
Herpesvirus Nuclear Egress across the Outer Nuclear Membrane

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
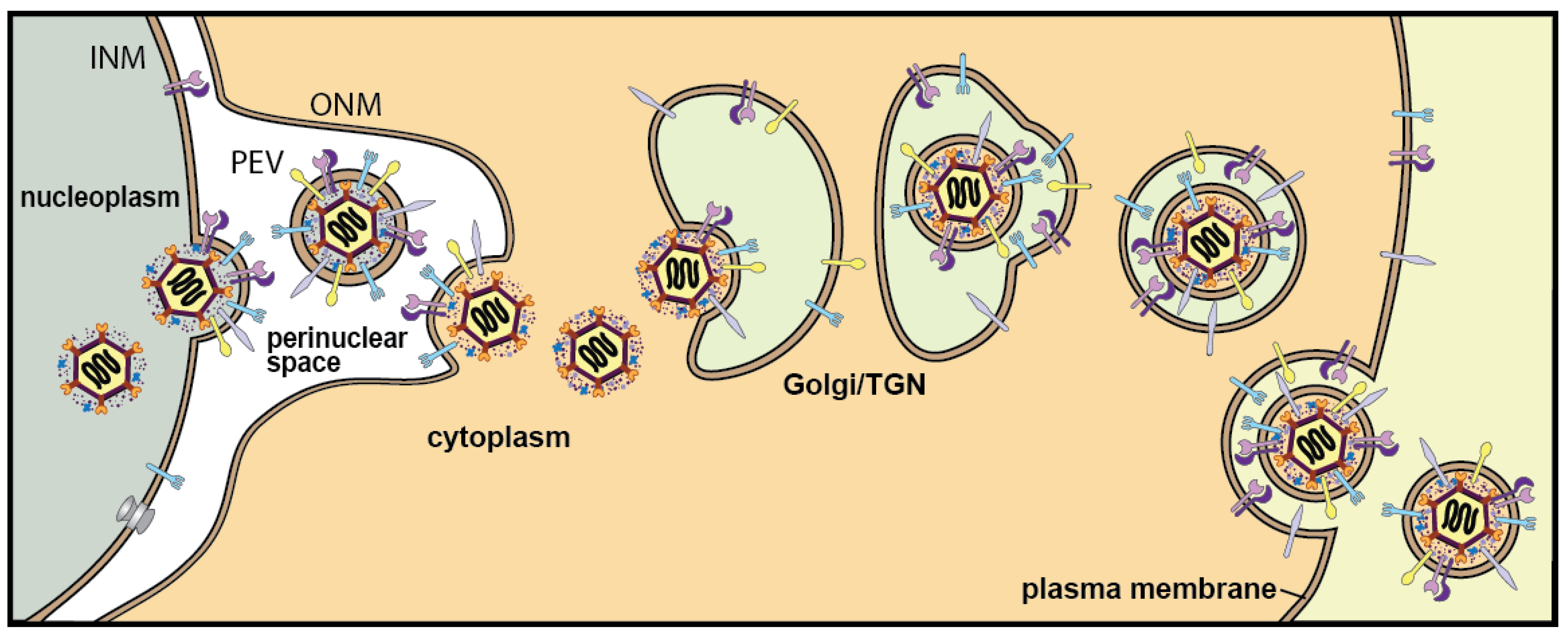
:1. Introduction
2. HSV Membrane Proteins That Promote De-Envelopment
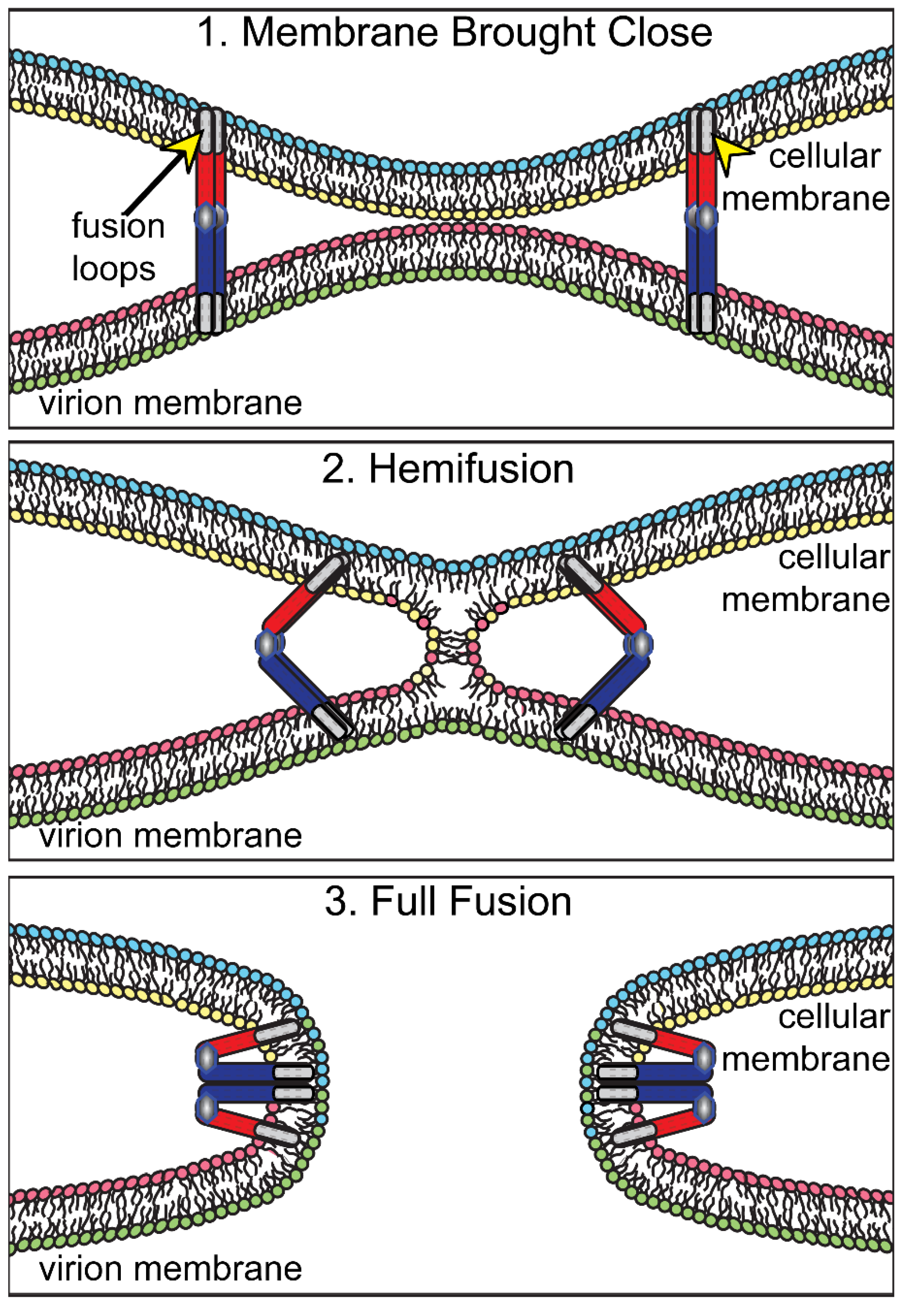
2.1. HSV Glycoproteins gB and gH/gL in Fusion of PEVs with the ONM
2.2. Caveats and Puzzles Associated with Observations That gB and gH/gL Promote De-Envelopment
2.3. gK and UL20, Other Membrane Proteins That May Regulate De-Envelopment Fusion
2.4. Summary of the Roles of HSV Membrane Proteins gB, gH/gL, gK and UL20 in Nuclear Egress
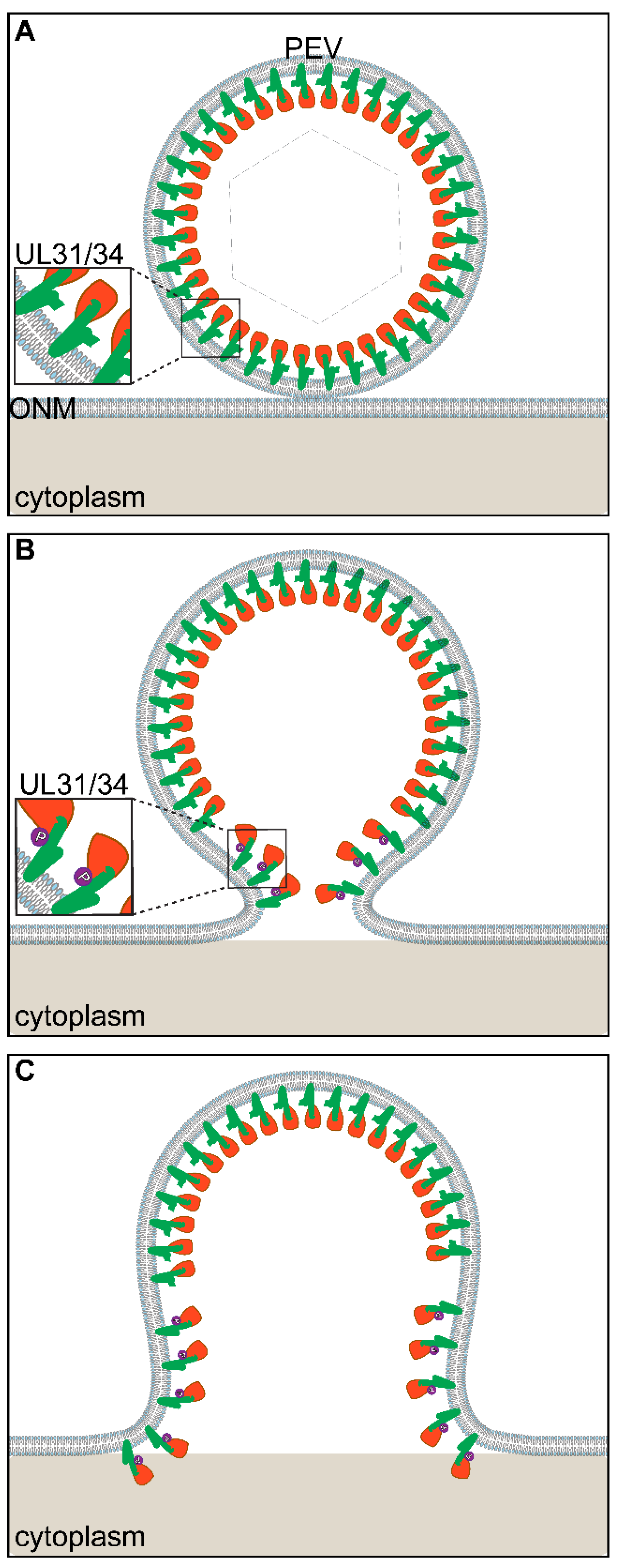
3. US3-Mediated Phosphorylation of NEC Proteins in De-Envelopment
The Role of US3 in De-Envelopment
4. Models for HSV De-Envelopment
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Campadelli-Fiume, G.; Farabegoli, F.; Di Gaeta, S.; Roizman, B. Origin of unenveloped capsids in the cytoplasm of cells infected with herpes simplex virus 1. J. Virol. 1991, 65, 1589–1595. [Google Scholar] [CrossRef] [Green Version]
- Wild, P.; Engels, M.; Senn, C.; Tobler, K.; Ziegler, U.; Schraner, E.M.; Loepfe, E.; Ackermann, M.; Mueller, M.; Walther, P. Impairment of nuclear pores in bovine herpesvirus 1-infected MDBK cells. J. Virol. 2005, 79, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Stackpole, C.W. Herpes-type virus of the frog renal adenocarcinoma. I. Virus development in tumor transplants maintained at low temperature. J. Virol. 1969, 4, 75–93. [Google Scholar] [CrossRef] [Green Version]
- Draganova, E.B.; Thorsen, M.K.; Heldwein, E.E. Nuclear Egress. Curr. Issues Mol. Biol. 2021, 41, 125–170. [Google Scholar] [CrossRef] [PubMed]
- Arii, J. Host and Viral Factors Involved in Nuclear Egress of Herpes Simplex Virus 1. Viruses 2021, 13, 754. [Google Scholar] [CrossRef] [PubMed]
- Draganova, E.B.; Zhang, J.; Zhou, Z.H.; Heldwein, E.E. Structural basis for capsid recruitment and coat formation during HSV-1 nuclear egress. Elife 2020, 9, e56627. [Google Scholar] [CrossRef] [PubMed]
- Crump, C. Virus Assembly and Egress of HSV. Adv. Exp. Med. Biol. 2018, 1045, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Akimov, S.A.; Volynsky, P.E.; Galimzyanov, T.R.; Kuzmin, P.I.; Pavlov, K.V.; Batishchev, O.V. Pore formation in lipid membrane I: Continuous reversible trajectory from intact bilayer through hydrophobic defect to transversal pore. Sci. Rep. 2017, 7, 12152. [Google Scholar] [CrossRef]
- Farnsworth, A.; Wisner, T.W.; Webb, M.; Roller, R.; Cohen, G.; Eisenberg, R.; Johnson, D.C. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc. Natl. Acad. Sci. USA 2007, 104, 10187–10192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef]
- Vollmer, B.; Grunewald, K. Herpesvirus membrane fusion—A team effort. Curr. Opin. Struct. Biol. 2020, 62, 112–120. [Google Scholar] [CrossRef]
- Hutchinson, L.; Browne, H.; Wargent, V.; Davis-Poynter, N.; Primorac, S.; Goldsmith, K.; Minson, A.C.; Johnson, D.C. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J. Virol. 1992, 66, 2240–2250. [Google Scholar] [CrossRef] [Green Version]
- Roop, C.; Hutchinson, L.; Johnson, D.C. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J. Virol. 1993, 67, 2285–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, T.; Sagou, K.; Arii, J.; Kawaguchi, Y. Effects of phosphorylation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase in vivo and in vitro. J. Virol. 2010, 84, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.C.; Wisner, T.W.; Hannah, B.P.; Eisenberg, R.J.; Cohen, G.H.; Johnson, D.C. Fusion between perinuclear virions and the outer nuclear membrane requires the fusogenic activity of herpes simplex virus gB. J. Virol. 2009, 83, 11847–11856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannah, B.P.; Heldwein, E.E.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 2007, 81, 4858–4865. [Google Scholar] [CrossRef] [Green Version]
- Ryckman, B.J.; Roller, R.J. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 2004, 78, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Mou, F.; Wills, E.; Baines, J.D. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J. Virol. 2009, 83, 5181–5191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisner, T.; Wright, C.; Kato, A.; Kawaguchi, Y.; Mou, F.; Baines, J.; Roller, R.; Johnson, D. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J. Virol. 2009, 83, 3115–3126. [Google Scholar] [CrossRef] [Green Version]
- Kato, A.; Arri, J.; Shiratori, I.; Akashi, H.; Arase, H.; Kawaguchi, Y. Herpes simplex virus 1 protein kinease US3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J. Virol. 2009, 83, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Newcomb, W.W.; Fontana, J.; Winkler, D.C.; Cheng, N.; Heymann, J.B.; Steven, A.C. The Primary Enveloped Virion of Herpes Simplex Virus 1: Its Role in Nuclear Egress. mBio 2017, 8, e00825-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, M.; Mori, Y. Entry of betaherpesviruses. Adv. Virus Res. 2019, 104, 283–312. [Google Scholar] [CrossRef] [PubMed]
- Cairns, T.M.; Connolly, S.A. Entry of Alphaherpesviruses. Curr. Issues Mol. Biol. 2021, 41, 63–124. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.; Howard, P.; Wisner, T.; Johnson, D. Human Cytomegalovirus gH/gL Forms a Stable Complex with the Fusion Protein gB in Virions. PLoS Pathog. 2016, 12, e1005564. [Google Scholar] [CrossRef] [Green Version]
- Cairns, T.M.; Atanasiu, D.; Saw, W.T.; Lou, H.; Whitbeck, J.C.; Ditto, N.T.; Bruun, B.; Browne, H.; Bennett, L.; Wu, C.; et al. Localization of the Interaction Site of Herpes Simplex Virus Glycoprotein D (gD) on the Membrane Fusion Regulator, gH/gL. J. Virol. 2020, 94, e00983-20. [Google Scholar] [CrossRef]
- Heldwein, E.; Lou, H.; Bender, F.; Cohen, G.; Eisenberg, R.; Harrison, S. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar] [CrossRef] [Green Version]
- Vollmer, B.; Prazak, V.; Vasishtan, D.; Jefferys, E.E.; Hernandez-Duran, A.; Vallbracht, M.; Klupp, B.G.; Mettenleiter, T.C.; Backovic, M.; Rey, F.A.; et al. The prefusion structure of herpes simplex virus glycoprotein B. Sci. Adv. 2020, 6, eabc1726. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Heim, K.P.; Che, Y.; Chi, X.; Qiu, X.; Han, S.; Dormitzer, P.R.; Yang, X. Prefusion structure of human cytomegalovirus glycoprotein B and structural basis for membrane fusion. Sci. Adv. 2021, 7, eabf3718. [Google Scholar] [CrossRef]
- Cai, W.H.; Gu, B.; Person, S. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J. Virol. 1988, 62, 2596–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligas, M.W.; Johnson, D.C. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by beta-galactosidase sequences binds to but is unable to penetrate into cells. J. Virol. 1988, 62, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Forrester, A.; Farrell, H.; Wilkinson, G.; Kaye, J.; Davis-Poynter, N.; Minson, T. Construction and properties of a mutant of herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J. Virol. 1992, 66, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Galdiero, S.; Vitiello, M.; D’Isanto, M.; Falanga, A.; Collins, C.; Raieta, K.; Pedone, C.; Browne, H.; Galdiero, M. Analysis of synthetic peptides from heptad-repeat domains of herpes simplex virus type 1 glycoproteins H and B. J. Gen. Virol. 2006, 87, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, S.; Falanga, A.; Vitiello, M.; Raiola, L.; Fattorusso, R.; Browne, H.; Pedone, C.; Isernia, C.; Galdiero, M. Analysis of a membrane interacting region of herpes simplex virus type 1 glycoprotein H. J. Biol. Chem. 2008, 283, 29993–30009. [Google Scholar] [CrossRef] [Green Version]
- Galdiero, S.; Falanga, A.; Vitiello, M.; D’Isanto, M.; Cantisani, M.; Kampanaraki, A.; Benedetti, E.; Browne, H.; Galdiero, M. Peptides containing membrane-interacting motifs inhibit herpes simplex virus type 1 infectivity. Peptides 2008, 29, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Kemble, G.W.; Danieli, T.; White, J.M. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell 1994, 76, 383–391. [Google Scholar] [CrossRef]
- Subramanian, R.P.; Geraghty, R.J. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc. Natl. Acad. Sci. USA 2007, 104, 2903–2908. [Google Scholar] [CrossRef] [Green Version]
- Klupp, B.; Altenschmidt, J.; Granzow, H.; Fuchs, W.; Mettenleiter, T.C. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J. Virol. 2008, 82, 6299–6309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrisi, M.R.; Di Lazzaro, C.; Pavan, A.; Pereira, L.; Campadelli-Fiume, G. Herpes simplex virus envelopment and maturation studied by fracture label. J. Virol. 1992, 66, 554–561. [Google Scholar] [CrossRef] [Green Version]
- Baines, J.D.; Wills, E.; Jacob, R.J.; Pennington, J.; Roizman, B. Glycoprotein M of herpes simplex virus 1 is incorporated into virions during budding at the inner nuclear membrane. J. Virol. 2007, 81, 800–812. [Google Scholar] [CrossRef] [Green Version]
- Padula, M.E.; Sydnor, M.L.; Wilson, D.W. Isolation and preliminary characterization of herpes simplex virus 1 primary enveloped virions from the perinuclear space. J. Virol. 2009, 83, 4757–4765. [Google Scholar] [CrossRef] [Green Version]
- Brack, A.R.; Dijkstra, J.M.; Granzow, H.; Klupp, B.G.; Mettenleiter, T.C. Inhibition of virion maturation by simultaneous deletion of glycoproteins E, I, and M of pseudorabies virus. J. Virol. 1999, 73, 5364–5372. [Google Scholar] [CrossRef] [Green Version]
- Browne, H.; Bell, S.; Minson, T. Analysis of the requirement for glycoprotein m in herpes simplex virus type 1 morphogenesis. J. Virol. 2004, 78, 1039–1041. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.C.; Wisner, T.W.; Wright, C.C. Herpes simplex virus glycoproteins gB and gD function in a redundant fashion to promote secondary envelopment. J. Virol. 2011, 85, 4910–4926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuRaine, G.; Johnson, D.C. Anterograde transport of alpha-herpesviruses in neuronal axons. Virology 2021, 559, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, L.; Goldsmith, K.; Snoddy, D.; Ghosh, H.; Graham, F.L.; Johnson, D.C. Identification and characterization of a novel herpes simplex virus glycoprotein, gK, involved in cell fusion. J. Virol. 1992, 66, 5603–5609. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, L.; Johnson, D.C. Herpes simplex virus glycoprotein K promotes egress of virus particles. J. Virol. 1995, 69, 5401–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayachandra, S.; Baghian, A.; Kousoulas, K.G. Herpes simplex virus type 1 glycoprotein K is not essential for infectious virus production in actively replicating cells but is required for efficient envelopment and translocation of infectious virions from the cytoplasm to the extracellular space. J. Virol. 1997, 71, 5012–5024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruyechan, W.T.; Morse, L.S.; Knipe, D.M.; Roizman, B. Molecular genetics of herpes simplex virus. II. Mapping of the major viral glycoproteins and of the genetic loci specifying the social behavior of infected cells. J. Virol. 1979, 29, 677–697. [Google Scholar] [CrossRef] [Green Version]
- Read, G.S.; Person, S.; Keller, P.M. Genetic studies of cell fusion induced by herpes simplex virus type 1. J. Virol. 1980, 35, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Bond, V.C.; Person, S. Fine structure physical map locations of alterations that affect cell fusion in herpes simplex virus type 1. Virology 1984, 132, 368–376. [Google Scholar] [CrossRef]
- Little, S.P.; Schaffer, P.A. Expression of the syncytial (syn) phenotype in HSV-1, strain KOS: Genetic and phenotypic studies of mutants in two syn loci. Virology 1981, 112, 686–702. [Google Scholar] [CrossRef]
- Dolter, K.E.; Ramaswamy, R.; Holland, T.C. Syncytial mutations in the herpes simplex virus type 1 gK (UL53) gene occur in two distinct domains. J. Virol. 1994, 68, 8277–8281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertel, P.E.; Spear, P.G. Modified entry and syncytium formation by herpes simplex virus type 1 mutants selected for resistance to heparin inhibition. Virology 1996, 226, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Turner, A.; Bruun, B.; Minson, T.; Browne, H. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 1998, 72, 873–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, L.; Graham, F.L.; Cai, W.; Debroy, C.; Person, S.; Johnson, D.C. Herpes simplex virus (HSV) glycoproteins B and K inhibit cell fusion induced by HSV syncytial mutants. Virology 1993, 196, 514–531. [Google Scholar] [CrossRef]
- Hutchinson, L.; Roop-Beauchamp, C.; Johnson, D.C. Herpes simplex virus glycoprotein K is known to influence fusion of infected cells, yet is not on the cell surface. J. Virol. 1995, 69, 4556–4563. [Google Scholar] [CrossRef] [Green Version]
- Avitabile, E.; Lombardi, G.; Campadelli-Fiume, G. Herpes simplex virus glycoprotein K, but not its syncytial allele, inhibits cell-cell fusion mediated by the four fusogenic glycoproteins, gD, gB, gH, and gL. J. Virol. 2003, 77, 6836–6844. [Google Scholar] [CrossRef] [Green Version]
- Chouljenko, V.N.; Iyer, A.V.; Chowdhury, S.; Kim, J.; Kousoulas, K.G. The herpes simplex virus type 1 UL20 protein and the amino terminus of glycoprotein K (gK) physically interact with gB. J. Virol. 2010, 84, 8596–8606. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.P.; Alvarez, X.; Kousoulas, K.G. Plasma membrane topology of syncytial domains of herpes simplex virus type 1 glycoprotein K (gK): The UL20 protein enables cell surface localization of gK but not gK-mediated cell-to-cell fusion. J. Virol. 2003, 77, 499–510. [Google Scholar] [CrossRef] [Green Version]
- McGeoch, D.J.; Dalrymple, M.A.; Davison, A.J.; Dolan, A.; Frame, M.C.; McNab, D.; Perry, L.J.; Scott, J.E.; Taylor, P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 1988, 69, 1531–1574. [Google Scholar] [CrossRef]
- Ward, P.L.; Campadelli-Fiume, G.; Avitabile, E.; Roizman, B. Localization and putative function of the UL20 membrane protein in cells infected with herpes simplex virus 1. J. Virol. 1994, 68, 7406–7417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, T.P.; Melancon, J.M.; Olivier, T.L.; Kousoulas, K.G. Herpes simplex virus type 1 glycoprotein K and the UL20 protein are interdependent for intracellular trafficking and trans-Golgi network localization. J. Virol. 2004, 78, 13262–13277. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.P.; Chouljenko, V.N.; Kousoulas, K.G. Functional and physical interactions of the herpes simplex virus type 1 UL20 membrane protein with glycoprotein K. J. Virol. 2008, 82, 6310–6323. [Google Scholar] [CrossRef] [Green Version]
- Baines, J.D.; Ward, P.L.; Campadelli-Fiume, G.; Roizman, B. The UL20 gene of herpes simplex virus 1 encodes a function necessary for viral egress. J. Virol. 1991, 65, 6414–6424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, T.P.; Melancon, J.M.; Baines, J.D.; Kousoulas, K.G. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J. Virol. 2004, 78, 5347–5357. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.L.; Taddeo, B.; Markovitz, N.S.; Roizman, B. Identification of a novel expressed open reading frame situated between genes U(L)20 and U(L)21 of the herpes simplex virus 1 genome. Virology 2000, 266, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, W.; Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. The UL20 gene product of pseudorabies virus functions in virus egress. J. Virol. 1997, 71, 5639–5646. [Google Scholar] [CrossRef] [Green Version]
- Avitabile, E.; Lombardi, G.; Gianni, T.; Capri, M.; Campadelli-Fiume, G. Coexpression of UL20p and gK inhibits cell-cell fusion mediated by herpes simplex virus glycoproteins gD, gH-gL, and wild-type gB or an endocytosis-defective gB mutant and downmodulates their cell surface expression. J. Virol. 2004, 78, 8015–8025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenaar, F.; Pol, J.M.; Peeters, B.; Gielkens, A.L.; de Wind, N.; Kimman, T.G. The US3-encoded protein kinase from pseudorabies virus affects egress of virions from the nucleus. J. Gen. Virol. 1995, 76, 1851–1859. [Google Scholar] [CrossRef]
- Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Effect of the pseudorabies virus US3 protein on nuclear membrane localization of the UL34 protein and virus egress from the nucleus. J. Gen. Virol. 2001, 82, 2363–2371. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Chen, S.; Zhu, D.; Liu, M.; Zhao, X.; et al. The Pivotal Roles of US3 Protein in Cell-to-Cell Spread and Virion Nuclear Egress of Duck Plague Virus. Sci. Rep. 2020, 10, 7181. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Kawaguchi, Y. Us3 Protein Kinase Encoded by HSV: The Precise Function and Mechanism on Viral Life Cycle. Adv. Exp. Med. Biol. 2018, 1045, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Proft, A.; Spiesschaert, B.; Izume, S.; Taferner, S.; Lehmann, M.J.; Azab, W. The Role of the Equine Herpesvirus Type 1 (EHV-1) US3-Encoded Protein Kinase in Actin Reorganization and Nuclear Egress. Viruses 2016, 8, 275. [Google Scholar] [CrossRef]
- Schumacher, D.; Tischer, B.K.; Trapp, S.; Osterrieder, N. The protein encoded by the US3 orthologue of Marek’s disease virus is required for efficient de-envelopment of perinuclear virions and involved in actin stress fiber breakdown. J. Virol. 2005, 79, 3987–3997. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, A.E.; Wills, E.G.; Roller, R.J.; Ryckman, B.J.; Baines, J.D. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 2002, 76, 8939–8952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purves, F.C.; Spector, D.; Roizman, B. UL34, the target of the herpes simplex virus Us3 protein kinase, is a membrane protein which in its unphosphorylated state associates with novel phosphoproteins. J. Virol. 1992, 66, 4295–4303. [Google Scholar] [CrossRef] [Green Version]
- Klupp, B.G.; Granzow, H.; Fuchs, W.; Keil, G.M.; Finke, S.; Mettenleiter, T.C. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 7241–7246. [Google Scholar] [CrossRef] [Green Version]
- Bigalke, J.M.; Heldwein, E.E. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J. 2015, 34, 2921–2936. [Google Scholar] [CrossRef]
- Bigalke, J.M.; Heuser, T.; Nicastro, D.; Heldwein, E.E. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat. Commun. 2014, 5, 4151. [Google Scholar] [CrossRef]
- Hagen, C.; Dent Kyle, C.; Zeev-Ben-Mordehai, T.; Grange, M.; Bosse Jens, B.; Whittle, C.; Klupp Barbara, G.; Siebert, C.A.; Vasishtan, D.; Bäuerlein Felix, J.B.; et al. Structural Basis of Vesicle Formation at the Inner Nuclear Membrane. Cell 2015, 163, 1692–1701. [Google Scholar] [CrossRef] [Green Version]
- Lye, M.F.; Sharma, M.; El Omari, K.; Filman, D.J.; Schuermann, J.P.; Hogle, J.M.; Coen, D.M. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J. 2015, 34, 2937–2952. [Google Scholar] [CrossRef] [Green Version]
- Walzer, S.A.; Egerer-Sieber, C.; Sticht, H.; Sevvana, M.; Hohl, K.; Milbradt, J.; Muller, Y.A.; Marschall, M. Crystal Structure of the Human Cytomegalovirus pUL50-pUL53 Core Nuclear Egress Complex Provides Insight into a Unique Assembly Scaffold for Virus-Host Protein Interactions. J. Biol. Chem. 2015, 290, 27452–27458. [Google Scholar] [CrossRef] [Green Version]
- Thorsen, M.K.; Lai, A.; Lee, M.W.; Hoogerheide, D.P.; Wong, G.C.L.; Freed, J.H.; Heldwein, E.E. Highly Basic Clusters in the Herpes Simplex Virus 1 Nuclear Egress Complex Drive Membrane Budding by Inducing Lipid Ordering. mBio 2021, 12, e0154821. [Google Scholar] [CrossRef] [PubMed]
- Bahnamiri, M.M.; Roller, R.J. Mechanism of Nuclear Lamina Disruption and the Role of pUS3 in HSV-1 Nuclear Egress. J. Virol. 2021, 95, e02432-20. [Google Scholar] [CrossRef]
- Gao, J.; Finnen, R.L.; Sherry, M.R.; Le Sage, V.; Banfield, B.W. Differentiating the Roles of UL16, UL21, and Us3 in the Nuclear Egress of Herpes Simplex Virus Capsids. J. Virol. 2020, 94, e00738-20. [Google Scholar] [CrossRef] [PubMed]
- Benedyk, T.H.; Muenzner, J.; Connor, V.; Han, Y.; Brown, K.; Wijesinghe, K.J.; Zhuang, Y.; Colaco, S.; Stoll, G.A.; Tutt, O.S.; et al. pUL21 is a viral phosphatase adaptor that promotes herpes simplex virus replication and spread. PLoS Pathog. 2021, 17, e1009824. [Google Scholar] [CrossRef] [PubMed]
- de Wind, N.; Wagenaar, F.; Pol, J.; Kimman, T.; Berns, A. The pseudorabies virus homology of the herpes simplex virus UL21 gene product is a capsid protein which is involved in capsid maturation. J. Virol. 1992, 66, 7096–7103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takakuwa, H.; Goshima, F.; Koshizuka, T.; Murata, T.; Daikoku, T.; Nishiyama, Y. Herpes simplex virus encodes a virion-associated protein which promotes long cellular processes in over-expressing cells. Genes Cells 2001, 6, 955–966. [Google Scholar] [CrossRef]
- Purves, F.C.; Longnecker, R.M.; Leader, D.P.; Roizman, B. Herpes simplex virus 1 protein kinase is encoded by open reading frame US3 which is not essential for virus growth in cell culture. J. Virol. 1987, 61, 2896–2901. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.E.; Van Sant, C.; Krug, P.W.; Sears, A.E.; Roizman, B. The null mutant of the UL31 gene of herpes simplex virus 1: Construction and phenotype of infected cells. J. Virol. 1997, 71, 8307–8315. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, W.; Klupp, B.G.; Granzow, H.; Osterrieder, N.; Mettenleiter, T.C. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 2002, 76, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J. Virol. 2000, 74, 10063–10073. [Google Scholar] [CrossRef] [Green Version]
- Roller, R.; Zhou, Y.; Schnetzer, R.; Ferguson, J.; Desalvo, D. Herpes simplex virus type 1 UL34 gene product is required for viral envelopment. J. Virol. 2000, 74, 117–129. [Google Scholar] [CrossRef] [Green Version]
- De Magistris, P.; Antonin, W. The Dynamic Nature of the Nuclear Envelope. Curr. Biol. 2018, 28, R487–R497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puhka, M.; Joensuu, M.; Vihinen, H.; Belevich, I.; Jokitalo, E. Progressive sheet-to-tubule transformation is a general mechanism for endoplasmic reticulum partitioning in dividing mammalian cells. Mol. Biol. Cell 2012, 23, 2424–2432. [Google Scholar] [CrossRef] [PubMed]
- Benetti, L.; Roizman, B. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 9411–9416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daikoku, T.; Yamashita, Y.; Tsurumi, T.; Maeno, K.; Nishiyama, Y. Purification and biochemical characterization of the protein kinase encoded by the US3 gene of herpes simplex virus type 2. Virology 1993, 197, 685–694. [Google Scholar] [CrossRef]
- Frame, M.C.; Purves, F.C.; McGeoch, D.J.; Marsden, H.S.; Leader, D.P. Identification of the herpes simplex virus protein kinase as the product of virus gene US3. J. Gen.Virol. 1987, 68, 2699–2704. [Google Scholar] [CrossRef]
- Jahn, R.; Lang, T.; Sudhoff, T.C. Membrane fusion. Cell 2003, 112, 519–533. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roller, R.J.; Johnson, D.C. Herpesvirus Nuclear Egress across the Outer Nuclear Membrane. Viruses 2021, 13, 2356. https://doi.org/10.3390/v13122356
Roller RJ, Johnson DC. Herpesvirus Nuclear Egress across the Outer Nuclear Membrane. Viruses. 2021; 13(12):2356. https://doi.org/10.3390/v13122356
Chicago/Turabian StyleRoller, Richard J., and David C. Johnson. 2021. "Herpesvirus Nuclear Egress across the Outer Nuclear Membrane" Viruses 13, no. 12: 2356. https://doi.org/10.3390/v13122356