
Cotton Leafroll Dwarf Virus US Genomes Comprise Divergent Subpopulations and Harbor Extensive Variability

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. The Plant Samples
2.2. Total RNA Isolation and RT-PCR Diagnostics
2.3. Determination of CLRDV Genomes
2.4. Sequencing of CLRDV 5′- and 3′-Untranslated Ends
2.5. Sequence Comparisons
2.6. Phylogenetic Analysis
2.7. Recombination Analysis
2.8. Genetic Variability and Selection
3. Results
3.1. CLRDV Complete Genome Sequences
3.2. Pairwise Sequence Comparisons
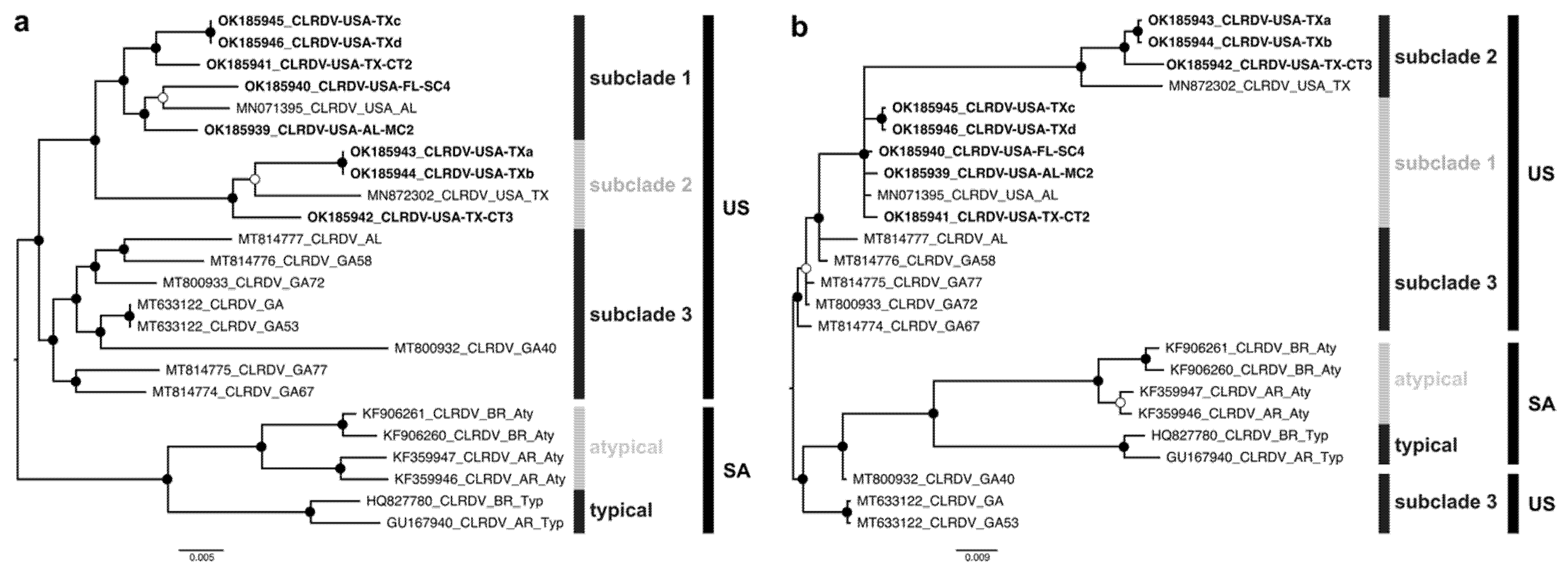
3.3. Phylogenetic Relationship
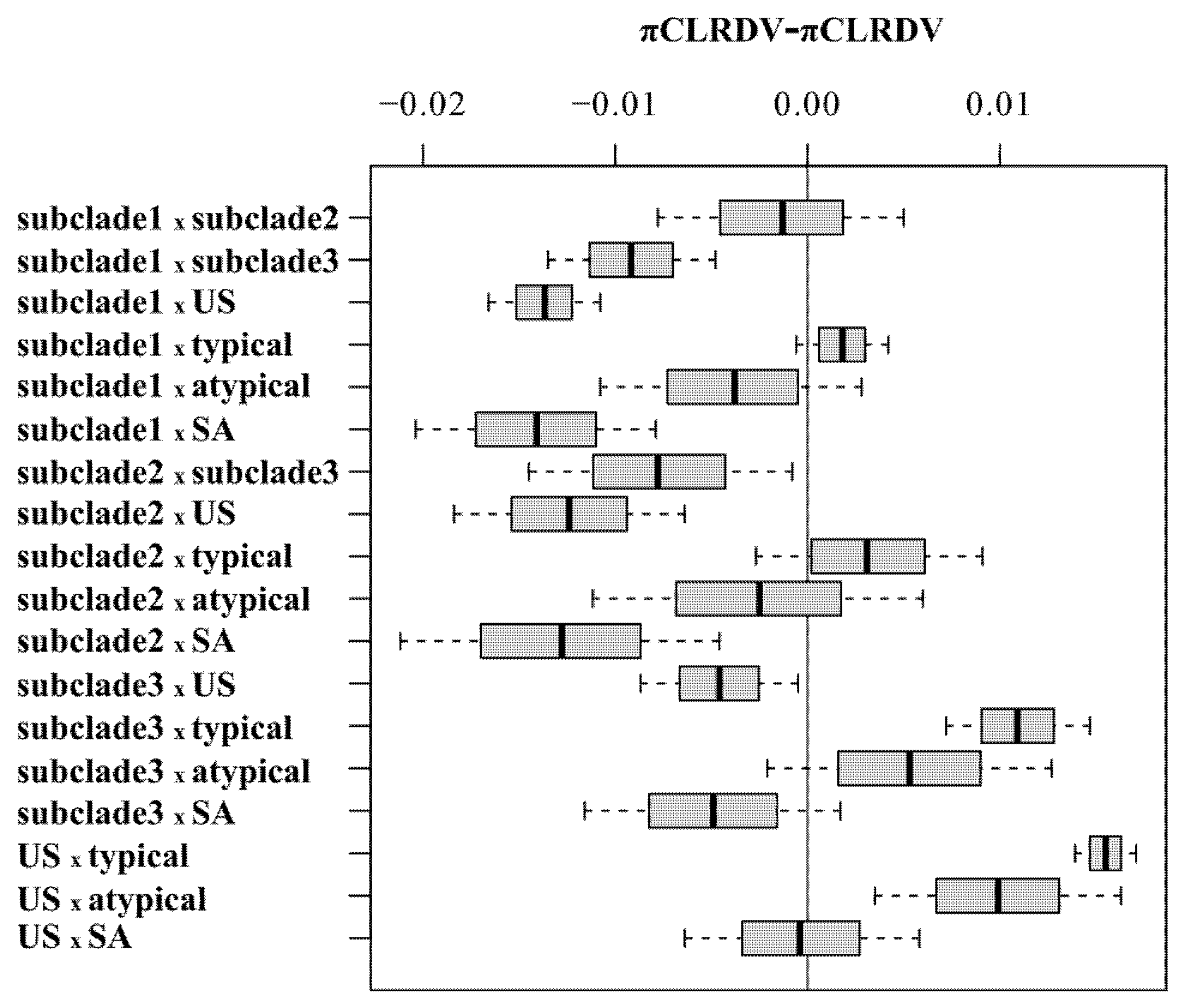
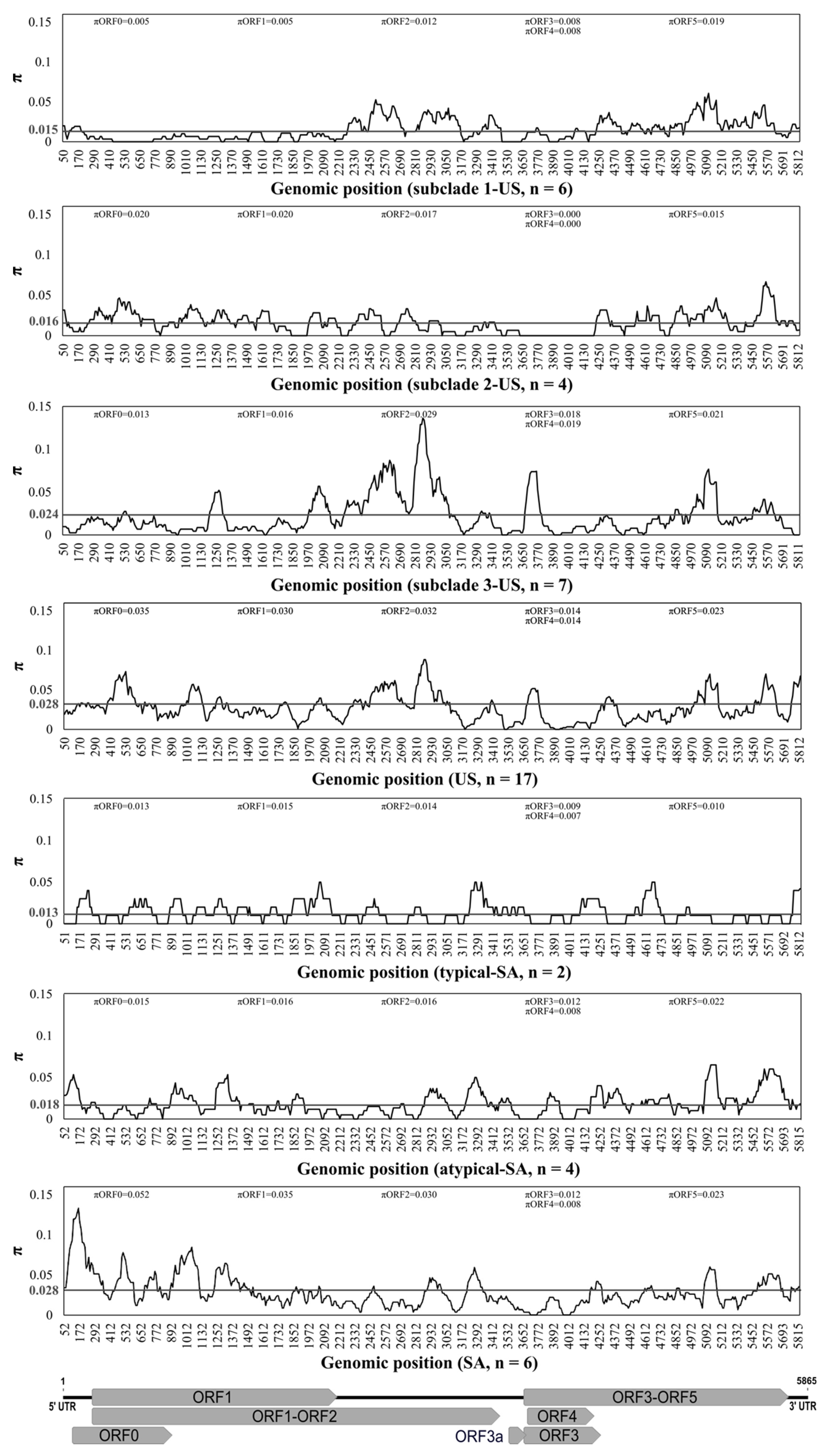
3.4. Population Genetics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmad, S.; Iqbal, M.; Muhammad, T.; Mehmood, A.; Ahmad, S.; Hasanuzzaman, M. Cotton productivity enhanced through transplanting and early sowing. Acta Sci. Biol. Sci. 2018, 40, 34610. [Google Scholar] [CrossRef]
- Ahmad, S.; Raza, I.; Ali, H.; Shahzad, A.N.; Rehman, A.U.; Sarwar, N. Response of cotton crop to exogenous application of glycinebetaine under sufficient and scarce water conditions. Braz. J. Bot. 2014, 37, 407–415. [Google Scholar] [CrossRef]
- Sunilkumar, G.; Campbell, L.M.; Puckhaber, L.; Stipanovic, R.D.; Rathore, K.S. Engineering cottonseed for use in human nutrition by tissue-specific reduction of toxic gossypol. Proc. Natl. Acad. Sci. USA 2006, 103, 18054–18059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FAO. FAOSTAT. 2019. Available online: http://www.fao.org/faostat/en/#home (accessed on 26 December 2019).
- USDA. Crop Values 2020 Summary. Available online: https://quickstats.nass.usda.gov/results/B7E6307C-4344-387D-B041-5BA186B72D77 (accessed on 22 July 2021).
- Chohan, S.; Perveen, R.; Abid, M.; Tahir, M.N.; Sajid, M. Cotton Diseases and Their Management. In Cotton Production and Uses; Springer: Dordrecht, The Netherlands, 2020; pp. 239–270. [Google Scholar]
- Cauquil, J. Etudes sur une maladie d’origine virale du cotonnier, la maladie bleue. Cot. Fib. Trop. 1977, 32, 259–278. [Google Scholar]
- Cauquil, J.; Vaissayre, M. La “Maladie Bleue” du Cotonnier en Afrique: Transmission de Cotonnier à Cotonnier par Aphis Gossypii Glover. Available online: http://agritrop.cirad.fr/455715/1/ID455715.pdf (accessed on 28 September 2021).
- Costa, A.S.; Carvalho, A.M.B. Moléstias de vírus do algodoeiro. Bragantia 1962, 21, 50–51. [Google Scholar]
- Costa, A.S.; Forster, R. Nota preliminar sobre uma nova moléstia de vírus do algodoeiro: Mosaico das nervuras. Revista de Agricultura 1938, 13, 187–191. [Google Scholar]
- Campagnac, N.A.; Bonacic Kresic, R.; Poisson, J. Mal de Misiones: Nueva enfermedad del algodón de probable origen virósico. Jorn. Fitosanit. Argent. 1986, 6, 503–511. [Google Scholar]
- Corrêa, R.L.; Silva, T.F.; Simões-Araújo, J.L.; Barroso, P.A.V.; Vidal, M.S.; Vaslin, M.F.S. Molecular characterization of a virus from the family Luteoviridae associated with cotton blue disease. Arch. Virol. 2005, 150, 1357–1367. [Google Scholar] [CrossRef]
- Ray, J.D.; Sharman, M.; Quintao, V.; Rossel, B.; Westaway, J.; Gambley, C. Cotton leafroll dwarf virus detected in Timor-Leste. Australas. Plant Dis. Notes 2016, 11, 29. [Google Scholar] [CrossRef] [Green Version]
- Sharman, M.; Lapbanjob, S.; Sebunruang, P.; Belot, J.L.; Galbieri, R.; Giband, M.; Suassuna, N. First report of Cotton leafroll dwarf virus in Thailand using a species-specific PCR validated with isolates from Brazil. Australas. Plant Dis. Notes 2015, 10, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.K.; Chahande, P.R.; Meshram, M.K.; Kranthi, K.R. First report of Polerovirus of the family Luteovir-idae infecting cotton in India. New Dis. Rep. 2012, 25, 22. [Google Scholar] [CrossRef] [Green Version]
- Quyen, L.Q.; Hai, N.T.; Mai, T.A.H.; Hao, V.; Binh, N.T.T.; Bu’u, D.N.; Dieu, D.X.; Underwood, E. Cotton production in Vietnam. In Environmental Risk Assessment of Genetically Modified Organisms: Challenges and Opportunities with bt Cotton in Vietnam; Andow, D.A., Hilbeck, A., Eds.; CABI Publishing: Wallingford, UK, 2008; Volume 4, pp. 24–63. [Google Scholar]
- Distéfano, A.J.; Kresic, I.B.; Hopp, H.E. The complete genome sequence of a virus associated with cotton blue disease, cotton leafroll dwarf virus, confirms that it is a new member of the genus Polerovirus. Arch. Virol. 2010, 155, 1849–1854. [Google Scholar] [CrossRef]
- Silva, T.F.; Corrêa, R.L.; Castilho, Y.; Silvie, P.; Bélot, J.-L.; Vaslin, M.F.S. Widespread distribution and a new re-combinant species of Brazilian virus associated with cotton blue disease. Virol. J. 2008, 5, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avelar, S.; Schrimsher, D.W.; Lawrence, K.; Brown, J.K. First Report of Cotton leafroll dwarf virus Associated with Cotton Blue Disease Symptoms in Alabama. Plant Dis. 2019, 103, 592. [Google Scholar] [CrossRef]
- Alabi, O.J.; Isakeit, T.; Vaughn, R.; Stelly, D.; Conner, K.N.; Gaytán, B.C.; Villegas, C.; Hitzelberger, C.; De Santiago, L.; Monclova-Santana, C.; et al. First Report of Cotton leafroll dwarf virus Infecting Upland Cotton (Gossypium hirsutum) in Texas. Plant Dis. 2020, 104, 998. [Google Scholar] [CrossRef]
- Ali, A.; Mokhtari, S. Complete Genome Sequence of Cotton Leafroll Dwarf Virus Isolated from Cotton in Texas, USA. Microbiol. Resour. Announc. 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Faske, T.R.; Stainton, D.; Ghanem-Sabanadzovic, N.A.; Allen, T.W. First Report of Cotton Leafroll Dwarf Virus from Upland Cotton (Gossypium hirsutum) in Arkansas. Plant Dis. 2020, 104. [Google Scholar] [CrossRef]
- Iriarte, F.B.; Dey, K.K.; Small, I.M.; Conner, K.; O’Brien, G.K.; Johnson, L.; Savery, C.; Carter, E.; Sprague, D.; Nichols, R.L.; et al. First Report of Cotton Leafroll Dwarf Virus in Florida. Plant Dis. 2020, 104. [Google Scholar] [CrossRef] [Green Version]
- Price, T.; Valverde, R.; Singh, R.; Davis, J.; Brown, S.; Jones, H. First Report of Cotton Leafroll Dwarf Virus in Louisiana. Plant Heal. Prog. 2020, 21, 142–143. [Google Scholar] [CrossRef]
- Tabassum, A.; Roberts, P.M.; Bag, S. Genome Sequence of Cotton Leafroll Dwarf Virus Infecting Cotton in Georgia, USA. Microbiol. Resour. Announc. 2020, 9, 00812-20. [Google Scholar] [CrossRef]
- Thiessen, L.D.; Schappe, T.L.; Zaccaron, M.; Conner, K.; Koebernick, J.; Jacobson, A.; Huseth, A. First Report of Cotton Leafroll Dwarf Virus in Cotton Plants Affected by Cotton Leafroll Dwarf Disease in North Carolina. Plant Dis. 2020, 104, 3275. [Google Scholar] [CrossRef]
- Wang, H.; Greene, J.; Mueller, J.D.; Conner, K.; Jacobson, A. First Report of Cotton Leafroll Dwarf Virus in Cotton Fields of South Carolina. Plant Dis. 2020, 104, 2532. [Google Scholar] [CrossRef] [Green Version]
- Aboughanem-Sabanadzovic, N.; Allen, T.W.; Wilkerson, T.H.; Conner, K.N.; Sikora, E.J.; Nichols, R.L.; Sabanadzovic, S. First Report of Cotton Leafroll Dwarf Virus in Upland Cotton (Gossypium hirsutum) in Mississippi. Plant Dis. 2019, 103, 1798. [Google Scholar] [CrossRef]
- Huseth, A.; Reisig, D.; Collins, G.; Thiessen, L. Detection of Cotton Leafroll Dwarf Virus (CLRDV) in North Carolina. NC State Extension. 2019. Available online: http://go.ncsu.edu/readext?639597 (accessed on 11 July 2021).
- Tabassum, A.; Bag, S.; Roberts, P.; Suassuna, N.; Chee, P.; Whitaker, J.R.; Conner, K.N.; Brown, J.; Nichols, R.L.; Kemerait, R.C. First Report of Cotton Leafroll Dwarf Virus Infecting Cotton in Georgia, U.S.A. Plant Dis. 2019, 103, 1803. [Google Scholar] [CrossRef]
- Mahas, J.W. Management of Aphis gossypii Populations and the Spread of Cotton leafroll dwarf virus in Southeastern Cotton Production Systems. Master’s Thesis, Department of Entomology and Plant pathology, Auburn University, Auburn, AL, USA, 2020. [Google Scholar]
- Avelar, S.; Ramos-Sobrinho, R.; Conner, K.; Nichols, R.L.; Lawrence, K.; Brown, J.K. Characterization of the Complete Genome and P0 Protein for a Previously Unreported Genotype of Cotton Leafroll Dwarf Virus, an Introduced Polerovirus in the United States. Plant Dis. 2020, 104, 780–786. [Google Scholar] [CrossRef]
- Bag, S.; Roberts, P.M.; Kemerait, R.C. Cotton Leafroll Dwarf Disease: An Emerging Virus Disease on Cotton in the U.S. Crop. Soils 2021, 54, 18–22. [Google Scholar] [CrossRef]
- Sedhain, N.P.; Bag, S.; Morgan, K.; Carter, R.; Triana, P.; Whitaker, J.; Kemerait, R.C.; Roberts, P.M. Natural host range, incidence on overwintering cotton and diversity of cotton leafroll dwarf virus in Georgia USA. Crop. Prot. 2021, 144, 105604. [Google Scholar] [CrossRef]
- ICTV.Taxonomy History: Cotton Leafroll Dwarf Virus. Available online: https://talk.ictvonline.org/taxonomy/p/taxonomy-history?taxnode_id=202003781 (accessed on 23 July 2021).
- Domier, L.L.; King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Family Luteoviridae. In Virus Taxonomy; Ninth report of the International Committee on Taxonomy of Viruses; Elsevier/Academic Press: London, UK, 2012; pp. 1045–1053. [Google Scholar] [CrossRef]
- Gray, S.; Gildow, F.E. Luteovirus-aphid interactions. Annu. Rev. Phytopathol. 2003, 41, 539–566. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chay, C.; Gildow, F.; Gray, S. Readthrough Protein Associated with Virions of Barley Yellow Dwarf Luteovirus and Its Potential Role in Regulating the Efficiency of Aphid Transmission. Virology 1995, 206, 954–962. [Google Scholar] [CrossRef] [Green Version]
- Takimoto, J.K.; Benetti Queiroz-Voltan, R.; Caram de Souza-Dias, J.A.; Cia, E. AlteraÇões anatômicas em algodoeiro infectado pelo vírus da doenÇa azul. Bragantia 2009, 68, 109–116. [Google Scholar]
- Cascardo, R.S.; Arantes, I.L.G.; Silva, T.F.; Sachetto-Martins, G.; Vaslin, M.F.S.; Corrêa, R.L. Function and diversity of P0 proteins among cotton leafroll dwarf virus isolates. Virol. J. 2015, 12, 1–10. [Google Scholar] [CrossRef]
- Miller, W.; Brown, C.; Wang, S. New Punctuation for the Genetic Code: Luteovirus Gene Expression. Semin. Virol. 1997, 8, 3–13. [Google Scholar] [CrossRef]
- Mayo, M.; Ziegler-Graff, V. Molecular Biology of Luteoviruses. Adv. Virus Res. 1996, 46, 413–460. [Google Scholar] [CrossRef] [PubMed]
- Agrofoglio, Y.C.; Delfosse, V.C.; Casse, M.F.; Hopp, H.E.; Kresic, I.B.; Ziegler-Graff, V.; Distéfano, A.J. P0 protein of cotton leafroll dwarf virus-atypical isolate is a weak RNA silencing suppressor and the avirulence determinant that breaks the cotton Cbd gene-based resistance. Plant Pathol. 2019, 68, 1059–1071. [Google Scholar] [CrossRef]
- Delfosse, V.C.; Agrofoglio, Y.C.; Casse, M.F.; Kresic, I.B.; Hopp, H.E.; Ziegler-Graff, V.; Distéfano, A.J. The P0 protein encoded by cotton leafroll dwarf virus (CLRDV) inhibits local but not systemic RNA silencing. Virus Res. 2014, 180, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Dunoyer, P.; Heim, F.; Richards, K.E.; Jonard, G.; Ziegler-Graff, V. P0 of beet Western yellows virus is a sup-pressor of posttranscriptional gene silencing. J. Virol. 2002, 76, 6815–6824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, E.; Firth, A.; Miller, W.A.; Scheidecker, D.; Brault, V.; Reinbold, C.; Rakotondrafara, A.M.; Chung, B.Y.-W.; Ziegler-Graff, V. Discovery of a Small Non-AUG-Initiated ORF in Poleroviruses and Luteoviruses That Is Required for Long-Distance Movement. PLoS Pathog. 2015, 11, e1004868. [Google Scholar] [CrossRef] [Green Version]
- Chay, C.; Gunasinge, U.; Kumarb, S.-; Miller, W.; Gray, S.M. Aphid Transmission and Systemic Plant Infection Determinants of Barley Yellow Dwarf Luteovirus-PAV are Contained in the Coat Protein Readthrough Domain and 17-kDa Protein, Respectively. Virology 1996, 219, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brault, V.; Heuvel, J.V.D.; Verbeek, M.; Ziegler-Graff, V.; Reutenauer, A.; Herrbach, E.; Garaud, J.; Guilley, H.; Richards, K.; Jonard, G. Aphid transmission of beet western yellows luteovirus requires the minor capsid read-through protein P74. EMBO J. 1995, 14, 650–659. [Google Scholar] [CrossRef]
- Miller, W.A.; Beckett, R.; Liu, S. Structure, Function, and Variation of the Barley Yellow Dwarf Virus and Cereal Yellow Dwarf Virus Genomes. In Barley Yellow Dwarf Disease: Recent Advances and Future Strategies, Proceedings of the an International Symposium held at El Batán, Texcoco, Mexico, 1–5 September 2002; Henry, M., McNab, A., Eds.; CIMMYT: Texcoco, Mexico, 2002. [Google Scholar]
- van der Wilk, F.; Verbeek, M.; Dullemans, A.; Heuvel, J.V.D. The Genome-Linked Protein of Potato Leafroll Virus Is Located Downstream of the Putative Protease Domain of the ORF1 Product. Virology 1997, 234, 300–303. [Google Scholar] [CrossRef] [Green Version]
- Pagán, I. The diversity, evolution and epidemiology of plant viruses: A phylogenetic view. Infect. Genet. Evol. 2018, 65, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C. The Evolution and Emergence of RNA Viruses; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon-Loriere, E.; Holmes, E. Why do RNA viruses recombine? Nat. Rev. Genet. 2011, 9, 617–626. [Google Scholar] [CrossRef]
- Hall, G. Selective constraint and genetic differentiation in geo- graphically distant barley yellow dwarf virus popula-tions. J. Gen. Virol. 2006, 87, 3067–3075. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Weiller, G.F. Evidence that a plant virus switched hosts to infect a vertebrate and then recombined with a vertebrate-infecting virus. Proc. Natl. Acad. Sci. USA 1999, 96, 8022–8027. [Google Scholar] [CrossRef] [Green Version]
- Tabassum, A.; Bag, S.; Suassuna, N.D.; Conner, K.N.; Chee, P.; Kemerait, R.C.; Roberts, P. Genome analysis of cotton leafroll dwarf virus reveals variability in the silencing suppressor protein, genotypes and genomic recombinants in the USA. PLoS ONE 2021, 16, e0252523. [Google Scholar] [CrossRef] [PubMed]
- Agrofoglio, Y.C.; Delfosse, C.; Casse, F.; Hopp, H.E.; Kresic, B.; Distéfano, A.J. Identification of a new cotton disease caused by an atypical cotton leafroll dwarf virus in Argentina. Phytopathology 2017, 107, 1–8. [Google Scholar]
- Da Silva, A.K.F.; Romanel, E.; da F Silva, T.; Castilhos, Y.; Schrago, C.; Galbieri, R.; Bélot, J.-L.; Vaslin, M.F.S. Complete genome sequences of two new virus isolates associated with cotton blue disease resistance breaking in Brazil. Arch. Virol. 2015, 160, 1371–1374. [Google Scholar] [CrossRef]
- Rott, M.; Jelkmann, W. Characterization and Detection of Several Filamentous Viruses of Cherry: Adaptation of an Alternative Cloning Method (DOP-PCR), and Modification of an RNA Extraction Protocol. Eur. J. Plant Pathol. 2001, 107, 411–420. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- NBSP; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; IEEE: New Orleans, LA, USA, 2010; pp. 1–8. [Google Scholar] [CrossRef] [Green Version]
- Rannala, B.; Yang, Z. Probability distribution of molecular evolutionary trees: A new method of phylogenetic in-ference. J Mol Evol 1996, 43, 304–311. [Google Scholar]
- Wright, S. The Genetical Structure of Populations. Ann. Eugen. 1949, 15, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2005, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, A.T.M.; Silva, J.C.F.; Silva, F.N.; Castillo-Urquiza, G.P.; Silva, F.F.; Seah, Y.M.; Mizubuti, E.S.G.; Duffy, S.; Zerbini, F.M. The diversification of begomovirus populations is predominantly driven by mutational dynamics. Virus Evol. 2017, 3, vex005. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Frost, S.D.W. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Nielsen, R. Estimating Synonymous and Nonsynonymous Substitution Rates Under Realistic Evolution-ary Models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [PubMed] [Green Version]
- Holsinger, K.E.; Weir, B.S. Genetics in geographically structured populations: Defining, estimating and interpreting FST. Nat. Rev. Genet. 2009, 10, 639–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knierim, D.; Deng, T.C.; Tsai, W.S.; Green, S.K.; Kenyon, L.C. Molecular identification of three distinct Polerovirus species and a recombinant Cucurbit aphid-borne yellows virus strain infecting cucurbit crops in Taiwan. Plant Pathol. 2010, 59, 991–1002. [Google Scholar] [CrossRef]
- Shang, Q.-X.; Xiang, H.-Y.; Han, C.-G.; Li, D.; Yu, J.-L. Distribution and molecular diversity of three cucurbit-infecting poleroviruses in China. Virus Res. 2009, 145, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, O.; Beuve, M.; Hauser, S.; Stevens, M.; Herrbach, E. Diversity and phylogeny of beet poleroviruses. In Proceedings of the 8. International Plant Virus Epidemiology Symposium, Ascherleben, Germany, 12–17 May 2002. [Google Scholar]
- Moonan, F.; Molina, J.; Mirkov, T.E. Sugarcane yellow leaf virus: An emerging virus that has evolved by recombination between luteoviral and poleroviral ancestors. Virology 2000, 269, 156–171. [Google Scholar] [PubMed]
- Guilley, H.; Richards, K.E.; Jonard, G. Nucleotide sequence of beet mild yellowing virus RNA. Arch. Virol. 1995, 140, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- García-Arenal, F.; Zerbini, F.M. Life on the Edge: Geminiviruses at the Interface Between Crops and Wild Plant Hosts. Annu. Rev. Virol. 2019, 6, 411–433. [Google Scholar] [CrossRef] [Green Version]
- Fraile, A.; García-Arenal, F.; Malpica, J.M. Variation and evolution of plant virus populations. Int. Microbiol. 2003, 6, 225–232. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Location | Symptoms | PCR-ORF0 | # of Reads | Coverage | Reference |
|---|---|---|---|---|---|---|
| CLRDV-USA-AL-MC2 1 | Alabama | Severe | Positive | 33,141 | 850 | This study |
| CLRDV-USA-AL-SC4 1 | Alabama | Mild | Positive | 16,964 | 435 | This study |
| CLRDV-USA-TX-CT2 | Texas | Severe | Positive | 13,043 | 336 | This study |
| CLRDV-USA-TX-CT3 1 | Texas | Severe | Positive | 147,639 | 3,780 | This study |
| CLRDV-USA-TXa | Texas | Severe | Positive | Sanger-derived | This study | |
| CLRDV-USA-TXb | Texas | Severe | Positive | Sanger-derived | This study | |
| CLRDV-USA-TXc | Texas | Severe | Positive | Sanger-derived | This study | |
| CLRDV-USA-TXd | Texas | Severe | Positive | Sanger-derived | This study | |
| CLRDV-USA-AL 1 | Alabama | Severe | Positive | - | - | Avelar et al., 2020 |
| Genomes—Nucleotide/Amino Acid | ORF0—Nucleotide/Amino Acid | ||||
|---|---|---|---|---|---|
| CLRDV-SA | CLRDV-US | CLRDV-SA | CLRDV-US | ||
| CLRDV-SA | 95.9–99.5/- 1 | CLRDV-SA | 91.1–99.5/87.0–98.1 | ||
| CLRDV-US | 94.0–96.2/- | 94.5–100.0/- | CLRDV-US | 89.4–94.3/81.6–90.4 | 92.1–100.0/90.8–100.0 |
| ORF1—Nucleotide/Amino Acid | ORF1-ORF2—Nucleotide/Amino Acid | ||||
| CLRDV-SA | CLRDV-US | CLRDV-SA | CLRDV-US | ||
| CLRDV-SA | 94.3–99.5/93.8–99.5 | CLRDV-SA | 95.5–99.4/93.3–99.1 | ||
| CLRDV-US | 92.4–95.5/90.3–95.0 | 93.8–100.0/91.6–100.0 | CLRDV-US | 92.9–96.2/87.8–94.7 | 92.5–100.0/87.5–100.0 |
| ORF3—Nucleotide/Amino Acid | ORF3a—nucleotide/amino acid | ||||
| CLRDV-SA | CLRDV-US | CLRDV-SA | CLRDV-US | ||
| CLRDV-SA | 97.9–100.0/98.5–100.0 | CLRDV-SA | 97.8–100.0/100.0 | ||
| CLRDV-US | 95.0–98.3/95.5–100.0 | 96.2–100.0/95.5–100.0 | CLRDV-US | 97.1–100.0/100.0 | 97.8–100.0/100.0 |
| ORF4—Nucleotide/Amino Acid | ORF5—Nucleotide/Amino Acid | ||||
| CLRDV-SA | CLRDV-US | CLRDV-SA | CLRDV-US | ||
| CLRDV-SA | 98.1–100.0/95.4–100.0 | CLRDV-SA | 96.7–99.7/96.8–100.0 | ||
| CLRDV-US | 94.9–98.9/89.7–97.1 | 96.0–100.0/92.5–100.0 | CLRDV-US | 94.8–96.8/94.1–98.3 | 96.5–100.0/95.7–100.0 |
| Subpopulations | Nst 1 | Fst 1 | |
|---|---|---|---|
| subclade 1-US | subclade 2-US | 0.52 | 0.51 |
| subclade 1-US | Subclade 3-US | 0.31 | 0.31 |
| subclade 1-US | US | 0.07 | 0.07 |
| subclade 1-US | typical-SA | 0.72 | 0.72 |
| subclade 1-US | atypical-SA | 0.67 | 0.67 |
| subclade 1-US | SA | 0.56 | 0.56 |
| subclade 2-US | subclade 3-US | 0.51 | 0.50 |
| subclade 2-US | US | 0.27 | 0.27 |
| subclade 2-US | typical-SA | 0.71 | 0.71 |
| subclade 2-US | atypical-SA | 0.66 | 0.66 |
| subclade 2-US | SA | 0.56 | 0.55 |
| subclade 3-US | US | 0.06 | 0.06 |
| subclade 3-US | typical-SA | 0.62 | 0.62 |
| subclade 3-US | atypical-SA | 0.56 | 0.55 |
| subclade 3-US | SA | 0.45 | 0.45 |
| US | typical-SA | 0.58 | 0.57 |
| US | atypical-SA | 0.53 | 0.52 |
| US | SA | 0.42 | 0.41 |
| typical-SA | atypical-SA | 0.60 | 0.60 |
| typical-SA | SA | 0.26 | 0.26 |
| atypical-SA | SA | − 0.07 | − 0.07 |
| Dataset | Gene | SLAC (dN/dS) | FEL (# of Sites) | |
|---|---|---|---|---|
| Negative | Positive | |||
| CLRDV-subclade1-US | ORF0 | 0.52 | 2 | 0 |
| ORF1 | 0.36 | 3 | 0 | |
| ORF2 | 0.15 | 33 | 0 | |
| ORF3 | 0.16 | 3 | 0 | |
| ORF4 | 1.19 | 1 | 0 | |
| ORF5 | 0.19 | 30 | 0 | |
| CLRDV-subclade2-US | ORF0 | 0.36 | 8 | 0 |
| ORF1 | 0.28 | 27 | 0 | |
| ORF2 | 0.19 | 54 | 1 | |
| ORF3 | − 1 | − | − | |
| ORF4 | − | − | − | |
| ORF5 | 0.24 | 20 | 0 | |
| CLRDV-subclade3-US | ORF0 | 0.67 | 3 | 0 |
| ORF1 | 0.55 | 17 | 0 | |
| ORF2 | 0.47 | 54 | 3 | |
| ORF3 | 0.50 | 6 | 0 | |
| ORF4 | 0.98 | 1 | 1 | |
| ORF5 | 0.38 | 29 | 0 | |
| CLRDV-atypical-SA 2 | ORF0 | 1.63 | 1 | 0 |
| ORF1 | 0.33 | 13 | 0 | |
| ORF2 | 0.25 | 32 | 0 | |
| ORF3 | 0.16 | 5 | 1 | |
| ORF4 | 1.26 | 1 | 0 | |
| ORF5 | 0.15 | 16 | 1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos-Sobrinho, R.; Adegbola, R.O.; Lawrence, K.; Schrimsher, D.W.; Isakeit, T.; Alabi, O.J.; Brown, J.K. Cotton Leafroll Dwarf Virus US Genomes Comprise Divergent Subpopulations and Harbor Extensive Variability. Viruses 2021, 13, 2230. https://doi.org/10.3390/v13112230
Ramos-Sobrinho R, Adegbola RO, Lawrence K, Schrimsher DW, Isakeit T, Alabi OJ, Brown JK. Cotton Leafroll Dwarf Virus US Genomes Comprise Divergent Subpopulations and Harbor Extensive Variability. Viruses. 2021; 13(11):2230. https://doi.org/10.3390/v13112230
Chicago/Turabian StyleRamos-Sobrinho, Roberto, Raphael O. Adegbola, Kathy Lawrence, Drew W. Schrimsher, Thomas Isakeit, Olufemi J. Alabi, and Judith K. Brown. 2021. "Cotton Leafroll Dwarf Virus US Genomes Comprise Divergent Subpopulations and Harbor Extensive Variability" Viruses 13, no. 11: 2230. https://doi.org/10.3390/v13112230