
Semi-Supervised Pipeline for Autonomous Annotation of SARS-CoV-2 Genomes

 ,
,  , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
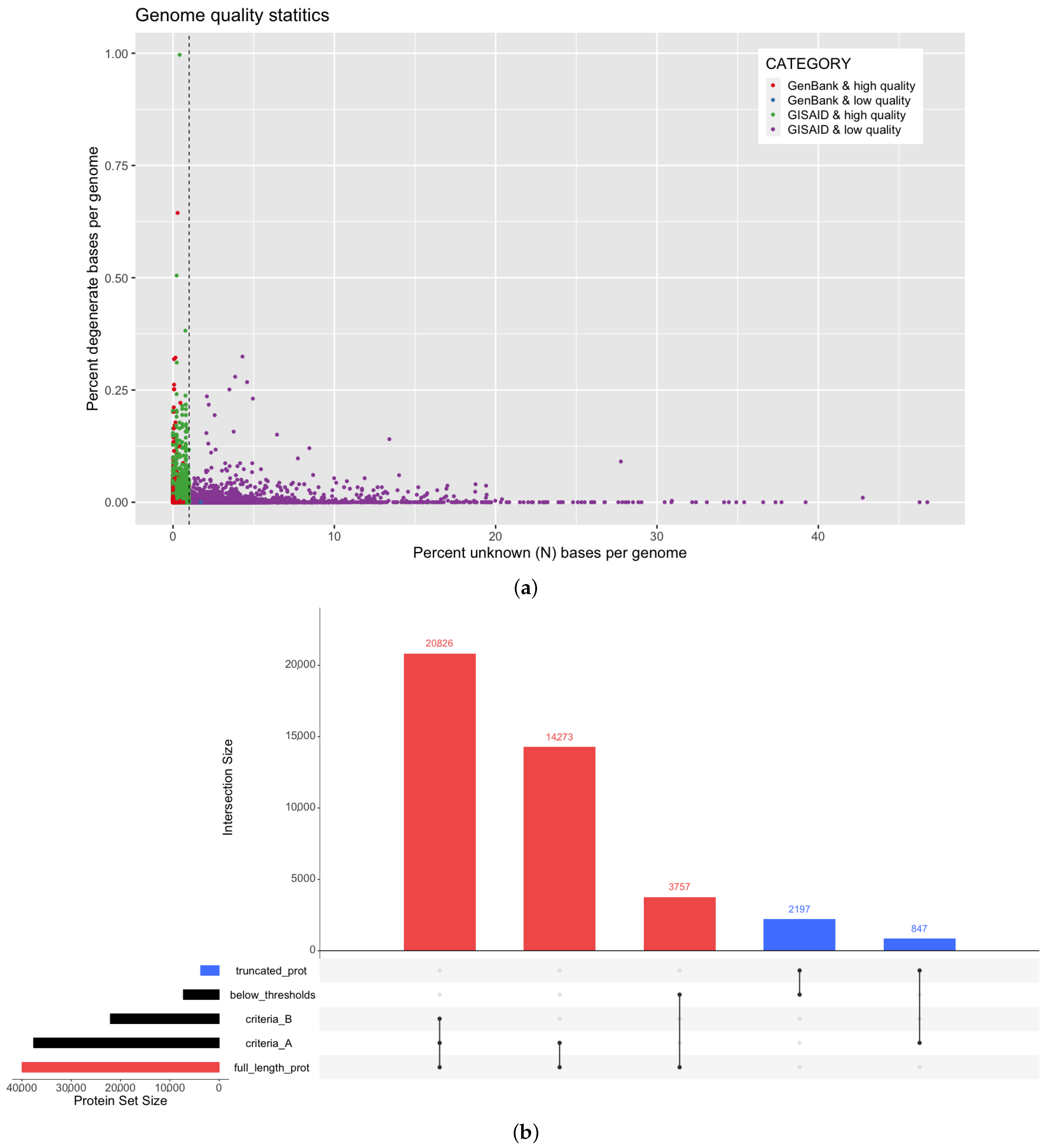
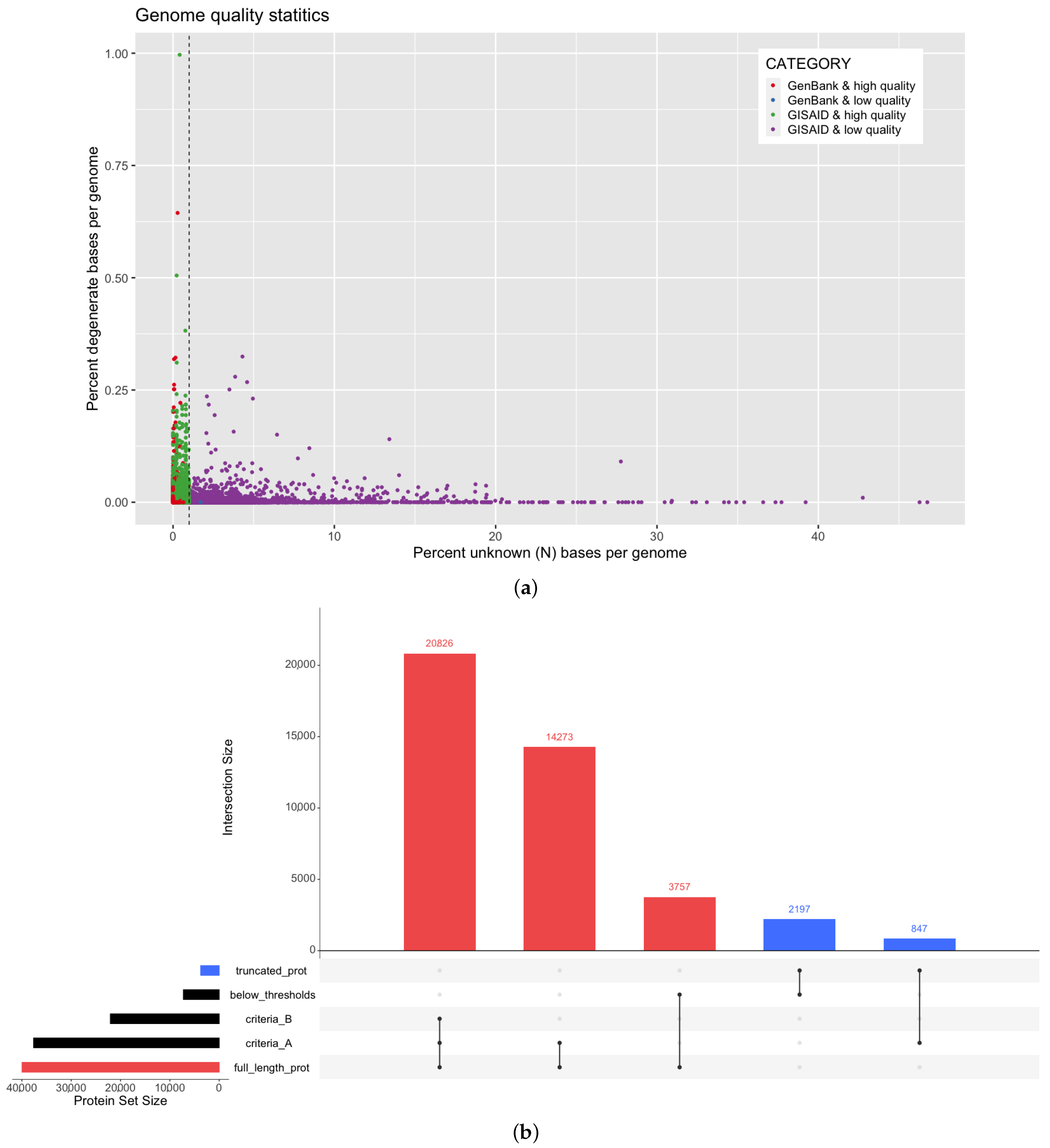
2.1. Genome Data Retrieval and Quality Thresholds
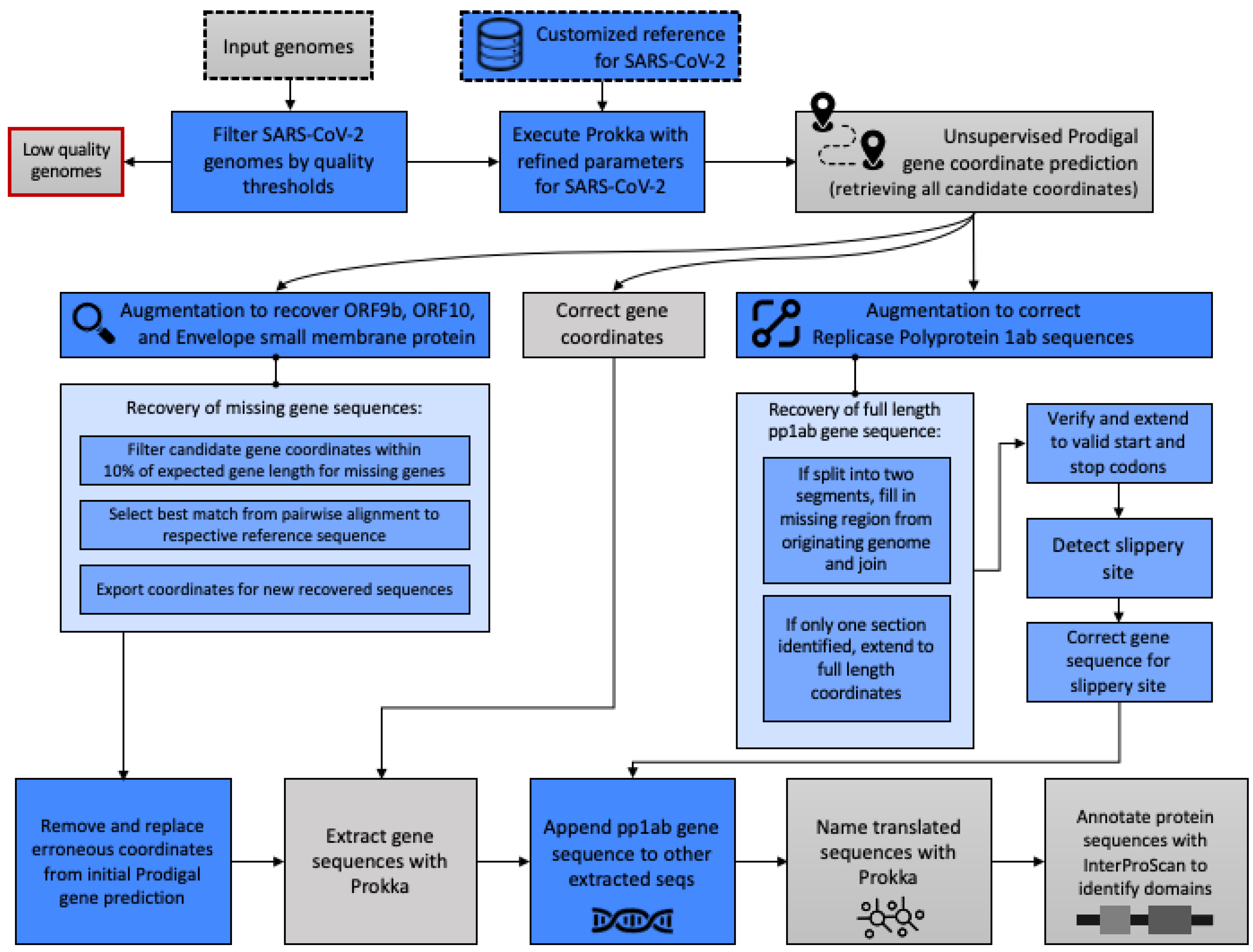
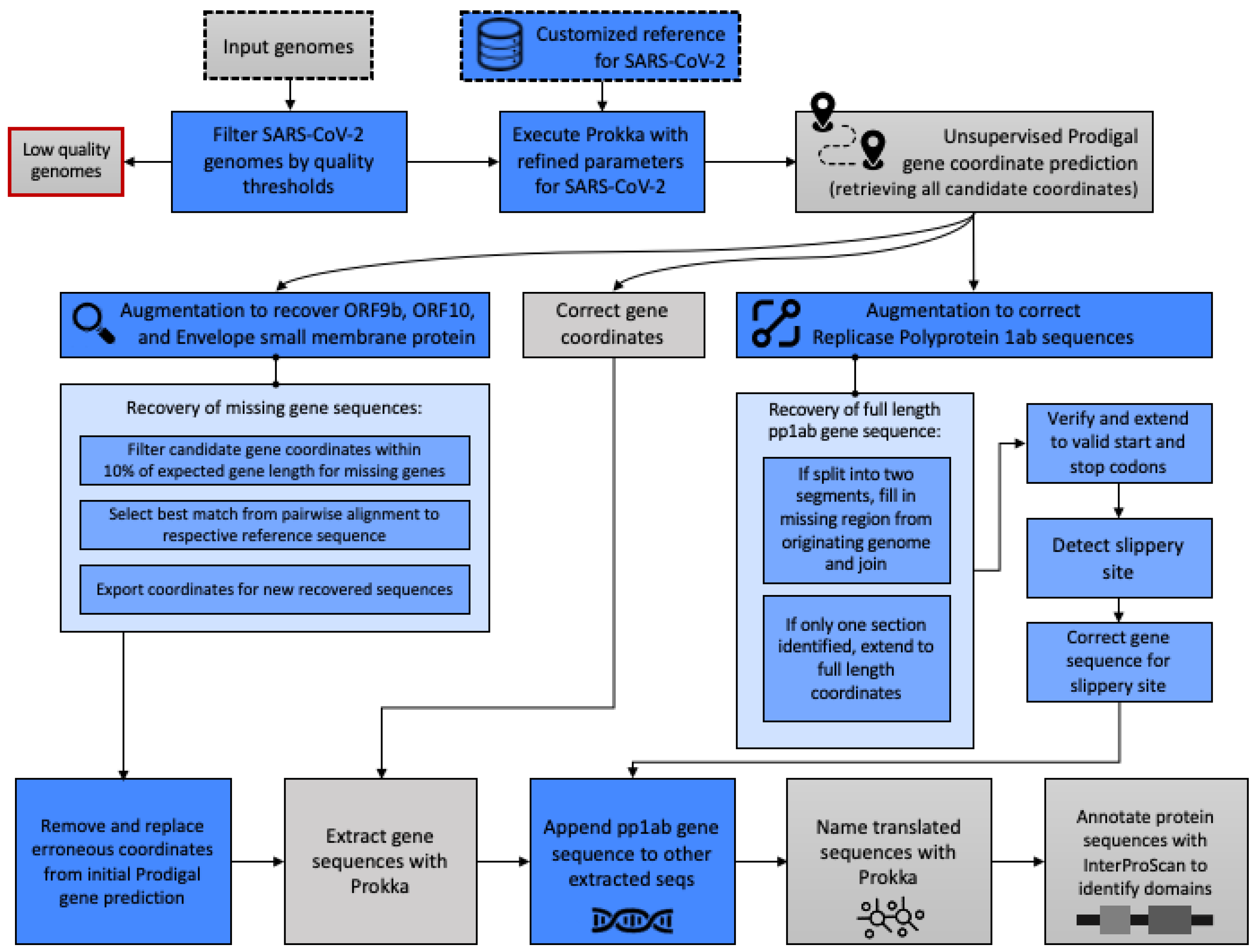
2.2. Gene and Protein Annotation
2.3. Protein Domain Annotation
2.4. Comparative Analysis
3. Results
3.1. Assessment of SARS-CoV-2 Genome Quality in Multiple Data Sources
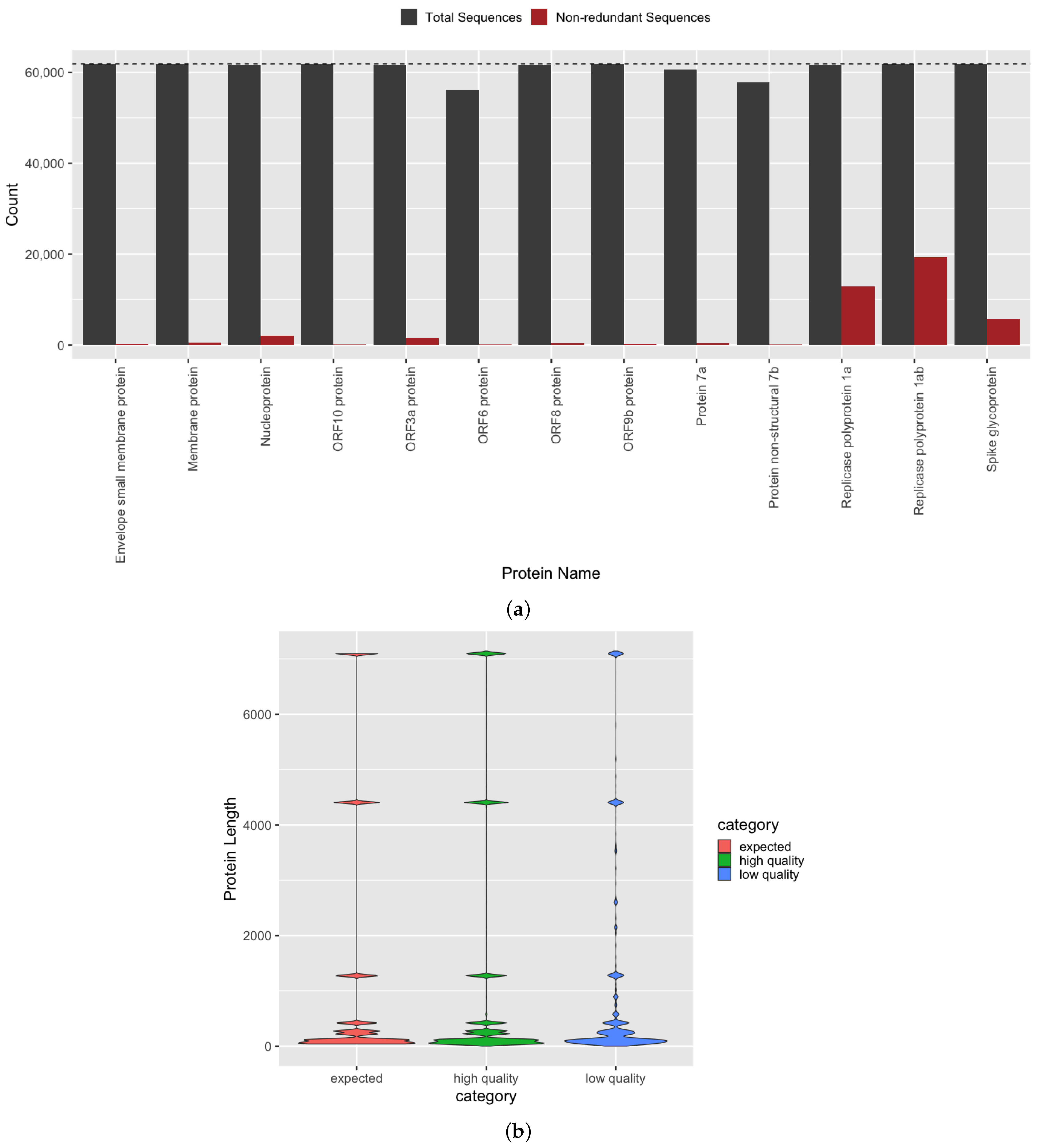
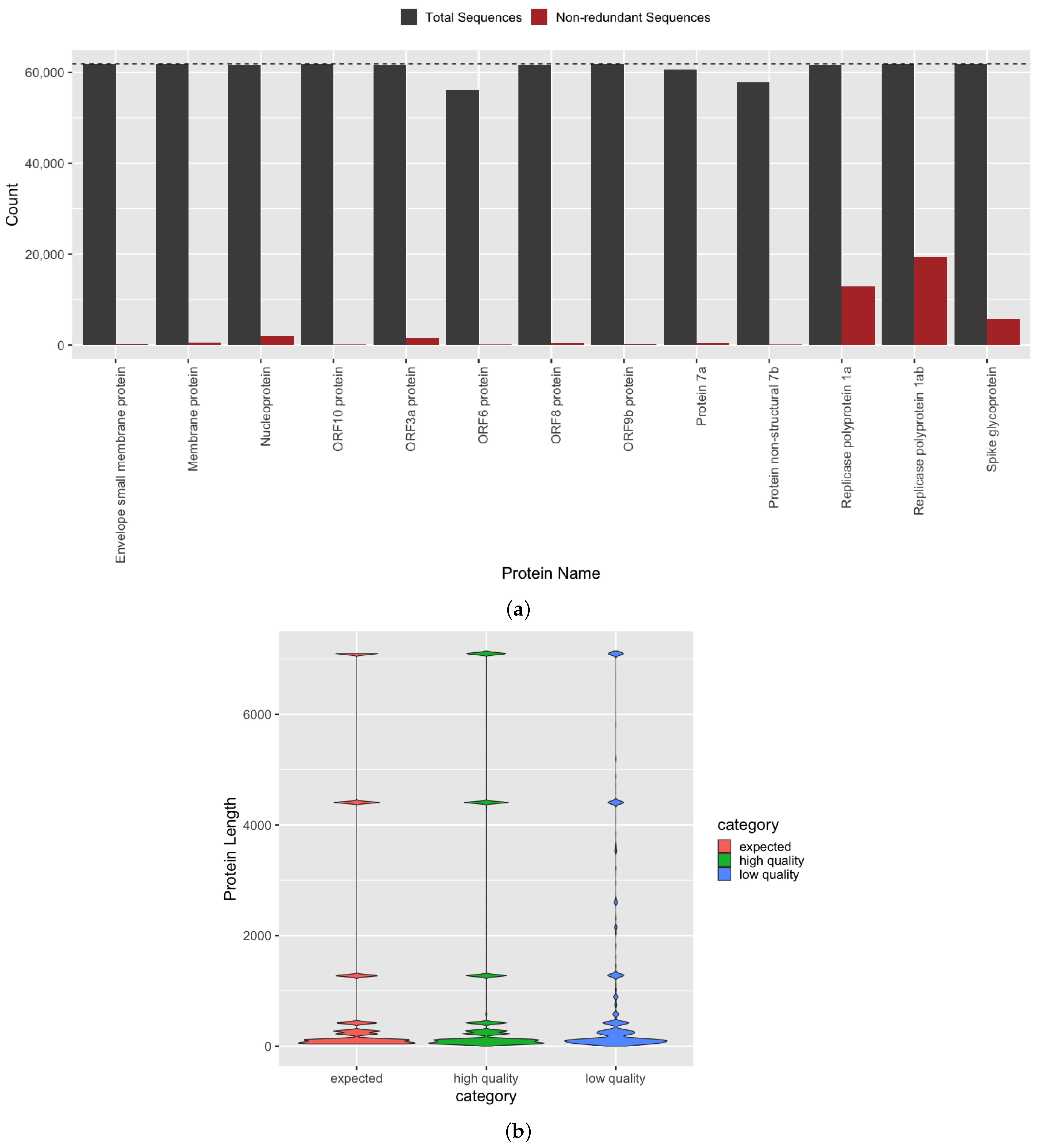
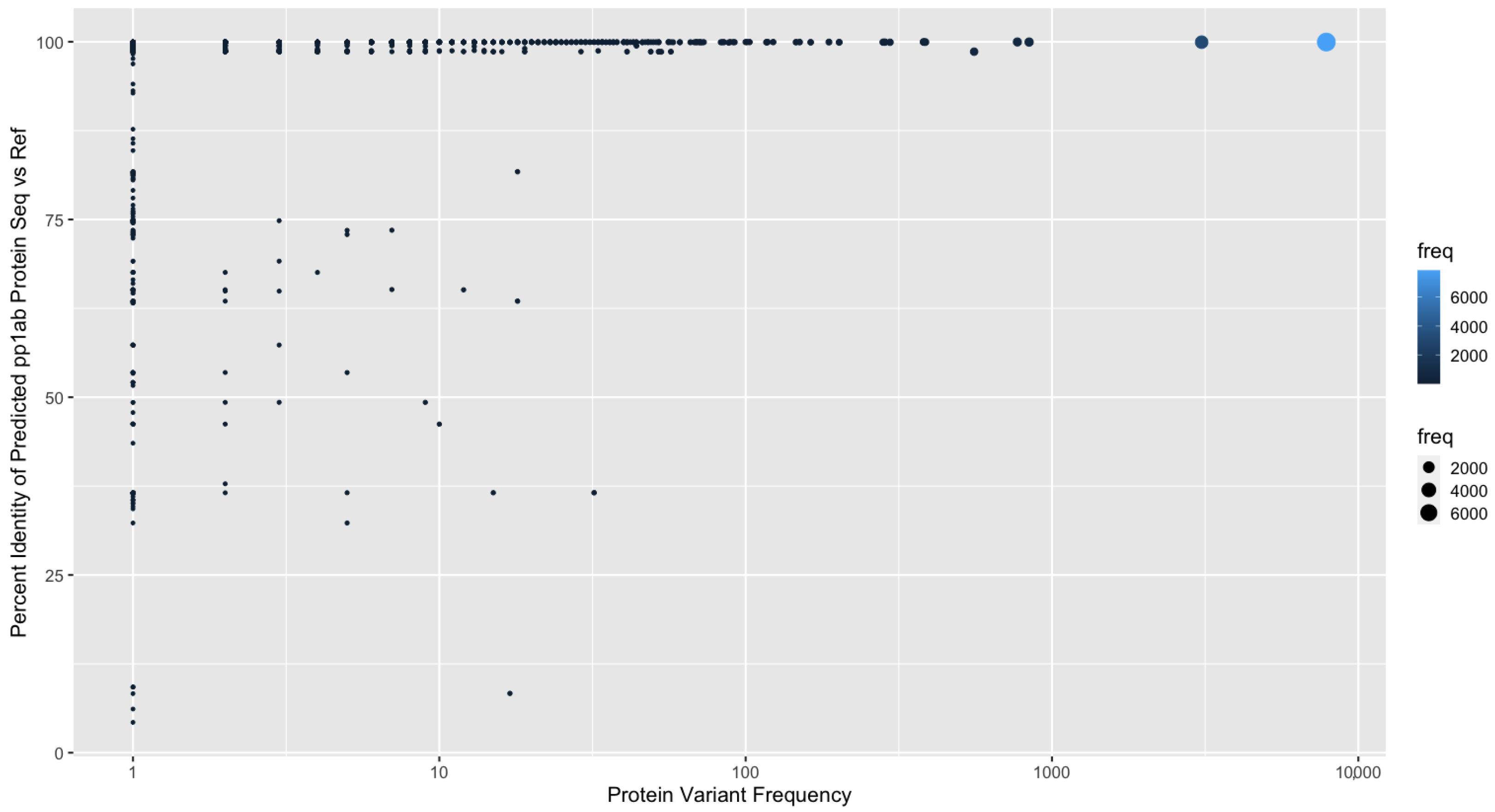
3.2. Quantification of Protein Sequence Prediction Accuracy
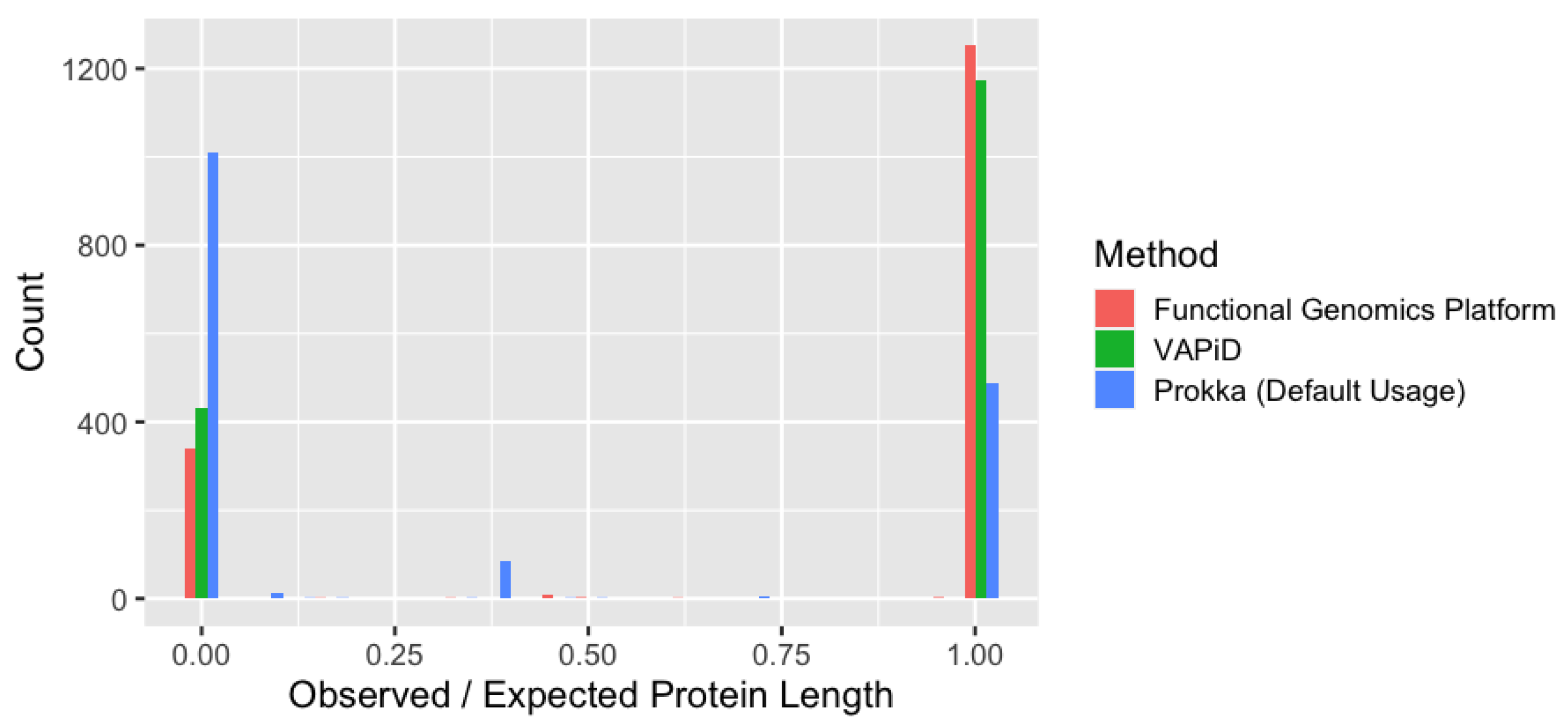
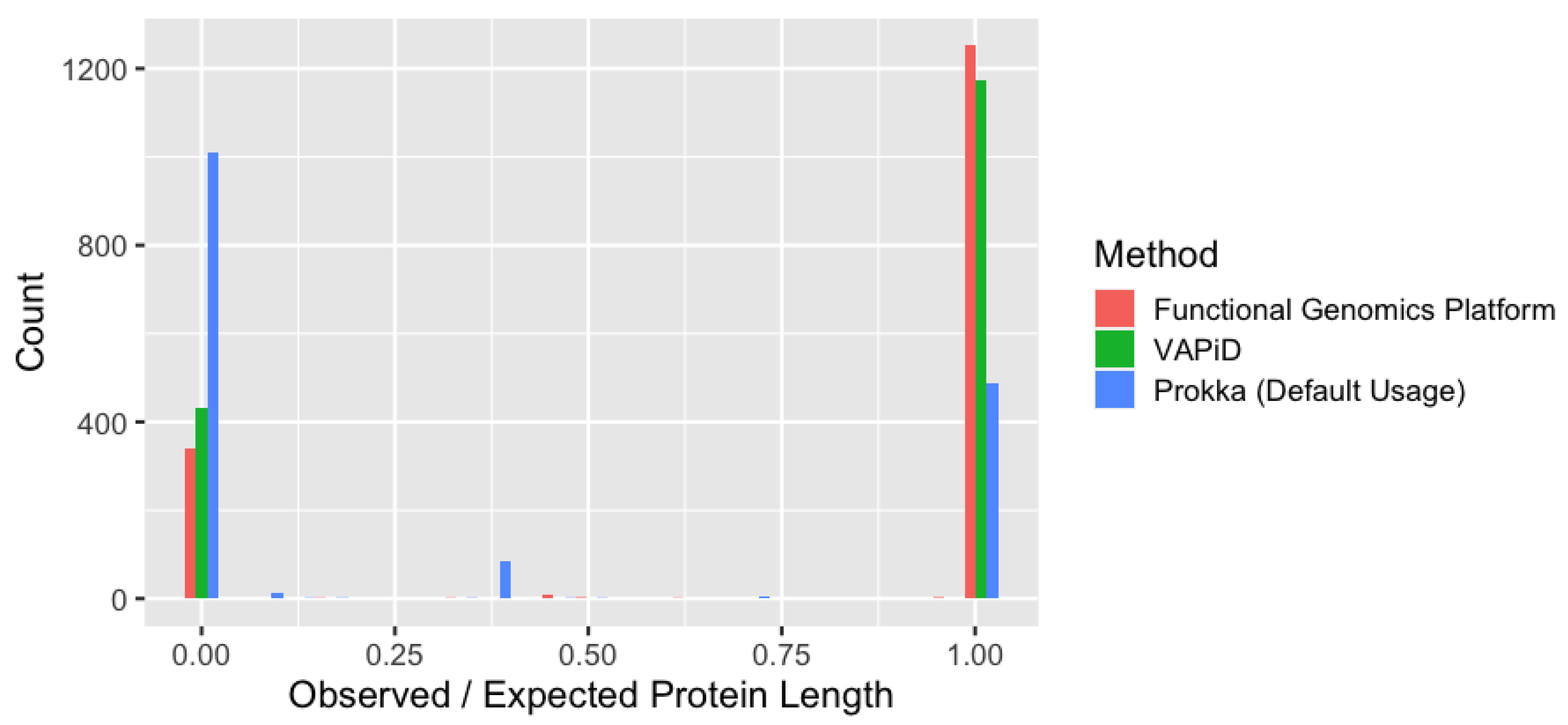
3.3. Comparative Analysis of Genome Annotation Methods
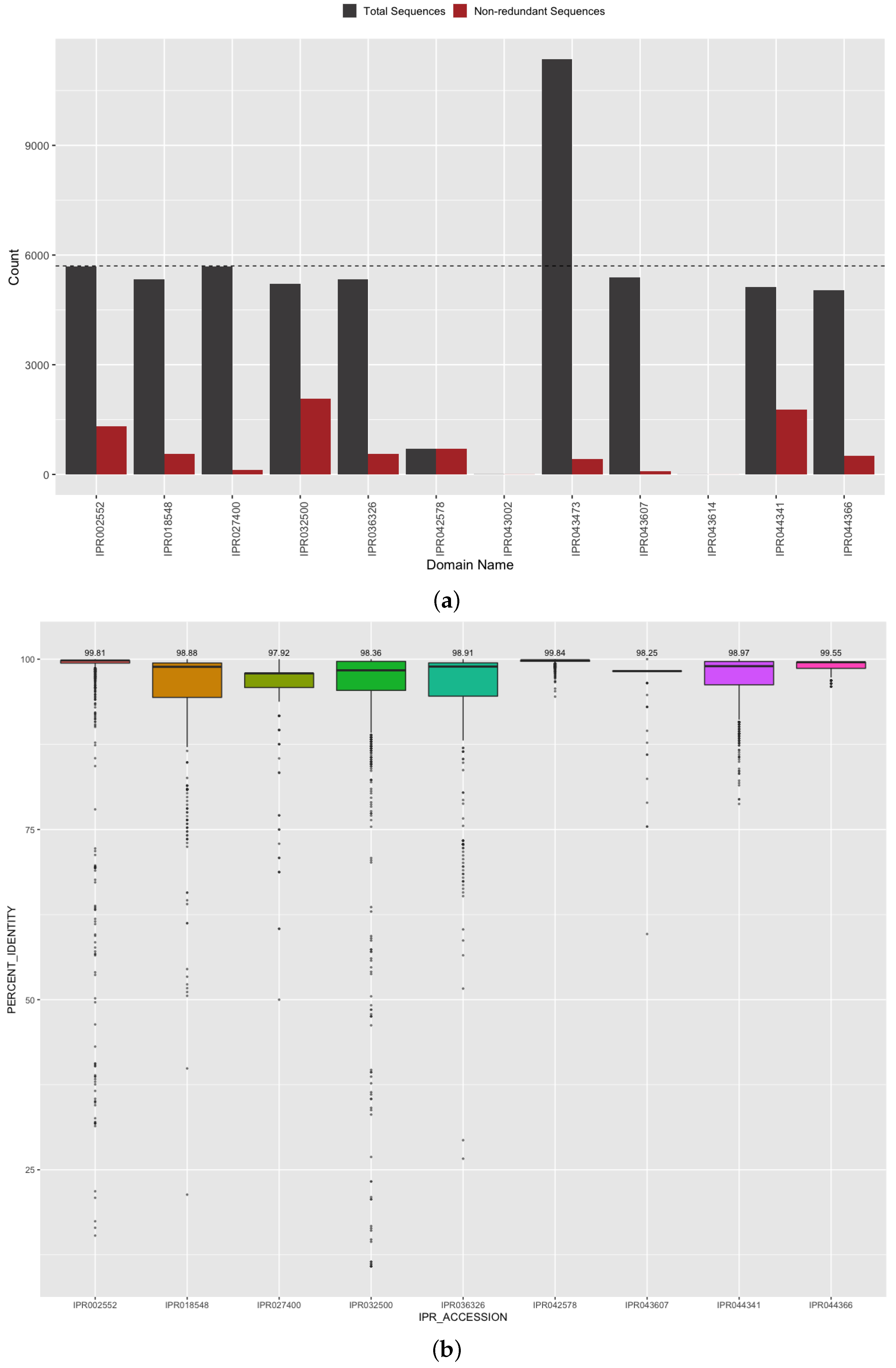
3.4. Investigation of Predicted SARS-CoV-2 Protein and Domain Sequences
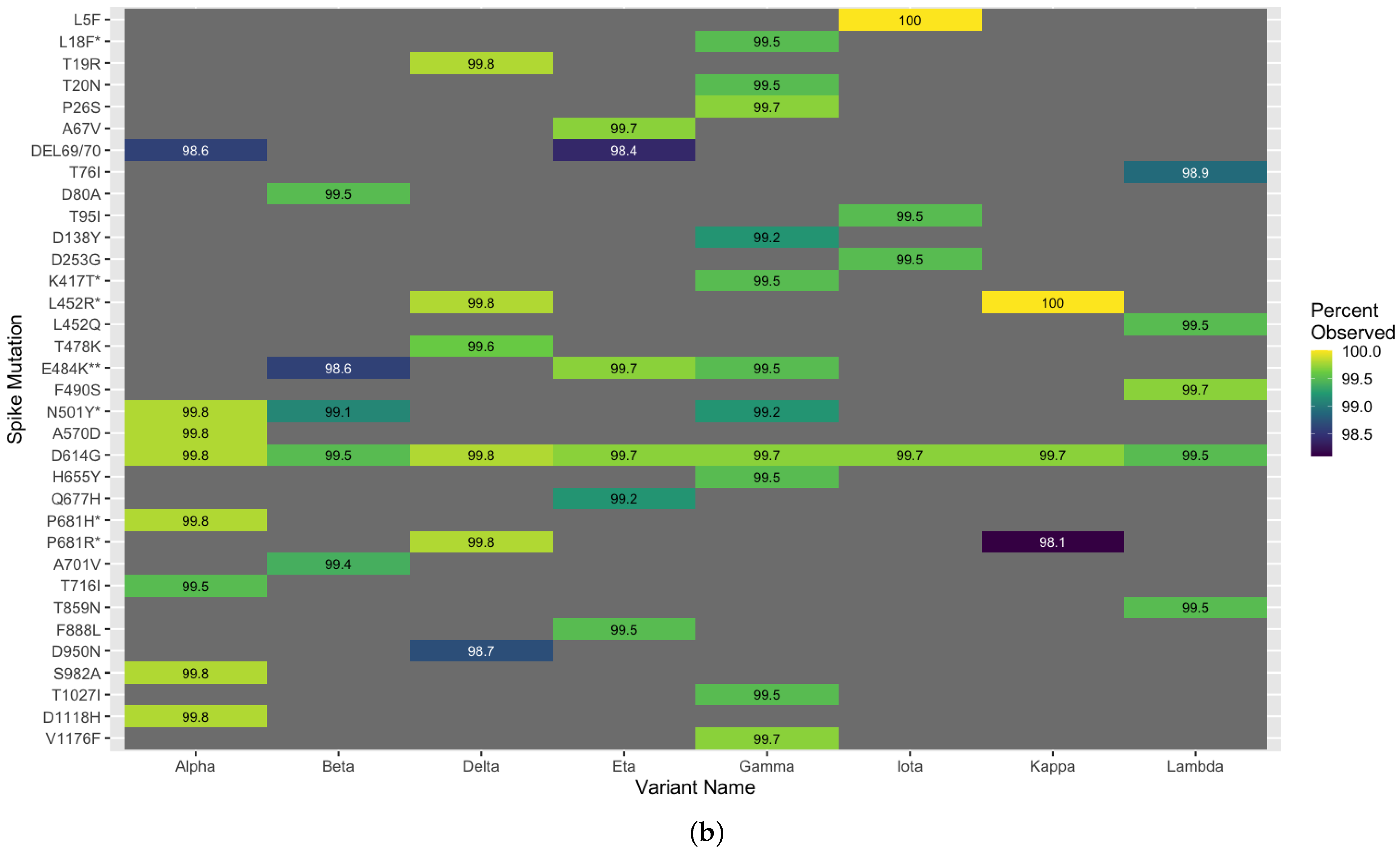
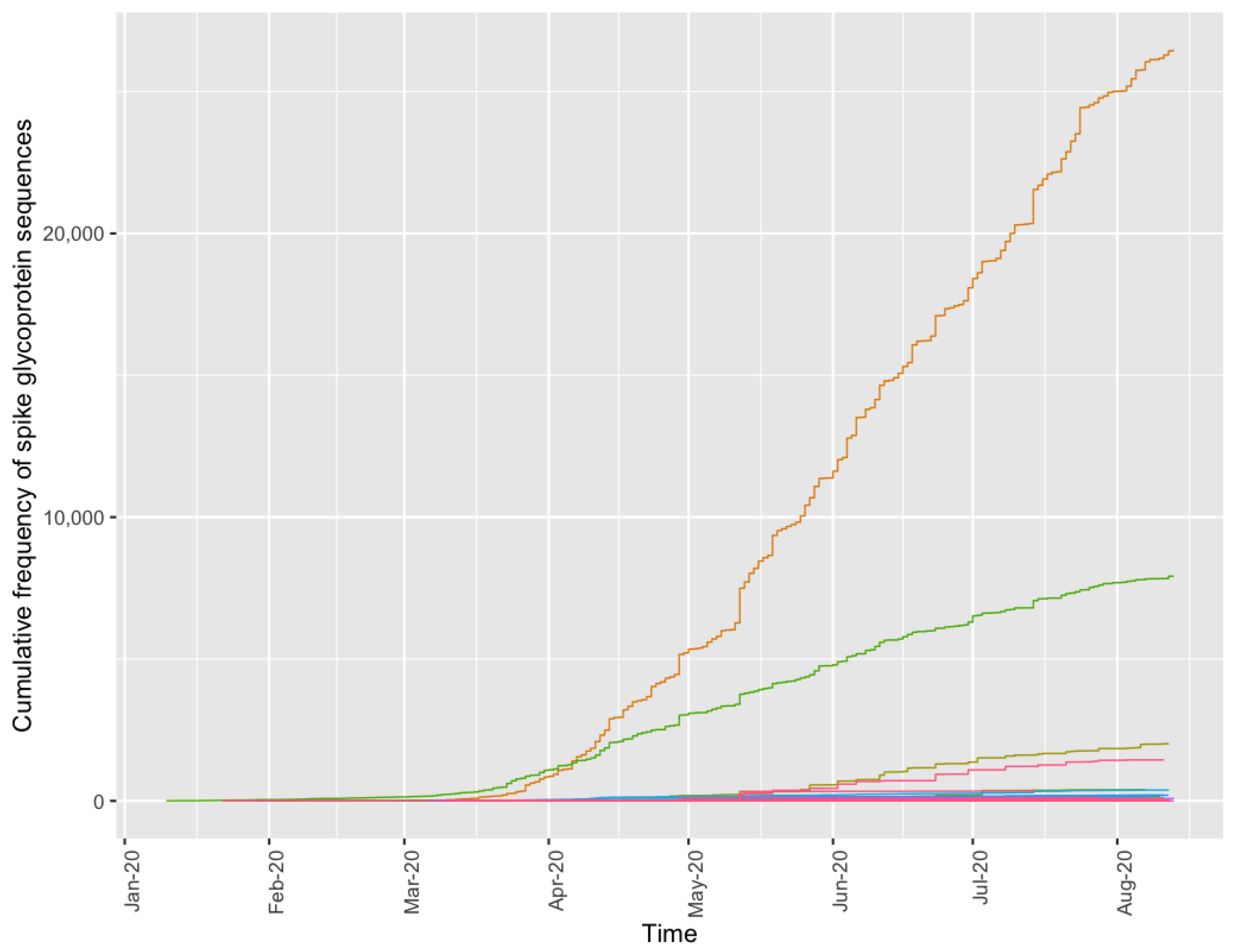
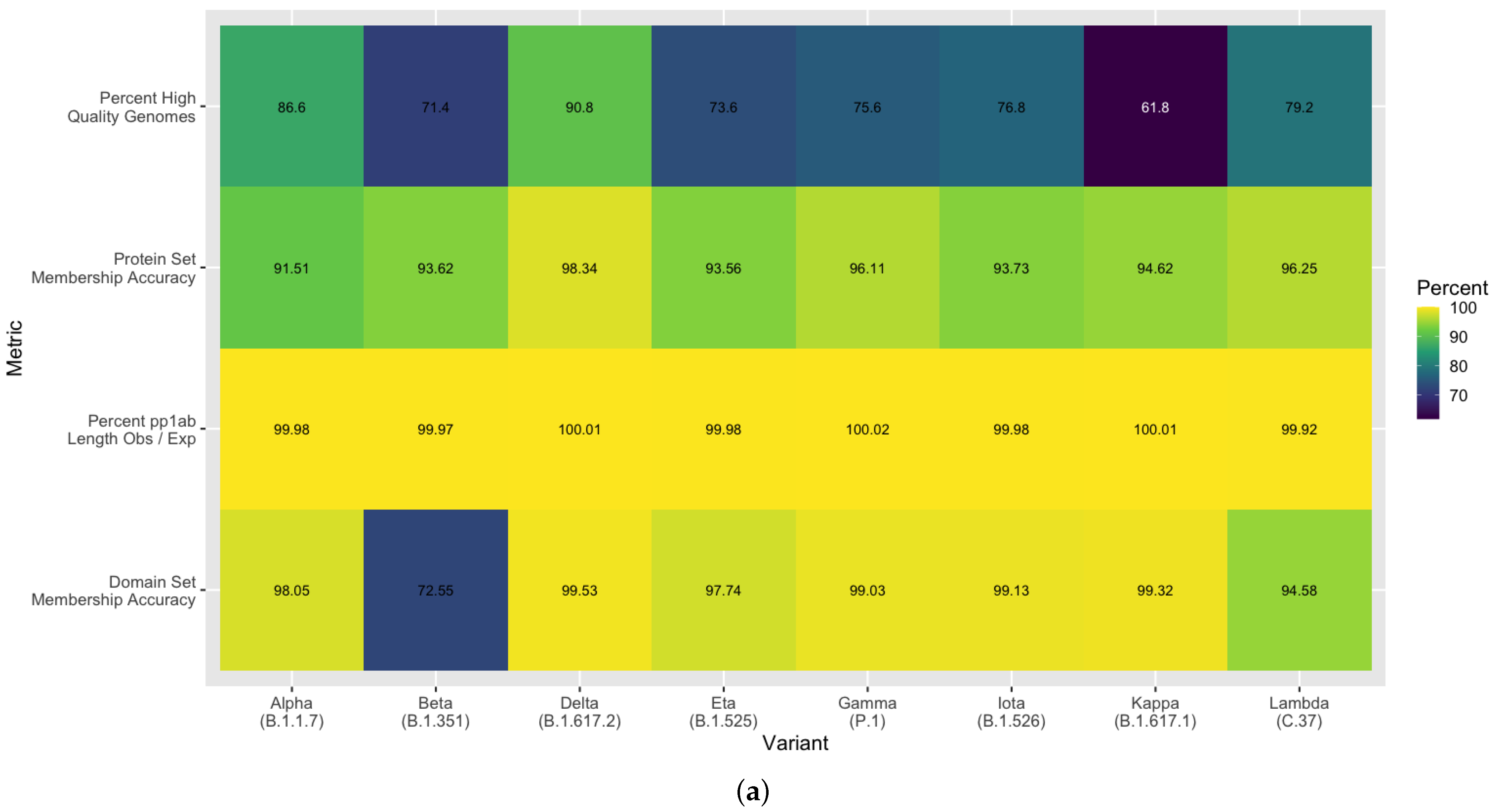
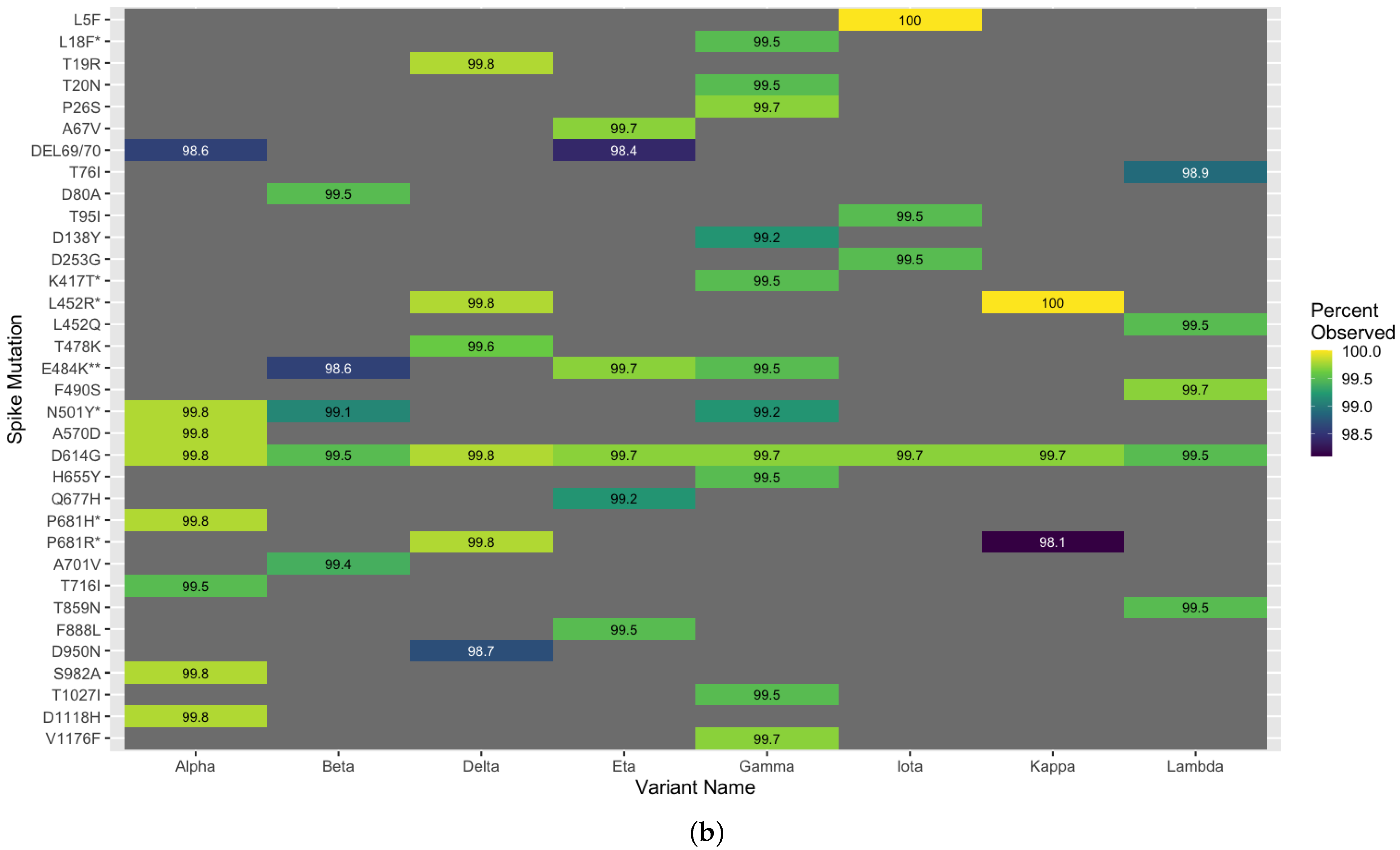
3.5. Methodological Robustness in Variant Diverse Genomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CDC | Centers for Disease Control |
| FGP | Functional Genomics Platform |
| GISAID | Global Initiative for Sharing All Influenza Data |
| IPR | InterPro |
| IUPAC | International Union of Pure and Applied Chemistry |
| NCBI | National Center for Biotechnology Information |
| ORF | Open Reading Frame |
| pp1ab | Replicase polyprotein 1ab |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| WHO | World Health Organization |
Appendix A. Methodological Details for Gene and Protein Annotation
Appendix A.1. Modifications to Accommodate SARS-CoV-2 Genome Attributes and Nascent State of Reference Data
- Modify minimum evidence level required from transcript level (evidence = 2) to predicted (evidence = 4) when selecting reference proteins. This change allows proteins with evidence levels—at the protein level (evidence = 1), at the transcript level (evidence = 2), inferred from homology (evidence = 3), or predicted (evidence = 4)—to be used when building references (but does not include protein uncertain, evidence = 5). This is to better accommodate the nascent state of SAR-CoV-2 protein references.
- Do not assign “hypothetical protein” to the recommended full names that start with the following regular expression:/^UPD\d|^Uncharacterized protein|^ORF|^Protein /as some valid SARS-CoV-2 proteins contain these prefixes, e.g., ORF3a protein and Uncharacterized protein 14.
- Accept proteins without a recommended full name as long as the entry includes a full name provided by the submitter, e.g., ORF10.
Appendix A.2. Modifications to Improve Complete and Accurate Protein Identification
- If Prodigal output two separate segments of the full gene sequence, we augmented and filled in the missing gap section from the originating genome based on the overall start and end coordinates to yield one contiguous gene sequence.
- If Prodigal output only one truncated segment of the full pp1ab gene sequence, we shifted the starting index to ensure that the full length sequence achieved the expected entire 21,289 bp known to be part of the reference sequence (UniProt ID: P0DTD1).
- In both cases, we verified that the gene sequence begins with an expected start codon (Methionine, ATG) and ends with a proper stop codon (TAA). When identifying the start codon, we verified that the expected first three nucleotides were in the predicted sequence and shifted the start index to ensure this was the start position if that was not the case. Then, if the sequence did not include the start codon, we subtracted 1 from the start index until the correct start codon (ATG) was the first three nucleotides. The same procedure was used to ensure that the sequence ended with a proper stop codon, as we added 1 to the end index until TAA were the last three nucleotides.
- Next, the slippery site as identified by Kelly et al. [6] was identified in the gene sequence, allowing for nucleotide degeneracy as indicated.
- At the point of the slippery site, the preceding base was repeated, and the remaining gene sequence was appended to yield the gene sequence, which was then translated to yield the full length pp1ab protein sequence.
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef]
- Fernandes, J.D.; Hinrichs, A.S.; Clawson, H.; Gonzalez, J.N.; Lee, B.T.; Nassar, L.R.; Raney, B.J.; Rosenbloom, K.R.; Nerli, S.; Rao, A.A.; et al. The UCSC SARS-CoV-2 Genome Browser. Nat. Genet. 2020, 52, 991–998. [Google Scholar] [CrossRef]
- Gussow, A.B.; Auslander, N.; Faure, G.; Wolf, Y.I.; Zhang, F.; Koonin, E.V. Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. Proc. Natl. Acad. Sci. USA 2020, 117, 15193–15199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Holmes, E.C. A Genomic Perspective on the Origin and Emergence of SARS-CoV-2. Cell 2020, 181, 223–227. [Google Scholar] [CrossRef]
- Kelly, J.A.; Olson, A.N.; Neupane, K.; Munshi, S.; Emeterio, J.S.; Pollack, L.; Woodside, M.T.; Dinman, J.D. Structural and functional conservation of the programmed -1 ribosomal frameshift signal of SARS coronavirus 2 (SARS-CoV-2). J. Biol. Chem. 2020, 295, 10741–10748. [Google Scholar] [CrossRef] [PubMed]
- Shean, R.C.; Makhsous, N.; Stoddard, G.D.; Lin, M.J.; Greninger, A.L. VAPiD: A lightweight cross-platform viral annotation pipeline and identification tool to facilitate virus genome submissions to NCBI GenBank. BMC Bioinform. 2019, 20, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Koyama, T.; Weeraratne, D.; Snowdon, J.L.; Parida, L. Emergence of Drift Variants That May Affect COVID-19 Vaccine Development and Antibody Treatment. Pathogens 2020, 9, 324. [Google Scholar] [CrossRef] [PubMed]
- Chand, M.; Hopkins, S.; Dabrera, G.; Allen, H.; Lamagni, T.; Edeghere, O.; Barclay, W.; Ferguson, N.; Volz, E.; Loman, N.; et al. Investigation of Novel SARS-CoV-2 Variant: Variant of Concern 202012/01 Technical Briefing 2; Technical Report; Public Health England: London, UK, 2020.
- Report 42—Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from Linking Epidemiological and Genetic Data; Faculty of Medicine, Imperial College London: London, UK, 2021.
- US COVID-19 Cases Caused by Variants; CDC: Atlanta, GA, USA, 2021.
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.; Huddleston, J.; Chaisson, M.J.; Hill, C.M.; Kronenberg, Z.N.; Munson, K.M.; Malig, M.; Raja, A.; Fiddes, I.; Hillier, L.D.W.; et al. Long-read sequence assembly of the gorilla genome. Science 2016, 352, aae0344. [Google Scholar] [CrossRef]
- Vezzi, F.; Cattonaro, F.; Policriti, A. e-RGA: Enhanced Reference Guided Assembly of Complex Genomes. EMBnet J. 2011, 17, 46–54. [Google Scholar] [CrossRef]
- Callaway, E. The coronavirus is mutating—Does it matter? Nature 2020, 585, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Neches, R.Y.; McGee, M.D.; Kyrpides, N.C. Recombination should not be an afterthought. Nat. Rev. Microbiol. 2020, 18, 606. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2009, 37, D26–D31. [Google Scholar] [CrossRef] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Seabolt, E.; Nayar, G.; Krishnareddy, H.; Agarwal, A.; Beck, K.L.; Kandogan, E.; Kuntomi, M.; Roth, M.; Terrizzano, I.; Kaufman, J.; et al. IBM Functional Genomics Platform, A Cloud-Based Platform for Studying Microbial Life at Scale. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020. [Google Scholar] [CrossRef]
- Hulo, C.; De Castro, E.; Masson, P.; Bougueleret, L.; Bairoch, A.; Xenarios, I.; Le Mercier, P. ViralZone: A knowledge resource to understand virus diversity. Nucleic Acids Res. 2011, 39, D576–D582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2016, 45, D158–D169. [Google Scholar] [CrossRef]
- Latif, A.A.; Mullen, J.L.; Alkuzweny, M.; Tsueng, G.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. Lineage Comparison. Available online: https://outbreak.info/compare-lineages?pango=Alpha&pango=Beta&pango=Delta&pango=Eta&pango=Gamma&pango=Iota&pango=Kappa&pango=Lambda&gene=S&threshold=95&dark=true.
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.C.; Tay, A.P.; Wilson, L.O.W.; Reti, D.; Hosking, C.; McAuley, A.J.; Pharo, E.; Todd, S.; Stevens, V.; Neave, M.J.; et al. Supporting pandemic response using genomics and bioinformatics: A case study on the emergent SARS-CoV-2 outbreak. Transbound. Emerg. Dis. 2020, 67, 1453–1462. [Google Scholar] [CrossRef]
- Lemoine, F.; Blassel, L.; Voznica, J.; Gascuel, O. COVID-Align: Accurate online alignment of hCoV-19 genomes using a profile HMM. Bioinformatics 2021, 37, 1761–1762. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, K.; Zhang, H.; Zhang, L.; Bian, Y.; Huang, L. CoV-Seq, a new tool for SARS-CoV-2 genome analysis and visualization: Development and usability study. J. Med. Internet Res. 2020, 22, e22299. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, L.; Pascal, K.E.; Tompkins-Tinch, C.; Nyalile, T.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; Veinotte, K.; et al. SARS-CoV-2 Spike protein variant D614G increases infectivity and retains sensitivity to antibodies that target the receptor binding domain. bioRxiv 2020. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef]
- Xie, X.; Liu, Y.; Liu, J.; Zhang, X.; Zou, J.; Fontes-Garfias, C.R.; Xia, H.; Swanson, K.A.; Cutler, M.; Cooper, D.; et al. Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat. Med. 2021, 27, 620–621. [Google Scholar] [CrossRef]
- Kupferschmidt, K. Fast-spreading U.K. virus variant raises alarms. Science 2021, 371, 9–10. [Google Scholar] [CrossRef]
- Vogel, C.; Bashton, M.; Kerrison, N.D.; Chothia, C.; Teichmann, S.A. Structure, function and evolution of multidomain proteins. Curr. Opin. Struct. Biol. 2004, 14, 208–216. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Pearson, W.R. Selecting the right similarity-scoring matrix. Curr. Protoc. Bioinform. 2013, 43, 3–5. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source 1 | Source 2 | Unique Sequences | Total Accessions |
|---|---|---|---|
| GISAID | NA | 47,908 | 55,708 |
| GENBANK | NA | 9398 | 11,196 |
| REFSEQ | NA | 1 | 1 |
| GISAID | GISAID | 2791 | 10,528 |
| GISAID | GENBANK | 5559 | 13,977 |
| GENBANK | GENBANK | 706 | 2504 |
| GISAID | REFSEQ | 1 | 43 |
| GENBANK | REFSEQ | 1 | 11 |
| Type | Total Count | Unique Count |
|---|---|---|
| Gene | 936,603 | 59,531 |
| Protein | 815,878 | 42,611 |
| Domain | 11,621,784 | 59,271 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beck, K.L.; Seabolt, E.; Agarwal, A.; Nayar, G.; Bianco, S.; Krishnareddy, H.; Ngo, T.A.; Kunitomi, M.; Mukherjee, V.; Kaufman, J.H. Semi-Supervised Pipeline for Autonomous Annotation of SARS-CoV-2 Genomes. Viruses 2021, 13, 2426. https://doi.org/10.3390/v13122426
Beck KL, Seabolt E, Agarwal A, Nayar G, Bianco S, Krishnareddy H, Ngo TA, Kunitomi M, Mukherjee V, Kaufman JH. Semi-Supervised Pipeline for Autonomous Annotation of SARS-CoV-2 Genomes. Viruses. 2021; 13(12):2426. https://doi.org/10.3390/v13122426
Chicago/Turabian StyleBeck, Kristen L., Edward Seabolt, Akshay Agarwal, Gowri Nayar, Simone Bianco, Harsha Krishnareddy, Timothy A. Ngo, Mark Kunitomi, Vandana Mukherjee, and James H. Kaufman. 2021. "Semi-Supervised Pipeline for Autonomous Annotation of SARS-CoV-2 Genomes" Viruses 13, no. 12: 2426. https://doi.org/10.3390/v13122426