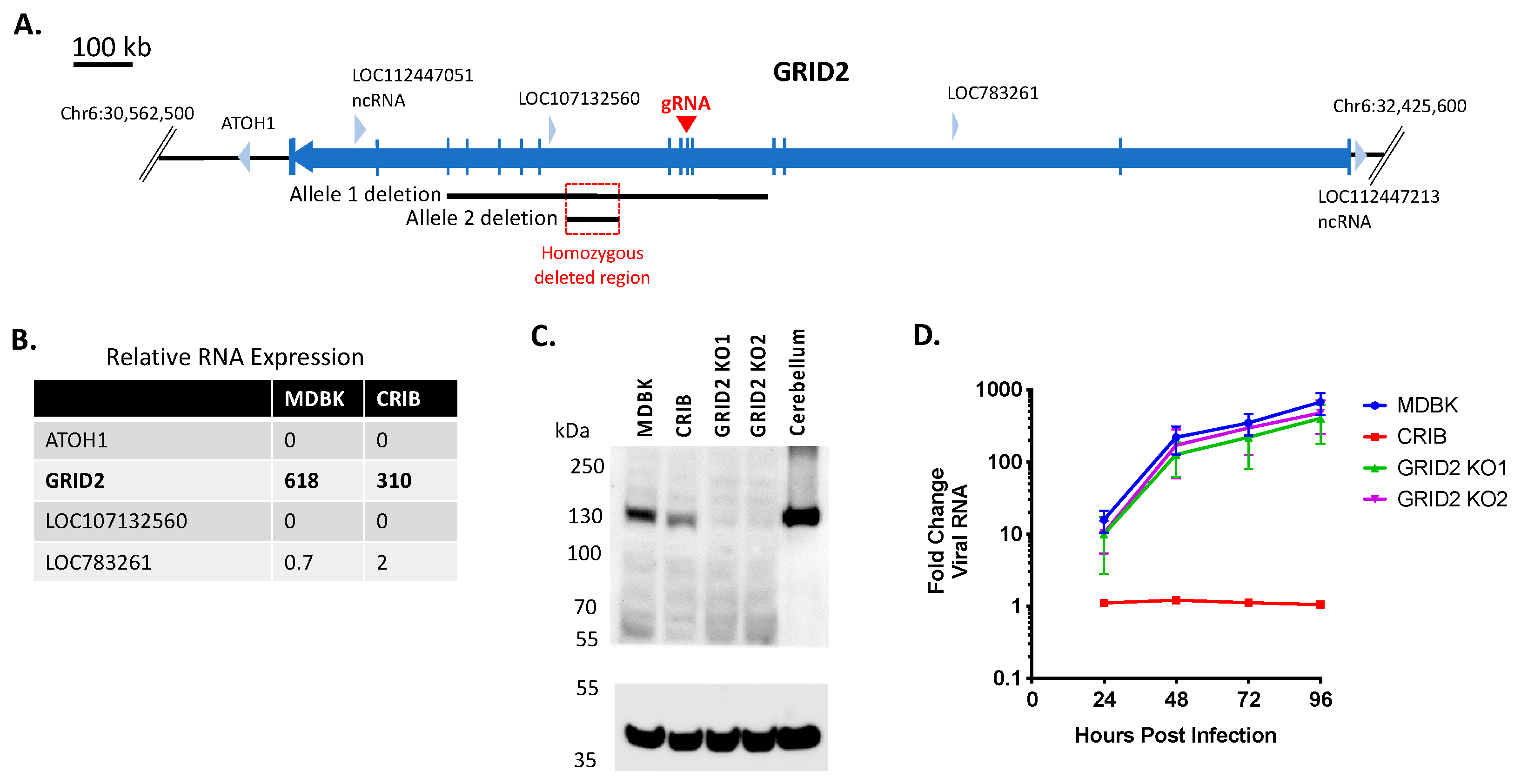
3.1. Identification of Three Large Genomic Deletions in the BVDV-Resistant CRIB Cell Line
3.2. Loss of PTPN12 Protein Expression Does Not Significantly Impact BVDV Infection in MDBK Cells
3.3. Loss of GRID2 Protein Expression Does Not Significantly Impact BVDV Infection in MDBK Cells
3.4. Disruption of the RABGAP1L Gene Does Not Significantly Impact BVDV Infection in MDBK Cells
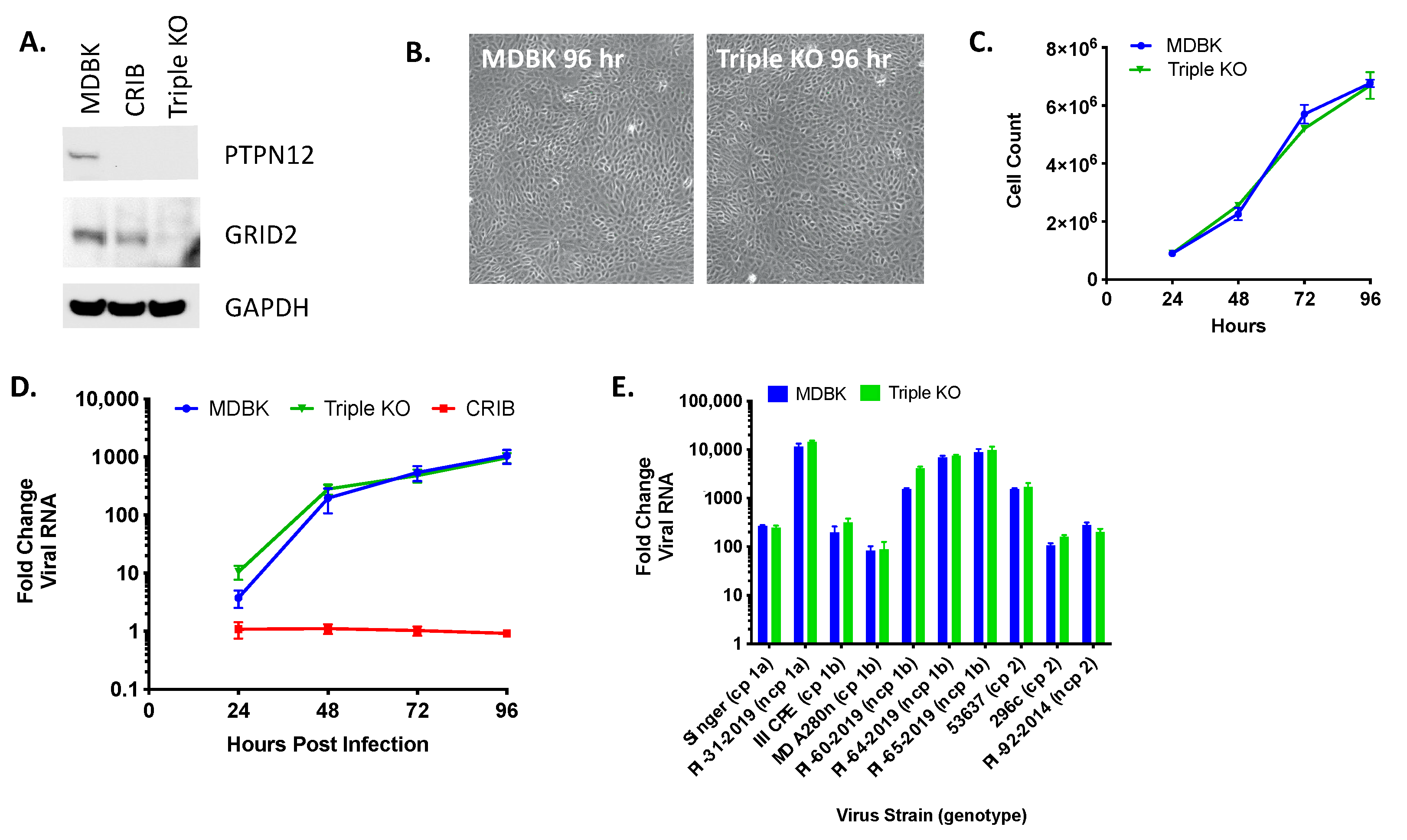
3.5. Triple KO of PTPN12, GRID2, and RABGAP1L Genes Does Not Impact BVDV Infection in MDBK Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Richter, V.; Bakran-Lebl, K.; Baumgartner, W.; Obritzhauser, W.; Käsbohrer, A.; Pinior, B. A systematic worldwide review of the direct monetary losses in cattle due to bovine viral diarrhoea virus infection. Vet. J. 2017, 220, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Olafson, P.; Maccallum, A.D.; Fox, F.H. An apparently new transmissible disease of cattle. Cornell Vet. 1946, 36, 205–213. [Google Scholar] [PubMed]
- Walz, P.H.; Chamorro, M.F.; Falkenberg, M.S.; Passler, T.; van der Meer, F.; Woolums, R.A. Bovine viral diarrhea virus: An updated American College of Veterinary Internal Medicine consensus statement with focus on virus biology, hosts, immunosuppression, and vaccination. J. Vet. Intern. Med. 2020, 34, 1690–1706. [Google Scholar] [CrossRef]
- McClurkin, A.W.; Littledike, E.T.; Cutlip, R.C.; Frank, G.H.; Coria, M.F.; Bolin, S.R. Production of cattle immunotolerant to bovine viral diarrhea virus. Can. J. Comp. Med. Rev. Can. Med. Comp. 1984, 48, 156–161. [Google Scholar]
- Hessman, B.E.; Fulton, R.W.; Sjeklocha, D.B.; Murphy, T.A.; Ridpath, J.F.; Payton, M.E. Evaluation of economic effects and the health and performance of the general cattle population after exposure to cattle persistently infected with bovine viral diarrhea virus in a starter feedlot. Am. J. Vet. Res. 2009, 70, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.A.; Pinior, B.; Larska, M.; Graham, D.; Schweizer, M.; Guidarini, C.; DeCaro, N.; Ridpath, J.; Gates, M.C. Global knowledge gaps in the prevention and control of bovine viral diarrhoea (BVD) virus. Transbound. Emerg. Dis. 2019, 66, 640–652. [Google Scholar] [CrossRef]
- Ridpath, J.F. Immunology of BVDV vaccines. Biologicals 2013, 41, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.; Flick-Smith, H.; McCauley, J.W. Interactions of bovine viral diarrhoea virus glycoprotein Erns with cell surface glycosaminoglycans. Microbiology 2000, 81, 451–459. [Google Scholar] [CrossRef]
- Iqbal, M.; McCauley, J.W. Identification of the glycosaminoglycan-binding site on the glycoprotein Erns of bovine viral diarrhoea virus by site-directed mutagenesis. J. Gen. Virol. 2002, 83, 2153–2159. [Google Scholar] [CrossRef]
- Hulst, M.M.; van Gennip, H.G.P.; Moormann, R.J.M. Passage of Classical Swine Fever Virus in Cultured Swine Kidney Cells Selects Virus Variants That Bind to Heparan Sulfate due to a Single Amino Acid Change in Envelope Protein Erns. J Virol. 2000, 74, 9553–9561. [Google Scholar] [CrossRef] [Green Version]
- Szillat, K.P.; Koethe, S.; Wernike, K.; Höper, D.; Beer, M. A CRISPR/Cas9 Generated Bovine CD46-knockout Cell Line—A Tool to Elucidate the Adaptability of Bovine Viral Diarrhea Viruses (BVDV). Viruses 2020, 12, 859. [Google Scholar] [CrossRef]
- Krey, T.; Himmelreich, A.; Heimann, M.; Menge, C.; Thiel, H.-J.; Maurer, K.; Rümenapf, T. Function of Bovine CD46 as a Cellular Receptor for Bovine Viral Diarrhea Virus Is Determined by Complement Control Protein 1. J. Virol. 2006, 80, 3912–3922. [Google Scholar] [CrossRef] [Green Version]
- Maurer, K.; Krey, T.; Moennig, V.; Thiel, H.-J.; Rümenapf, T. CD46 Is a Cellular Receptor for Bovine Viral Diarrhea Virus. J. Virol. 2004, 78, 1792–1799. [Google Scholar] [CrossRef] [Green Version]
- Lecot, S.; Belouzard, S.; Dubuisson, J.; Rouillé, Y. Bovine viral diarrhea virus entry is dependent on clathrin-mediated endocytosis. J. Virol. 2005, 79, 10826–10829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krey, T.; Thiel, H.J.; Rümenapf, T. Acid-resistant bovine pestivirus requires activation for pH-triggered fusion during entry. J. Virol. 2005, 79, 4191–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, E.F.; Donis, R.O. Isolation of a mutant MDBK cell line resistant to bovine viral diarrhea virus infection due to a block in viral entry. Virology 1995, 208, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, E.F.; Kreutz, L.C.; Donis, R.O. Swine and ruminant pestiviruses require the same cellular factor to enter bovine cells. J. Gen. Virol. 1996, 77, 1295–1303. [Google Scholar] [CrossRef]
- Krey, T.; Moussay, E.; Thiel, H.-J.; Rümenapf, T. Role of the Low-Density Lipoprotein Receptor in Entry of Bovine Viral Diarrhea Virus. J. Virol. 2006, 80, 10862–10867. [Google Scholar] [CrossRef] [Green Version]
- Workman, A.M.; Harhay, G.P.; Smith, T.P.L.; Grotelueschen, D.M.; Sjeklocha, D.; Brodersen, B.; Petersen, J.L.; Chitko-McKown, C.G.; Heaton, M.P. Resolving Bovine viral diarrhea virus subtypes from persistently infected U.S. beef calves with complete genome sequence. J. Vet. Diagn. Invest. 2016, 28, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Heaton, M.P.; Smith, T.P.; Carnahan, J.K.; Basnayake, V.; Qiu, J.; Simpson, B.; Kalbfleisch, T.S. Using diverse U.S. beef cattle genomes to identify missense mutations in EPAS1, a gene associated with pulmonary hypertension. F1000Research 2016, 5, 2003. [Google Scholar] [CrossRef]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. GigaScience 2020, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Bae, S.; Kim, J.-S. Cas-Designer: A web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics 2015, 31, 4014–4016. [Google Scholar] [CrossRef] [Green Version]
- Mahlum, C.E.; Haugerud, S.; Shivers, J.L.; Rossow, K.D.; Goyal, S.M.; Collins, J.; Faaberg, K.S. Detection of Bovine Viral Diarrhea Virus by TaqMan® Reverse Transcription Polymerase Chain Reaction. J. Vet. Diagn. Invest. 2002, 14, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lee, C.; Rhee, I. Important roles of protein tyrosine phosphatase PTPN12 in tumor progression. Pharmacol. Res. 2019, 144, 73–78. [Google Scholar] [CrossRef]
- Yamasaki, M.; Miyazaki, T.; Azechi, H.; Abe, M.; Natsume, R.; Hagiwara, T.; Aiba, A.; Mishina, M.; Sakimura, K.; Watanabe, M. Glutamate Receptor δ2 Is Essential for Input Pathway-Dependent Regulation of Synaptic AMPAR Contents in Cerebellar Purkinje Cells. J. Neurosci. 2011, 31, 3362–3374. [Google Scholar] [CrossRef]
- Qu, F.; Lorenzo, D.N.; King, S.J.; Brooks, R.; Bear, E.J.; Bennett, V. Ankyrin-B is a PI3P effector that promotes polarized α5β1-integrin recycling via recruiting RabGAP1L to early endosomes. eLife 2016, 5, e20417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szatmári, Z.; Sass, M. The autophagic roles of Rab small GTPases and their upstream regulators: A review. Autophagy 2014, 10, 1154–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigau, M.; Juan, D.; Valencia, A.; Rico, D. Intronic CNVs and gene expression variation in human populations. PLoS Genet. 2019, 15, e1007902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Carlson, D.F.; Tan, W.; Hackett, P.B.; Fahrenkrug, S.C. Editing livestock genomes with site-specific nucleases. Reprod. Fertil. Dev. 2013, 26, 74–82. [Google Scholar] [CrossRef]
- Burkard, C.; Lillico, S.G.; Reid, E.; Jackson, B.; Mileham, A.J.; Ait-Ali, T.; Whitelaw, B.; Archibald, A.L. Precision engineering for PRRSV resistance in pigs: Macrophages from genome edited pigs lacking CD163 SRCR5 domain are fully resistant to both PRRSV genotypes while maintaining biological function. PLOS Pathog. 2017, 13, e1006206. [Google Scholar] [CrossRef] [PubMed]
- Wells, K.D.; Bardot, R.; Whitworth, K.M.; Trible, B.R.; Fang, Y.; Mileham, A.; Kerrigan, M.A.; Samuel, M.S.; Prather, R.S.; Rowland, R.R.R. Replacement of Porcine CD163 Scavenger Receptor Cysteine-Rich Domain 5 with a CD163-Like Homolog Confers Resistance of Pigs to Genotype 1 but Not Genotype 2 Porcine Reproductive and Respiratory Syndrome Virus. J. Virol. 2017, 91, e01521-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, D.; Lancto, A.C.; Zang, B.; Kim, E.-S.; Walton, M.; Oldeschulte, D.; Seabury, C.; Sonstegard, T.S.; Fahrenkrug, S.C. Production of hornless dairy cattle from genome-edited cell lines. Nat. Biotechnol. 2016, 34, 479–481. [Google Scholar] [CrossRef]
- Pipek, O.; Ribli, D.; Molnár, J.; Póti, Á.; Krzystanek, M.; Bodor, A.; Tusnády, G.E.; Szallasi, Z.; Csabai, I.; Szüts, D. Fast and accurate mutation detection in whole genome sequences of multiple isogenic samples with IsoMut. BMC Bioinform. 2017, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.B.; Sougnez, C.; Gabriel, S.B.; Meyerson, M.L.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Zhang, Q.; Cao, X. Epigenetic regulation of the innate immune response to infection. Nat. Rev. Immunol. 2019, 19, 417–432. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | gRNA(s) (5′-3′) | ssODN (5′-3′) |
|---|---|---|
| PTPN12 | GAUUUUUGGAGGAUGAUAU | GACCTTTAGCAAATACGGTAATAGATTTTTGGAGGATGATATAAAGCTTTGGGAGTACAATGTTGTAGTAAGTATTGTATGAAATGGCAT |
| GRID2 | GGAUCCAUUUGCUCAGAAUA | TATCTTCAACATTGTGTGATCCAAAGGATCCATTTGCTCAGTAAAGCTTAATATGGAGGTATATTCTAAGCACCCAGATATTTCTCTAAG |
| RABGAP1L | Targeting 5′: GGGAUUAUGCAGAAGAGGUTargeting 3′: GGAAUGUCACGUAGGCCUA | CACAAGAAATTATTAAGAGTAATTTTGGGATTATGCAGAAGAAAGCTTCTATGGCCTATCCTTCCATGCAACCAATAATTCTCTTTCCCT |
| Gene | Cellular Localization and Expression | Gene Function |
|---|---|---|
| PTPN12 | Cytosol; ubiquitously expressed | Member of the protein tyrosine phosphatase (PTP) family; plays role in cytoskeletal structure, cell adhesion, cell shape and mobility, and cell-cell junctions [32] |
| GRID2 | Plasma membrane; predominantly expressed in the cerebellum | Ionotropic glutamate receptor in the mammalian brain; plays a role in synaptogenesis, synaptic plasticity, and endocytosis of the AMPA receptor [33]. |
| RABGAP1L | Cytoplasm, endosome; ubiquitously expressed | Rab GTPase activating protein 1 like; plays a role in endocytosis, endosome maturation, and autophagosome formation [34,35]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Workman, A.M.; Heaton, M.P.; Webster, D.A.; Harhay, G.P.; Kalbfleisch, T.S.; Smith, T.P.L.; Falkenberg, S.M.; Carlson, D.F.; Sonstegard, T.S. Evaluating Large Spontaneous Deletions in a Bovine Cell Line Selected for Bovine Viral Diarrhea Virus Resistance. Viruses 2021, 13, 2147. https://doi.org/10.3390/v13112147
Workman AM, Heaton MP, Webster DA, Harhay GP, Kalbfleisch TS, Smith TPL, Falkenberg SM, Carlson DF, Sonstegard TS. Evaluating Large Spontaneous Deletions in a Bovine Cell Line Selected for Bovine Viral Diarrhea Virus Resistance. Viruses. 2021; 13(11):2147. https://doi.org/10.3390/v13112147
Chicago/Turabian StyleWorkman, Aspen M., Michael P. Heaton, Dennis A. Webster, Gregory P. Harhay, Theodore S. Kalbfleisch, Timothy P. L. Smith, Shollie M. Falkenberg, Daniel F. Carlson, and Tad S. Sonstegard. 2021. "Evaluating Large Spontaneous Deletions in a Bovine Cell Line Selected for Bovine Viral Diarrhea Virus Resistance" Viruses 13, no. 11: 2147. https://doi.org/10.3390/v13112147
APA StyleWorkman, A. M., Heaton, M. P., Webster, D. A., Harhay, G. P., Kalbfleisch, T. S., Smith, T. P. L., Falkenberg, S. M., Carlson, D. F., & Sonstegard, T. S. (2021). Evaluating Large Spontaneous Deletions in a Bovine Cell Line Selected for Bovine Viral Diarrhea Virus Resistance. Viruses, 13(11), 2147. https://doi.org/10.3390/v13112147