
Development of a Kit for Rapid Immunochromatographic Detection of Sacbrood Virus Infecting Apis cerana (AcSBV) Based on Polyclonal and Monoclonal Antibodies Raised against Recombinant VP1 and VP2 Expressed in Escherichia coli



Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of VP1 and VP2 DNA and Construction of Gene Expression Vector
2.2. Inducible Expression and Purification of Recombinant Protein and SDS-PAGE
2.3. Immunization and Production of Polyclonal Antibody
2.4. ELISA, SDS-PAGE, and Western Blot Analysis
2.5. Preparation of Splenocyte and Hybridoma Production and Selection
2.6. Generation of Ascities Fluids
2.7. Purification of mAbs, SDS-PAGE, and Western Blot Analysis
2.8. Preparation of Colloidal Gold and Colloidal Gold−pAb Conjugate
2.9. Preparation of IC Strip Device and Procedure for the Test
2.10. Real-Time RT-PCR
2.11. Detection Limit (Sensitivity) of the IC strips and Validation (Specificity) of the IC Strips Using Field Samples
3. Results
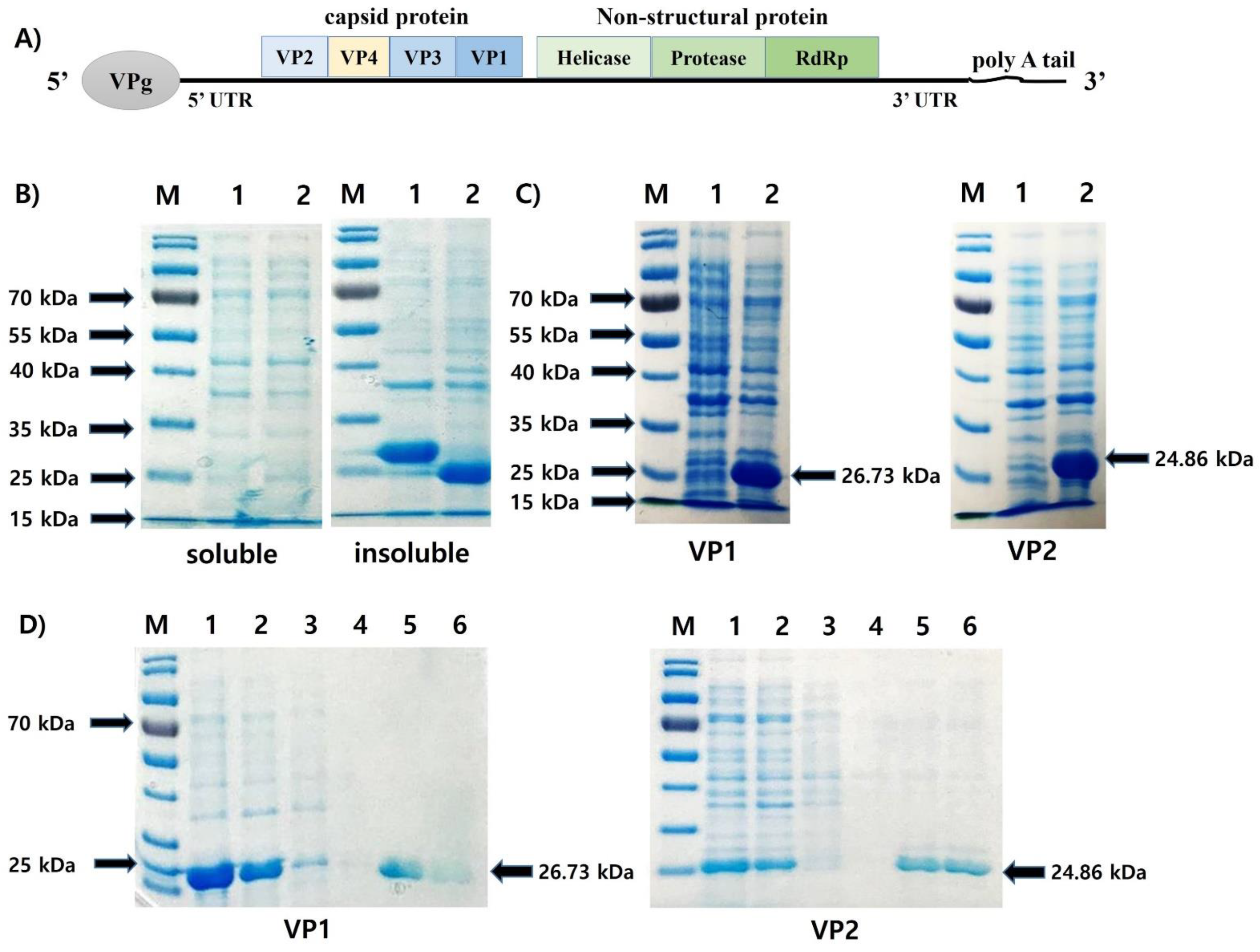
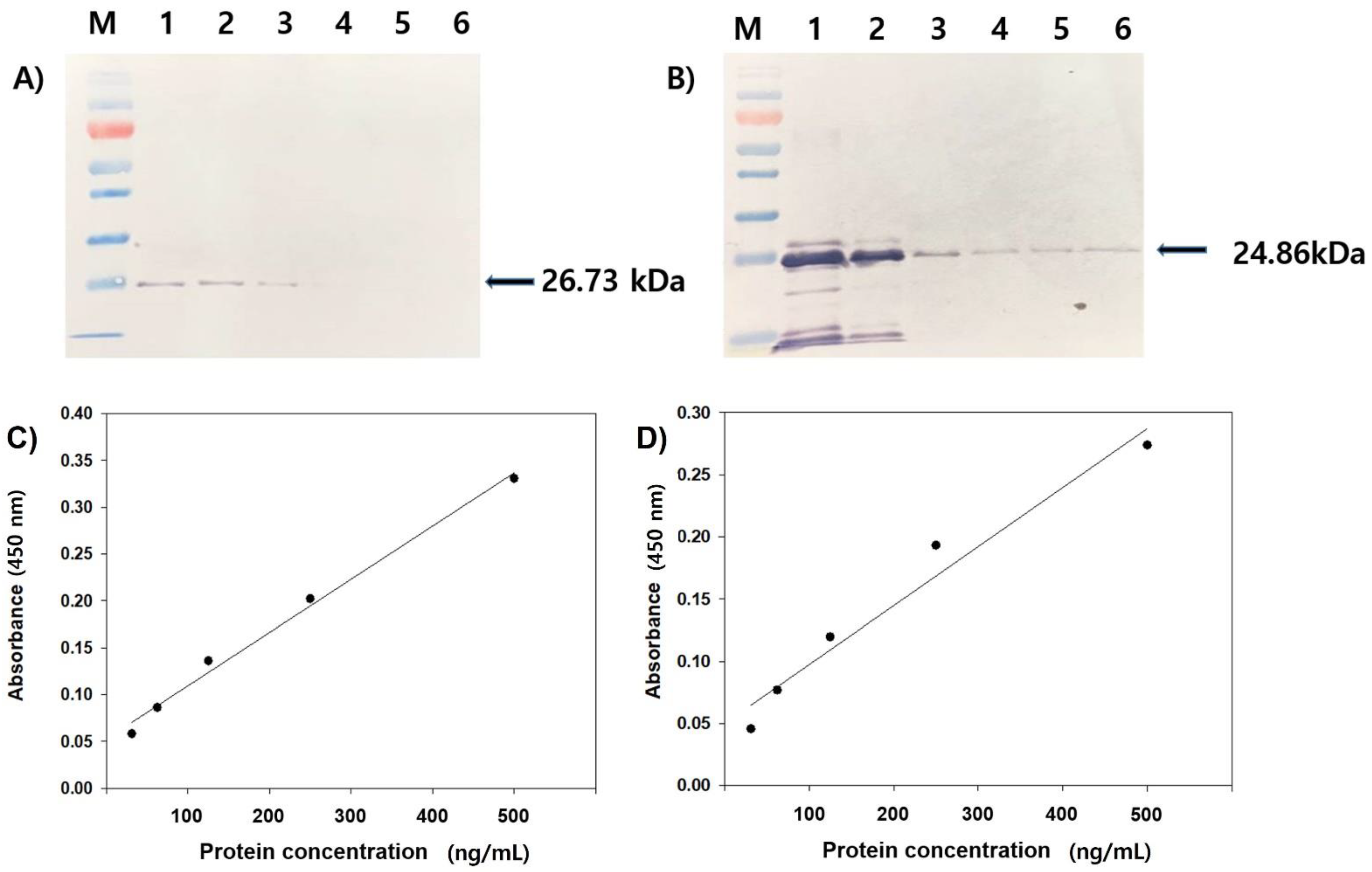
3.1. Expression and Purification of the Recombinant Protein VP1 (rVP1) and VP2 (rVP2)
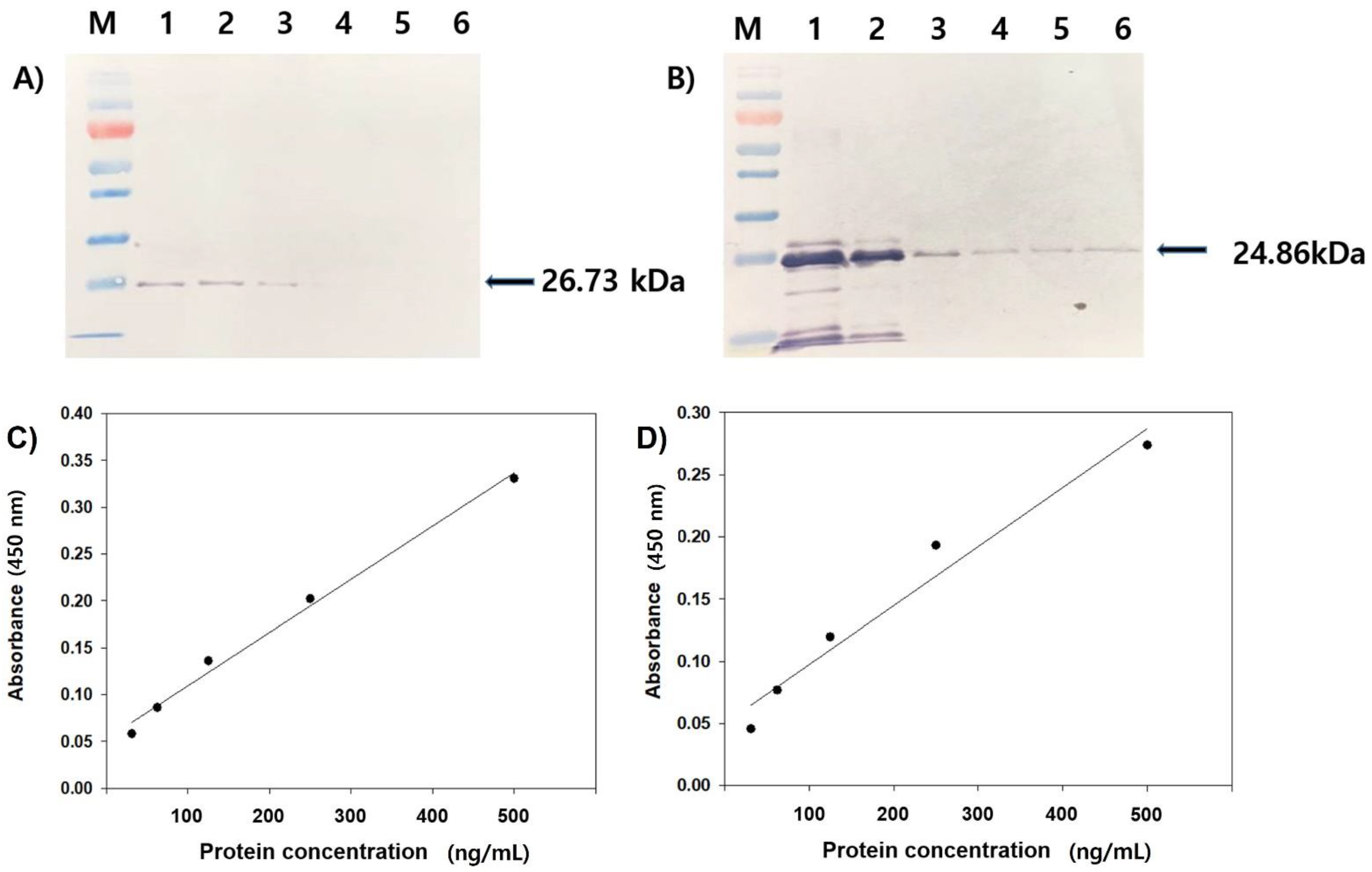
3.2. Reactivity Test of Antiserum by Western Blot and ELISA
3.3. The Specificity and Efficiency of Polyclonal Antibodies
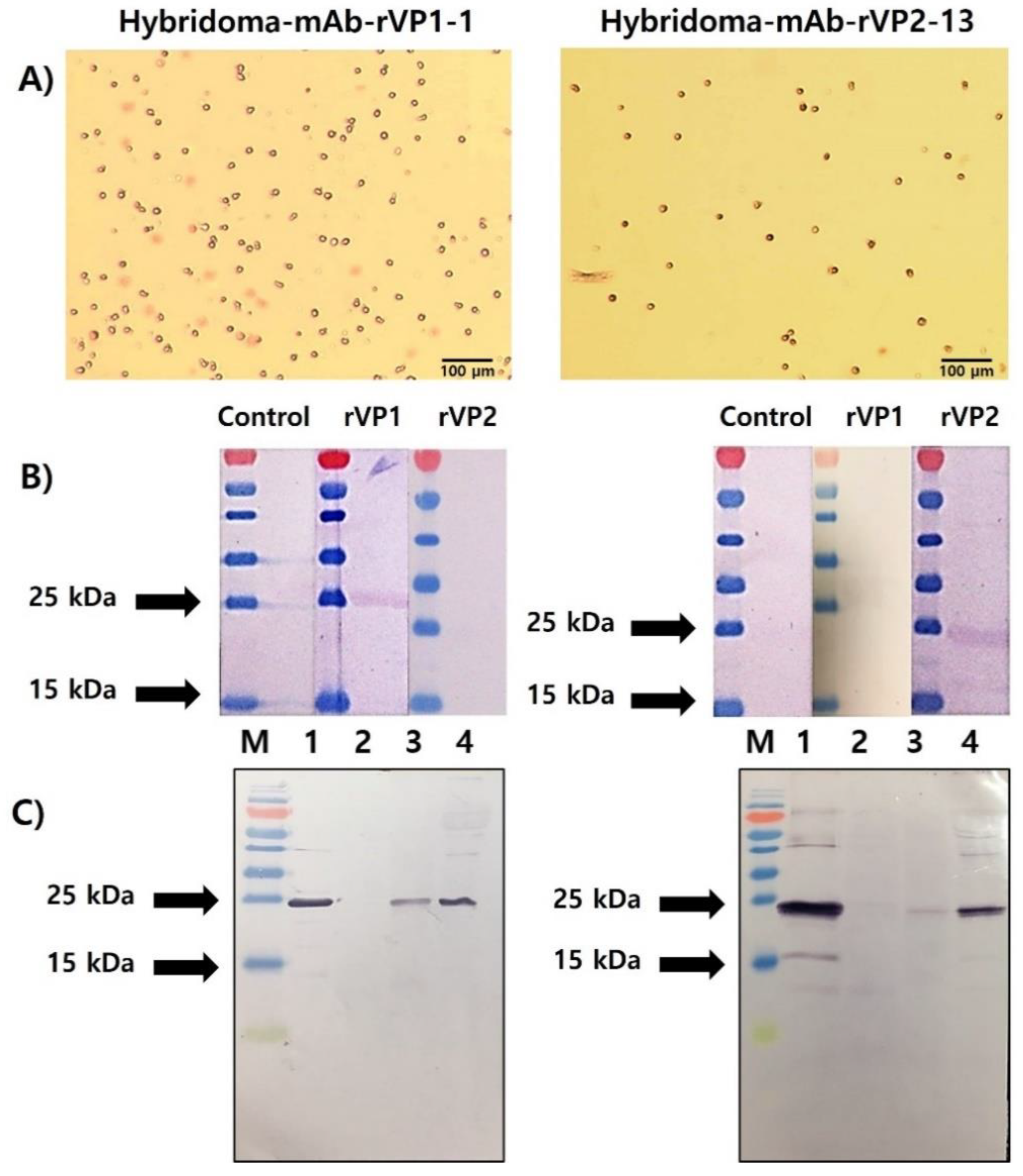
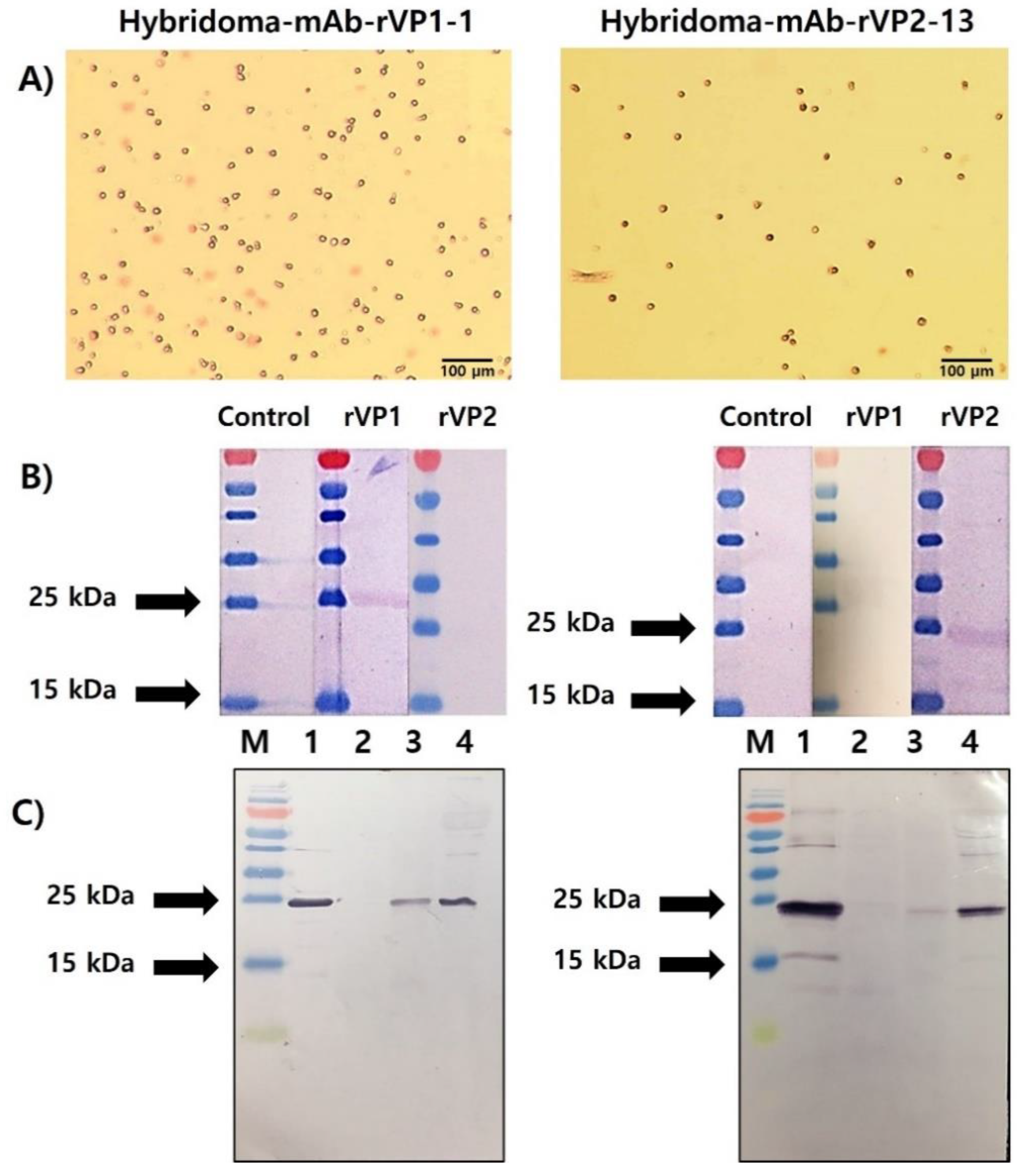
3.4. Selection of rVP1- and rVP2-Specific mAbs and Their Reactivity to Recombinant Protein and Honeybee Samples
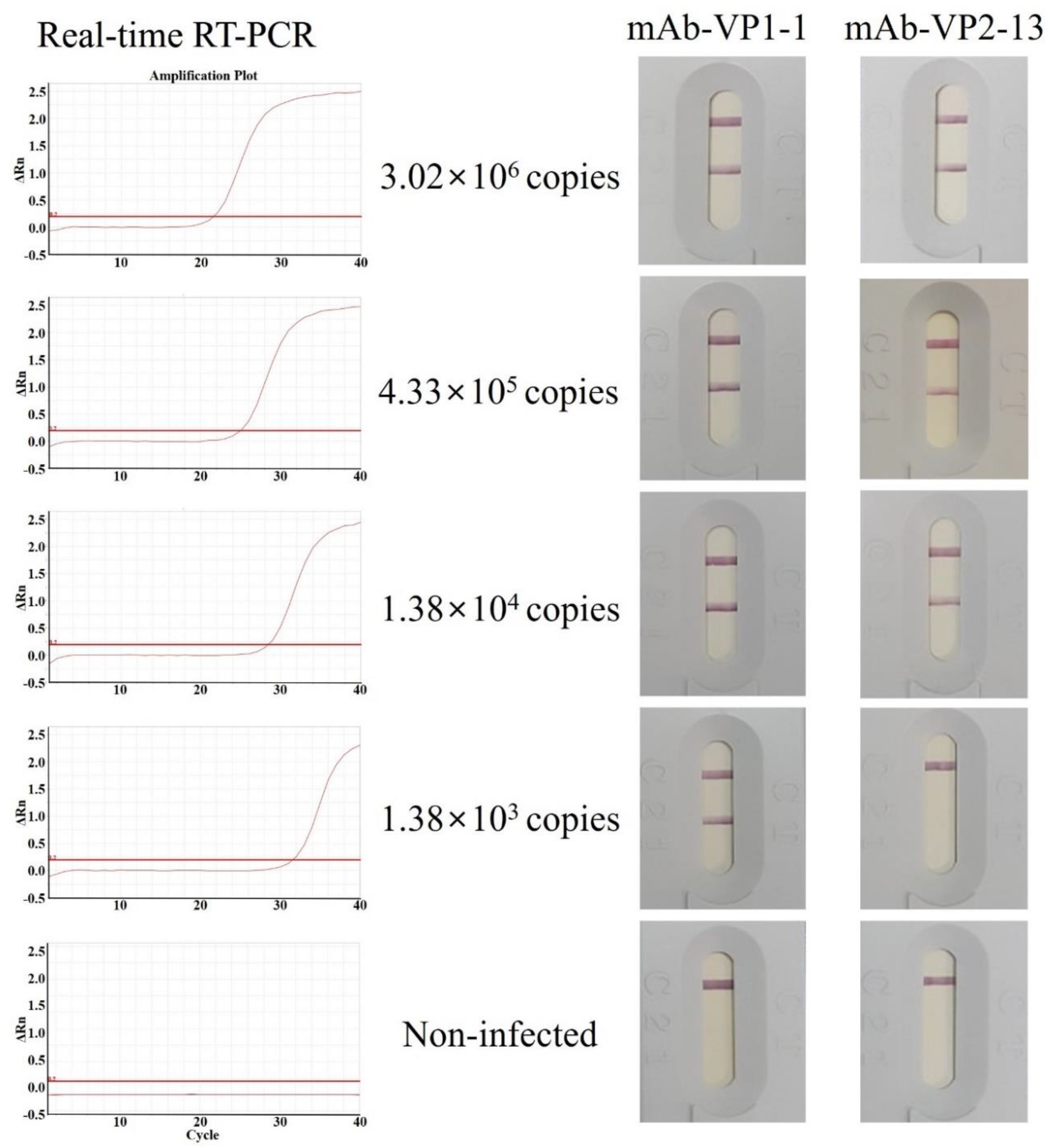
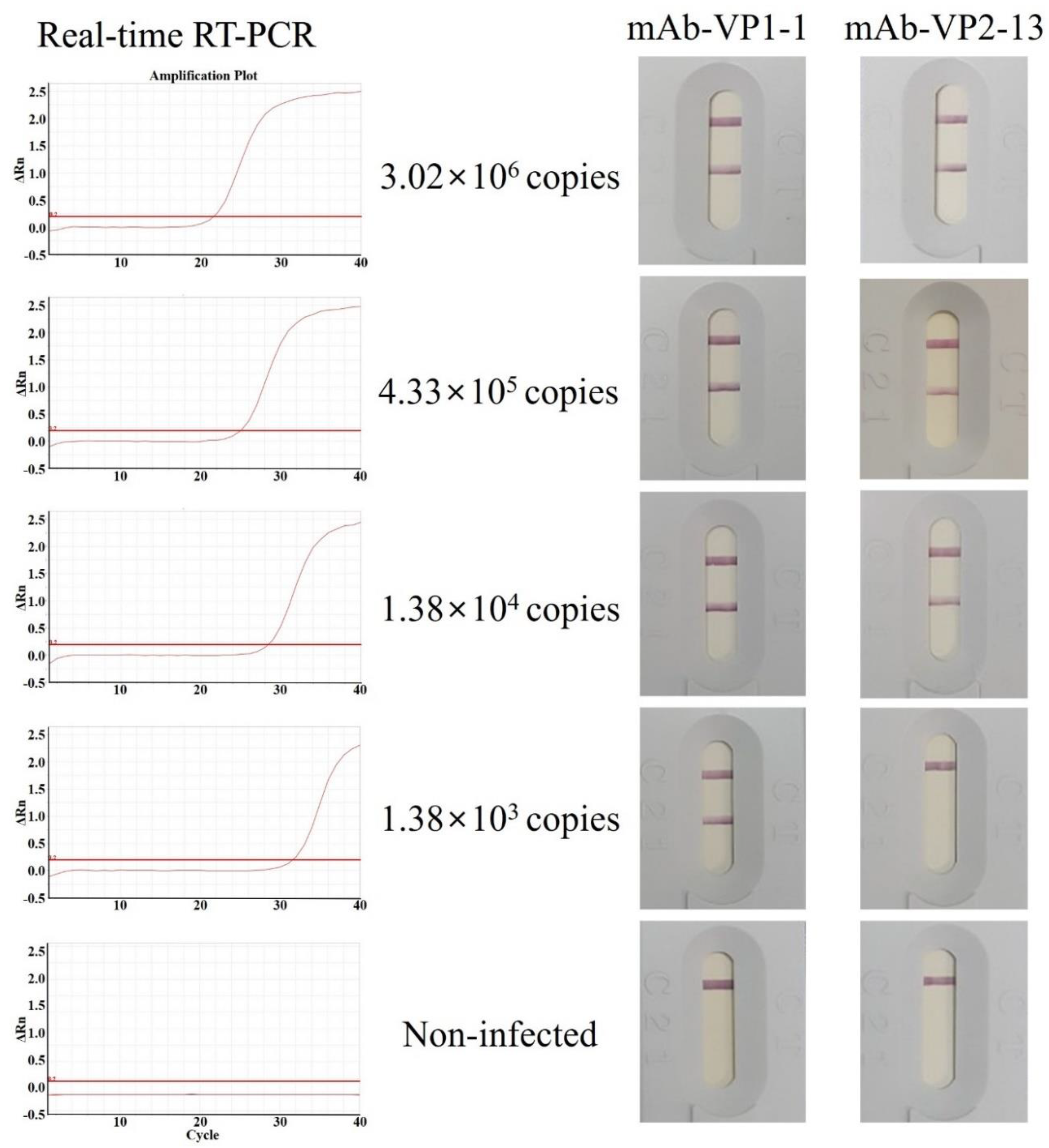
3.5. Antigen Detection Limit (Sensitivity) of the IC Strips Using pAb as a Detector Antibody and mAb as a Capture Antibody
3.6. Validation (Specificity) of the IC Strips Using pAb as a Detector Antibody and mAb as a Capture Antibody
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ai, H.; Yan, X.; Han, R. Occurrence and prevalence of seven bee viruses in Apis mellifera and Apis cerana apiaries in China. J. Invertebr. Pathol. 2012, 109, 160–164. [Google Scholar] [CrossRef]
- Forsgren, E.; Wei, S.; Guiling, D.; Zhiguang, L.; Tran, T.V.; Tang, P.T.; Truong, T.A.; Dinh, T.Q.; Fries, I. Preliminary observation on possible pathogen spill-over from Apis mellifera to Apis cerana. Apidologie 2015, 46, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Yanez, O.; Zheng, H.Q.; Su, X.L.; Hu, F.L.; Neumann, P.; Dietemann, V. Potential for virus transfer between the honey bees Apis mellifera and A. cerana. J. Apic. Res. 2015, 54, 179–191. [Google Scholar] [CrossRef]
- Thu, H.T.; Lien, N.T.K.; Linh, M.T.; Le, T.H.; Hoa, N.T.; Thai, P.H.; Reddy, K.E.; Yoo, M.S.; Kim, Y.-H.; Cho, Y.S.; et al. Prevalence of bee viruses among Apis cerana population in Vietnam. J. Apic. Res. 2016, 55, 379–385. [Google Scholar] [CrossRef]
- Li, Y.; Zeng, Z.J.; Wang, Z.L. Phylogenetic analysis of the honey bee scabrood virus. J. Apic. Sci. 2016, 60, 31–38. [Google Scholar]
- Thai, P.H.; Huyen, N.T.; Toan, T.V.; Jung, C. Apis cerana beekeeping and sacbrood disease management in Vietnam: Review. J. Apic. 2018, 33, 269–275. [Google Scholar] [CrossRef]
- Ko, C.-Y.; Chiang, Z.-L.; Liao, R.-J.; Chang, Z.-T.; Chang, J.-C.; Kuo, T.-Y.; Chen, Y.-W.; Nai, Y.-S. Dynamics of Apis cerana sacbrood virus (AcSBV) prevalence in Apis cerana (Hymenoptera: Apidae) in northern Taiwan and demonstration of its infection in Apis mellifera (Hymenoptera: Apidae). J. Econ. Entomol. 2019, 112, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. Managed honeybee colonies and honey production in China grew during the last five decades. J. Apic. Sci. 2011, 55, 77–85. [Google Scholar]
- Choi, Y.S.; Lee, M.Y.; Hong, I.P.; Kim, N.S.; Kim, H.K.; Lee, K.G.; Lee, M.L. Occurrence of sacbrood virus in Korean apiaries from Apis cerana (Hymenoptera: Apidae). J. Apic. 2010, 25, 187–191. [Google Scholar]
- Choe, S.E.; Nguyen, T.T.D.; Hyun, B.H.; Noh, J.H.; Lee, H.S.; Kang, S.W. Genetic and phylogenetic analysis of South Korean sacbrood virus isolates from infected honey bees (Apis cerana). Vet. Microbiol. 2012, 157, 32–40. [Google Scholar] [CrossRef]
- Theisen-Jones, H.; Bienefeld, K. The Asian honey bee (Apis cerana) is significantly in decline. Bee World 2016, 93, 90–97. [Google Scholar] [CrossRef]
- Vung, N.N.; Lee, M.-L.; Lee, M.-Y.; Kim, H.K.; Kang, E.J.; Kim, J.E.; Choi, Y.-S. Breeding and selection for resistance to sacbrood virus for Apis cerana. J. Apic. 2017, 32, 345–352. [Google Scholar] [CrossRef]
- Procházková, M.; Füzik, T.; Škubnik, K.; Moravcová, J.; Ubiparip, Z.; Přidal, A.; Plevka, P. Virion structure and genome delivery mechanism of sacbrood honeybee virus. Proc. Natl. Acad. Sci. USA 2018, 115, 7759–7764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.M.K.; Anderson, D.L. A novel strain of sacbrood virus of interest to world apiculture. J. Invertebr. Pathol. 2014, 118, 71–74. [Google Scholar] [CrossRef]
- Gong, H.-R.; Chen, X.-X.; Chen, Y.P.; Hu, F.-L.; Zhang, J.-L.; Lin, Z.-G.; Yu, J.-W.; Zheng, H.-Q. Evidence of Aphis cerana Sacbrrod virus infection in Apis mellifera. Appl. Environ. Microbiol. 2016, 82, 2256–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aruna, R.; Srinivasan, M.R.; Balasubramanian, V.; Selvarajan, R. Complete genome sequence of sacbrood virus isolated from Asiatic honey bee Apis cerana indica in India. Virus Dis. 2018, 28, 453–460. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, G.; Huang, X.; Han, R. Proteomic analysis of Apis cerana and Apis mellifera larvae fed with heterospecific royal jelly and by CSBV challenge. PLoS ONE 2014, 9, e102663. [Google Scholar] [CrossRef] [Green Version]
- Reddy, K.E.; Thu, H.T.; Yoo, M.S.; Ramya, M.; Reddy, B.A.; Lien, N.T.K.; Trang, N.T.P.; Duong, B.T.T.; Lee, H.-J.; Kang, S.-W.; et al. Comparative genomic analysis for genetic variation in Sacbrood virus of Apis cerana and Apis mellifera honeybees from different regions of Vietnam. J. Insect Sci. 2017, 17, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, T.; Evans, J.D.; Rose, R.; Zhao, Y.; Li, Z.; Li, J.; Huang, S.; Heerman, M.; Rodríguez-García, C.; et al. The phylogeny and pathogenesis of Sacbrood virus (SBV) infection in European honey bees, Apis mellifera. Viruses 2019, 11, 61. [Google Scholar] [CrossRef] [Green Version]
- Bostina, M. Monoclonal antibodies point to Achilles’ heel in picornavirus capsid. PLoS Biol. 2019, 17, e3000232. [Google Scholar] [CrossRef] [Green Version]
- Oh, S. Detection of sacbrood virus infecting Apis cerana (AcSBV) using polyclonal antibody raised against recombinant VP3 capsid protein expressed in Escherichia coli. J. Apic. 2019, 34, 149–156. [Google Scholar] [CrossRef]
- Lee, J.-G.; Nguyen, P.V.; Soo, B.J.; Lim, H.-Y.; Yoon, B.-S. Detection of capsid protein of Sacbrood virus in honeybee using monoclonal antibodies. J. Apic. 2013, 28, 113–120. [Google Scholar]
- Yan, X.; Chen, J.-H.; Han, R.-C. Detection of Chinese sacbrood virus (CSBV) in Apis cerana by RT-PCR method. Sociobiology 2009, 53, 687–694. [Google Scholar]
- Yang, J.-L.; Yang, R.; Shen, K.-F.; Peng, X.-W.; Xion, T.; Liu, Z.-H. Rapid detection of sacbrood virus (SBV) by one-step reverse transcription loop-mediated isothermal amplification assay. Virol. J. 2012, 9, 47. [Google Scholar]
- Kukielka, D.; Sanchez-Vizcaino, J.M. One-step real-time quantitative PCR for the detection and field study of Sacbrood honeybee and Acute bee paralysis viruses. J. Virol. Methods 2009, 161, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Thi, K.C.N.; Yoo, M.S.; Kang, M.H.; Han, S.H.; Yun, C.H.; Yoon, B.-S. Development of real time PCR assay for the detection of Sacbrood virus in honeybee (Apis mellifera L.). J. Apic. 2009, 24, 15–21. [Google Scholar]
- Rana, R.; Rana, B.S.; Kaushal, N.; Kumar, D.; Kaundal, P.; Khan, M.A.; Gwande, S.J.; Sharma, H.K. Identification of sacbrood virus disease in honeybee, Apis mellifera L. by using ELISA and RT-PCR techniques. Indian J. Biotechnol. 2011, 10, 274–284. [Google Scholar]
- Yoo, M.-S.; Thi, K.C.N.; Van Nguyen, P.; Han, S.-H.; Kwon, S.-H.; Yoon, B.S. Rapid detection of sacbrood virus in honeybee using ultra-rapid real-time polymerase chain reaction. J. Virol. Methods 2012, 179, 195–200. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, X.; Xu, Z.F.; Han, R.C.; Chen, J.H. Differential gene transcription in honeybee (Apis cerana) larvae challenged by Chinese sacbrood virus (CSBV). Sociobiology 2013, 60, 413–420. [Google Scholar] [CrossRef] [Green Version]
- McMenamin, A.J.; Flenniken, M.L. Recently identified bee viruses and their impact on bee pollinators. Curr. Opin. Insect Sci. 2018, 26, 120–129. [Google Scholar] [CrossRef]
- Jain, J.; Okabayashi, T.; Kaur, N.; Nakayama, E.; Shioda, T.; Gaind, R.; Kurosu, T.; Sunil, S. Evaluation of an immunochromatography rapid diagnosis kit for detection of chikungunya virus antigen in India, a dengue-endemic country. Virol. J. 2018, 15, 84. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Singh, M.; Upmanyu, V.; Chander, V.; Verma, S.; Chakrovarty, S.; Sharma, G.K.; Dhanze, H.; Singh, P.; Shrivastava, S.; et al. Development and evaluation of a gold nanoparticle-based immunochromatographic strip test for the detection of canine parvovirus. Arch. Virol. 2018, 163, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-T.; Bui, C.T.; Kim, D.T.H.; Nguyen, A.V.T.; Trinh, T.T.T.; Yeo, S.-J. Clinical evaluation of rapid flurescent diagnostic immunochromatographic test for influenza A virus (H1N1). Sci. Rep. 2018, 8, 13468. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wen, Y.; An, T.; Duan, G.; Sun, M.; Ge, J.; Li, X.; Yang, K.; Cai, X. Development of an immunochromatographic strip for rapid detection of canine adenovirus. Front. Microbiol. 2019, 10, 2882. [Google Scholar] [CrossRef]
- Yale, G.; Ginson, A.D.; Mani, R.S.; Harsha, P.K.; Costa, N.C.; Corfmat, J.; Otter, I.; Otter, N.; Handel, I.G.; Bronsvoort, B.M.; et al. Evaluation of an immunochromatographic assay as a canine rabies surveillance tool in Goa, India. Viruses 2019, 11, 649. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-L.; Wang, L.; Hasi, C.-L.; Wang, Y.-P.; Khan, A.; Ren, B.-Z.; Liu, Z.-Z.; Hou, S.-L.; Yang, L.-H.; Zhang, L.-Y.; et al. A rapid colloidal gold immunochromatographic assay for the diagnosis of coronavirus disease 2019. Chin. Med. J. 2020, 133, 1986–1988. [Google Scholar] [CrossRef]
- Janwan, P.; Intapan, P.M.; Sadaow, L.; Rodpai, R.; Yamasaki, H.; Boonroumkaew, P.; Sanpool, O.; Thanchomnang, T.; Sadee, P.; Maleewong, W. Development of immunochromatographic test kit for rapid detection of specific IgG4 antibody in whole-blood samples for diagnosis of human gnathostomiasis. Diagnostics 2021, 11, 862. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.-Y.; Wang, C.-R.; Xie, C.-M.; Wang, X.-G.; Liang, R.-L.; Xu, W.-W. Rapid and sensitive lateral flow immunoassay method for procalcitonin (PCT) based on time-resolved immunochromatography. Sensor 2017, 17, 480. [Google Scholar] [CrossRef] [Green Version]
- Ge, W.; Suryoprabowo, S.; Wu, X.; Zheng, Q.; Kuang, H. Rapid immnochromatographic test strip detection of mabuterol and its cross-reactivity with mapenterol. Food Agric. Immunol. 2018, 29, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, L.; Xu, L.; Guo, X.; Yang, H.; Zhuang, L.; Li, Y.; Wang, Z.; Gu, B. Rapid and sensitive detection of Shigella flexneri using fluorescent microspheres as label for immunochromatographic test strip. Ann. Transl. Med. 2019, 7, 565. [Google Scholar] [CrossRef]
- Moongkarndi, P.; Rodpai, E.; Kanarat, S. Evaluation of an immunochromatographic assay for rapid detection of Salmonella enterica serovars Typhimurium and Enteritidis. J. Vet. Diagn. Investig. 2011, 23, 797–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelista, V.O.; Pelegrini, P.B.; Mulinari, F.; de Moura, R.; Cardoso, L.P.V.; Bührer-Sékula, S.; Miller, R.; Pinto, E.R.; Grossi-de-Sá, M.F. A novel immunochromatographic strip test for rapid detection of Cry1Ac and Cry8Ka5 proteins in genetically modified crops. Anal. Methods 2015, 7, 9331. [Google Scholar]
- Isanga, J.; Tochi, B.N.; Mukunzi, D.; Chen, Y.; Liu, L.; Kuang, H.; Xu, C. Development of a specific monoclonal antibody assay and a rapid testing strip for the detection of apramycin residues in food samples. Food Agric. Immunol. 2017, 28, 49–66. [Google Scholar] [CrossRef] [Green Version]
- Qriouet, Z.; Cherrah, Y.; Sefrioui, H.; Qmichou, Z. Monoclonal antibodies application in lateral flow immunochromatographic assays for drugs of abuse detection. Molecules 2021, 26, 1058. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, W.; Wang, P.; Su, X. A paper-based competitive lateral flow immunoassay for multi β-agonist residues by using a single monoclonal antibody labelled with red fluorescent nanoparticles. Microchim. Acta 2018, 185, 191. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Song, S.; Zheng, Q.; Luo, P.; Wu, X.; Kuang, H. Development of a sandwich ELISA and immunochromatographic strip for the detection of shrimp tropomyosin. Food Agric. Immunol. 2019, 30, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Liu, I.-L.; Lin, Y.-C.; Lin, Y.-C.; Jian, C.-Z.; Cheng, I.C.; Chen, H.-W. A novel immunochromatographic strip for antigen detection of avian infectious bronchitis virus. Int. J. Mol. Sci. 2019, 20, 2216. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Kim, J.; Shin, S.G.; Hwang, S. Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. J. Biotechnol. 2006, 123, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Fei, D.; Zhang, H.; Diao, Q.; Jiang, L.; Wang, Q.; Zhong, Y.; Fan, Z.; Ma, M. Codon optimization, expression in Escherichia coli, and immunogenicity of recombinant Chinese sacbrood virus (CSBV) structural proteins VP1, VP2, VP3. PLoS ONE 2015, 10, e0128486. [Google Scholar] [CrossRef]
- Kantere, M.C.; Athanasiou, L.V.; Spyrou, V.; Kyriakis, C.S.; Kontos, V.; Chatzopoulos, D.C.; Tsokana, C.N.; Billinis, C. Diagnostic performance of a rapid in clinic test for the detection of Canine Parvovirus under different storage conditions and vaccination status. J. Virol. Methods 2015, 215, 52–55. [Google Scholar] [CrossRef]
- Cepica, A.; Yason, C.; Ralling, G. The use of ELISA for detection of the antibody induced conformational change in a viral protein and its intermolecular spread. J. Virol. Methods 1990, 28, 1–14. [Google Scholar] [CrossRef]
- Wong, Y.-P.; Othman, S.; Lau, Y.-L.; Radu, S.; Chee, H.-Y. Loop-mediated isothermal amplification (LAMP): A versatile technique for detection of micro-organism. J. Appl. Microbiol. 2018, 124, 626–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Real-Time RT-PCR | Rapid Strip Test | ||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive 1 | 50 | 0 | 50 |
| Negative | 0 | 50 | 50 |
| Total | 50 | 50 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.H.; Oh, T.-K.; Oh, S.; Kim, S.; Noh, H.B.; Vinod, N.; Lee, J.Y.; Moon, E.S.; Choi, C.W. Development of a Kit for Rapid Immunochromatographic Detection of Sacbrood Virus Infecting Apis cerana (AcSBV) Based on Polyclonal and Monoclonal Antibodies Raised against Recombinant VP1 and VP2 Expressed in Escherichia coli. Viruses 2021, 13, 2439. https://doi.org/10.3390/v13122439
Lee SH, Oh T-K, Oh S, Kim S, Noh HB, Vinod N, Lee JY, Moon ES, Choi CW. Development of a Kit for Rapid Immunochromatographic Detection of Sacbrood Virus Infecting Apis cerana (AcSBV) Based on Polyclonal and Monoclonal Antibodies Raised against Recombinant VP1 and VP2 Expressed in Escherichia coli. Viruses. 2021; 13(12):2439. https://doi.org/10.3390/v13122439
Chicago/Turabian StyleLee, Song Hee, Tae-Kyun Oh, Sung Oh, Seongdae Kim, Han Byul Noh, Nagarajan Vinod, Ji Yoon Lee, Eun Sun Moon, and Chang Won Choi. 2021. "Development of a Kit for Rapid Immunochromatographic Detection of Sacbrood Virus Infecting Apis cerana (AcSBV) Based on Polyclonal and Monoclonal Antibodies Raised against Recombinant VP1 and VP2 Expressed in Escherichia coli" Viruses 13, no. 12: 2439. https://doi.org/10.3390/v13122439
APA StyleLee, S. H., Oh, T.-K., Oh, S., Kim, S., Noh, H. B., Vinod, N., Lee, J. Y., Moon, E. S., & Choi, C. W. (2021). Development of a Kit for Rapid Immunochromatographic Detection of Sacbrood Virus Infecting Apis cerana (AcSBV) Based on Polyclonal and Monoclonal Antibodies Raised against Recombinant VP1 and VP2 Expressed in Escherichia coli. Viruses, 13(12), 2439. https://doi.org/10.3390/v13122439