1. Introduction
COVID-19, which is caused by SARS-CoV-2, poses a great threat to public health and the global economy [
1]. Patients with COVID-19 generally raise antibodies against SARS-CoV-2 following infection, and the antibody level is positively correlated to the severity of disease [
2]. Although it was believed that antibodies, particularly neutralizing antibodies played a pivotal role in inhibiting SARS-CoV-2 replication in patients, it has also been argued that they may also exacerbate COVID-19 through antibody-dependent enhancement (ADE) [
3].
ADE has been documented to other viruses including dengue virus (DENV), respiratory syncytial virus (RSV), measles virus, and feline infectious peritonitis virus (FIPV) [
4,
5,
6,
7]. In these cases, ADE increased the severity of diseases either by enhanced antibody-mediated virus uptake into Fc gamma receptor (FcγR)-expressing phagocytic cells, leading to increased viral infection and replication (type I ADE), or by excessive antibody Fc-mediated effector functions or immune complex formation causing enhanced inflammation and immunopathology (type II ADE) [
3]. The type I ADE normally requires viral productive infection of target immune cells, for example, macrophages or monocytes in the case of FIPV in cats [
6]. The type II ADE can occur without the need of viral productive infection albeit causing dysregulated immune activation of target cells [
3]. In humans, FcγR is expressed broadly among the various leukocyte subsets including macrophages, monocytes, B cells, and others, and modulates downstream immune responses upon binding to the Fc domain of an IgG antibody [
8]. Subsequently, these leukocytes could be potential targets of virus induced ADE.
ADE has been well characterized in cats infected with FIPV, a feline betacoronavirus. Experimental infection of FIPV antibody positive cats resulted in more severe diseases, regardless of naturally acquired or vaccine acquired antibodies [
6]. This ADE is also closely related to more viral replication and more inflammatory responses in viral target cells including monocytes and macrophages in an aminopeptidase N (APN)-independent, FcγR-dependent manner [
9,
10]. Likewise, it is not unexpected that SARS-CoV-1 and MERS-CoV could induce ADE in FcγR-expression Raji B cells or HEK293T cells in vitro [
11,
12]. It is also postulated that SARS-CoV-2 may also induce ADE in some leukocytes.
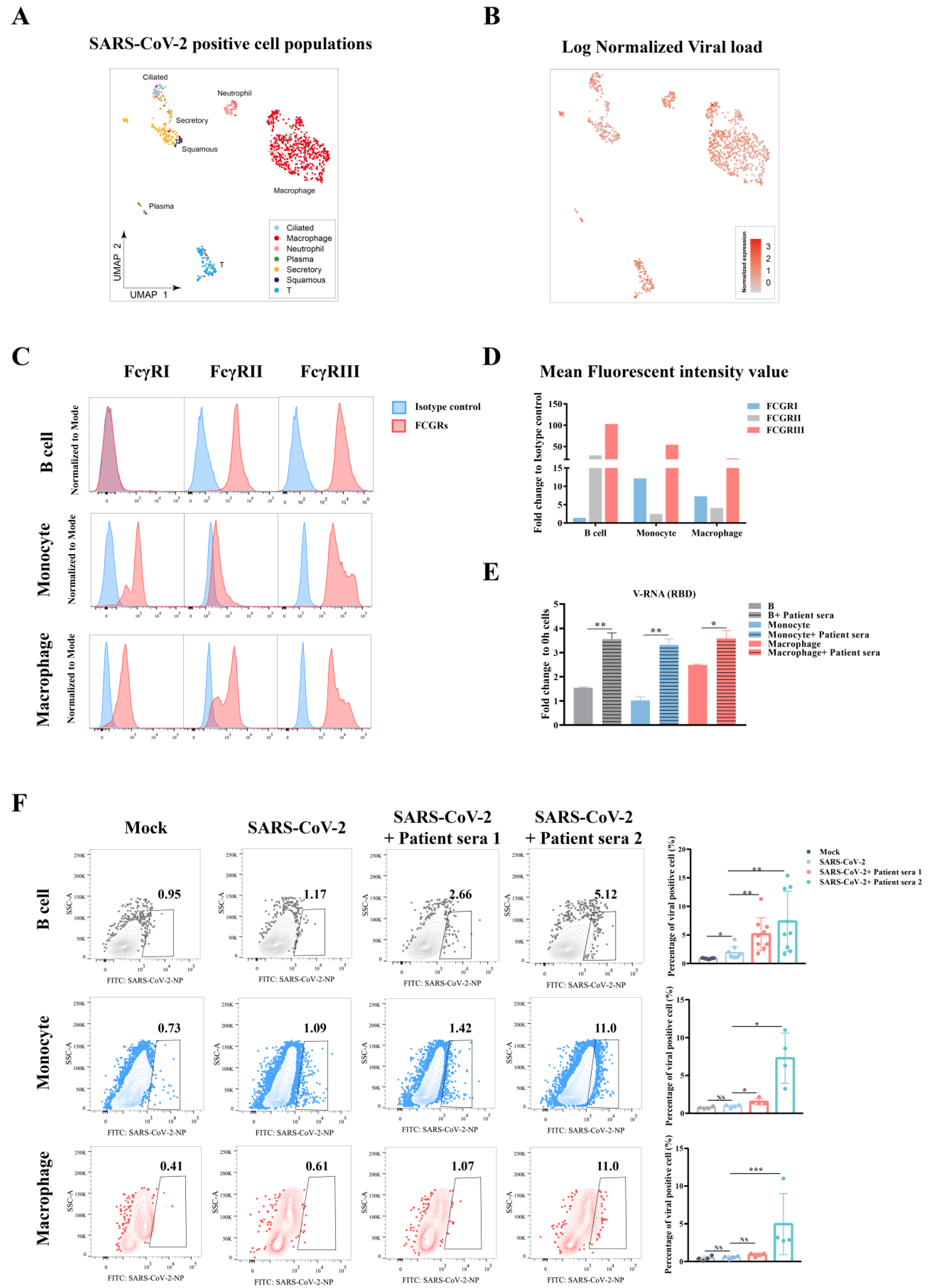
Severe patients with COVID-19 normally generated high levels of SARS-CoV-2 antibodies, and the antibody titer was positively related to the severity of disease, which showed less neutralization potency [
13]. This phenomenon suggests that ADE induced by non-neutralizing antibodies could play an important role in the pathogenesis of SARS-CoV-2 in patients. Moreover, it was argued that immune cells, which normally express low or no ACE2 receptors, could also be infected. Viral RNA positive or antigen positive have been reported in a few single-cell analysis of patient BALF or in postmortem analysis of COVID-19 patients. Whether this positivity is caused by ADE or a direct ACE2-independent infection is still unknown.
In this study, we tested SARS-CoV-2 induced ADE in vitro using convalescent COVID-19 patient serum samples in a list of FcγR-expression leukocytes. Our results contributed to the understanding of the pathogenesis of SARS-CoV-2 in the context of viral treatment and control.
2. Materials and Methods
2.1. Primary Immune Cells Preparation and SARS-CoV-2 Infection
This study obtained informed consent from all subjects. The blood samples from healthy donors were treated with Ficoll-Paque Plus (17144002; Cytiva, Danaher Corporation, WDC, USA). Briefly, 3 mL of Ficoll was added to a centrifuge tube, then 4 mL of whole blood was gently added before being centrifuged at 400× g for 30 min at 20 °C. The PBMC layer was carefully taken with a pipette. Magnetic beads conjugated with different cell markers were used to sort immune cells: CD19 microbeads (130-050-301; Miltenyi Biotec--, Bergisch Gladbach, Germany) for B cells, CD14 microbeads for monocytes (130-050-201; Miltenyi Biotec, --Bergisch Gladbach, Germany), and CD11b-beads for macrophages (130-049-601; Miltenyi Biotec, --Bergisch Gladbach, Germany). Primary immune cells after sorting were cultured in Roswell Park Memorial Institute 1640 culture medium (RPMI1640, C22400500BT; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, 10099141; Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA ).
For infection, primary B cells, monocytes, and macrophages were seeded into 24-well plates or 48-well plates at a density of 1 × 106 cells/mL. Cells were infected by SARS-CoV-2 at a moi of 0.1. 0 h samples were harvested once mixed cells with virus. Cells were washed three times using RPMI1640 and harvested for qRT-PCR or flow cytometry detection. Remaining cells were cultured at 37 °C supplied with 5% CO2 for 24 h or 48 h before collecting for further analysis.
For detection of antibody-dependent enhancement (ADE), virus (moi = 0.1) was incubated with equal-volume convalescent sera from COVID-19 patients (for no-sera group, virus were incubated with equal-volume RPMI1640) at 37 °C for 30 min. Mixture were added to primary cells and samples were harvested at 0 h, 24 h, or 48 h post infection.
2.2. Cell Lines and Virus Culture
Primary B cell, monocyte, macrophage, and Raji in RPMI-1640 (C22400500BT; Thermo Fisher Scientific, Waltham, MA, USA) + 10% FBS (10099141; Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA), or Vero E6 and Caco-2 in DMEM + 10% FBS (Gibco, C 11995500BT) were cultured at 37 °C in a humidified atmosphere of 5% CO2. All cell lines were tested free of mycoplasma contamination and applied to species identification and authenticated by microscopic morphologic evaluation. None of the cell lines was on the list of commonly misidentified cell lines (by ICLAC). The SARS-CoV-2 isolate WIV04 (GISAID accession number EPI_ISL_402124) was used in this study. WIV04 was isolated from Huh7 cells from the original sample and was passaged in Caco-2 cells. Viral titer (TCID50/mL) was determined in Vero E6 cells.
2.3. Proteins and Antibodies for SARS-CoV-2
NP and predicted RBD of SARS-CoV-2 strain WIV04 were inserted into pCAGGS vector with an N-terminal S-tag. Constructed plasmids were transiently transfected into HEK293T-17 cells. The supernatant collected for protein purification was purified using S-tag resin and the purity and yield was tested using anti-S-tag mAb (generated in-house). Rabbits were immunized with purified NP proteins or RBD protein three times at a dose of 700 ng/each at a two week interval. Rabbit serum was collected at 10 days after the final injection. Antibody titer was determined in an ELISA using purified NP protein or RBD protein as the detection antigen.
2.4. B Cell Line Infection
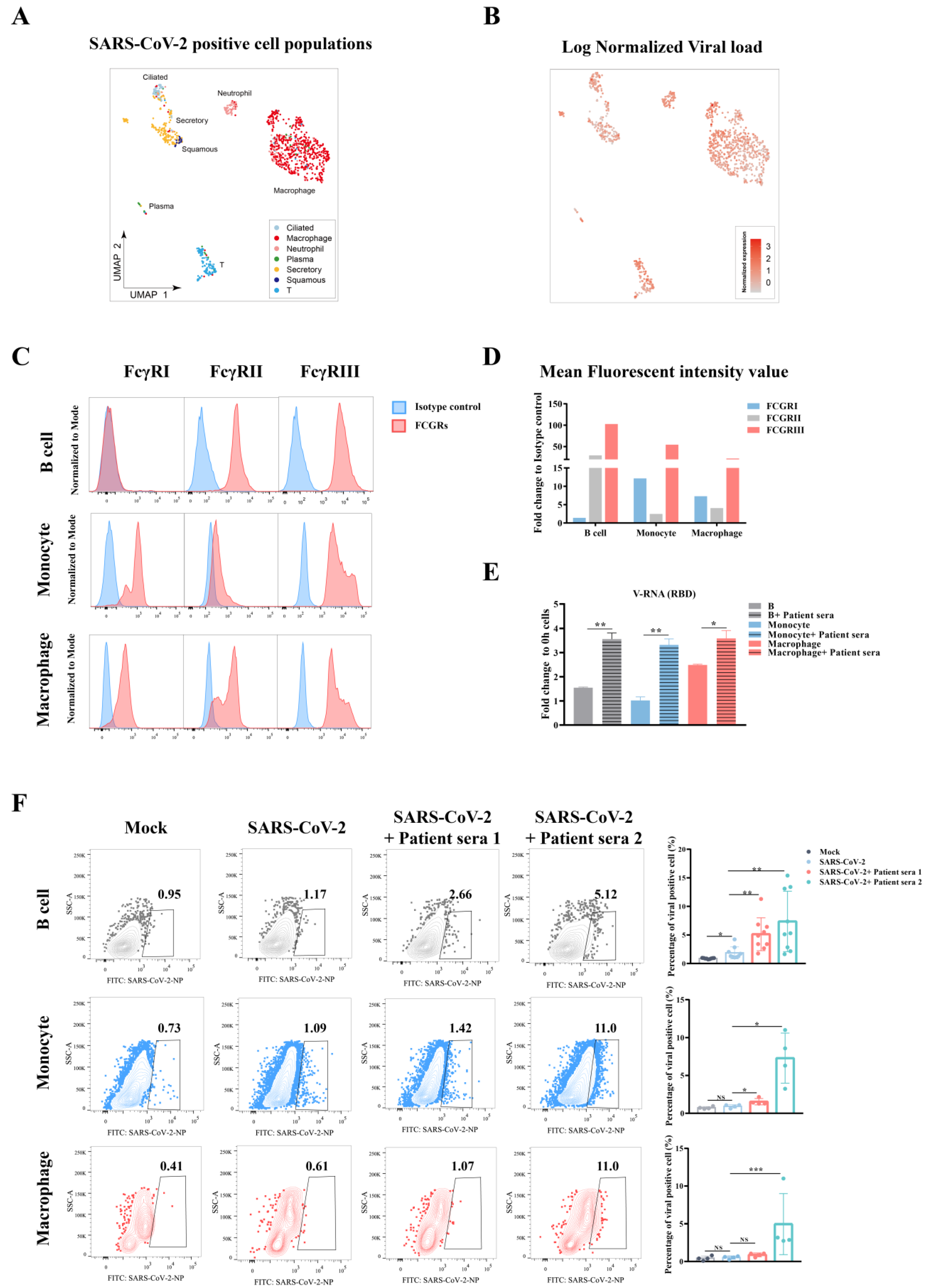
Raji B cells were infected with SARS-CoV-2 at a moi of 0.01, 0.1, or 0.2 depending on the purpose of the experiment. Infected cells were harvested at 0, 24, or 48 h after three washes with RPMI1640. Cellular viral RNA or sg-RNA expression was determined in qPCR or RNA-Seq. GAPDH was used in qPCR as the internal control. For detection of antibody-dependent enhancement (ADE), virus (moi = 0.01, 0.1, or 0.2) were incubated with equal-volume convalescent sera from COVID-19 patients (for no-sera group, virus were incubated with equal-volume RPMI1640) at 37 °C for 30 min. Mixture was added to primary cells and samples were harvested at 0 h, 24 h, or 48 h post infection.
2.5. Flow Cytometry Analysis of Human Peripheral Blood Samples
For FCGRs detection, primary B cells, monocytes, and macrophages were incubated with fluorochrome-labeled antibodies specific for humans before fixation: FITC mouse anti-human CD32 (552883; BD Pharmingen, San Diego, CA, USA), APC mouse anti-human CD64 (561189; BD Pharmingen, San Diego, CA, USA ), CD16 Rabbit PAb (16559-1-AP; Proteintech,--). FITC-anti-Rabbit IgG (H+L) (SA00003-2; Proteintech, Chicago, IL, USA) was used as the secondary antibody for CD16.
For SARS-CoV-2 infected primary immune cells, surface staining was conducted before fixation with AF700-anti-CD45 (368514; BioLegend, San Diege, CA, USA), BV650-anti-CD11b (101239; BioLegend, San Diege, CA, USA), PE-anti-CD68 (333808; Biolegend, San Diege, CA, USA), and Percp Cy5.5-anti-CD14 (367110; Biolegend, San Diege, CA, USA). Antibody stained cells were fixed overnight with 4% PFA at 4 °C and taken out of the BSL3 lab for downstream analysis. Cells were stained further with in house made SARS-CoV NP pAb (1:500) at 4 °C for 30 min after permeabilization. Then, cells were stained with FITC-anti-Rabbit IgG (H + L) (SA00003-2; Proteintech, Chicago, IL, USA) at room temperature for 30 min.
2.6. RNA Extraction and qRT-PCR
Whenever commercial kits were used, the manufacturer’s instructions were followed without modification. Viral RNA was extracted from 140 μL of samples with the QIAamp
® Vival RNA Mini Kit (52906; QIAGEN, Hilden, Germany). RNA was eluted in 50 μL of elution buffer and used as the template for qRT–PCR. The QPCR detection method based on the 2019-nCoV S gene can be found in the previous study (Zhou et al., 2020). Two microliters of RNA were used as a template for the amplification of selected genes by real-time quantitative PCR using HiSxript
® II One step qRT-PCR SYBR
® Green Kit. (Q221-01; Vazyme Biotech Co., Ltd, Nanjing, Jiangsu, China). The 10 μL qPCR reaction mix contained 1.9 μL of nuclease free water, 5 μL of 2× One Step SYBR Green Mix, 0.5 μL One Step SYBR Green Enzyme Mix, 0.2 μL of 50× ROX Reference Dye 1, 0.2 μL of each primer (10 μM), and 2 μL of template RNA. Amplification was performed as follows: 50 °C for 3 min, 95 °C for 30 s followed by 40 cycles consisting of 95 °C for 10 s, 60 °C for 30 s, and a default melting curve step in an Step-One Plus Real-time PCR machine (ABI) [
14].
2.7. Transcriptome Analysis
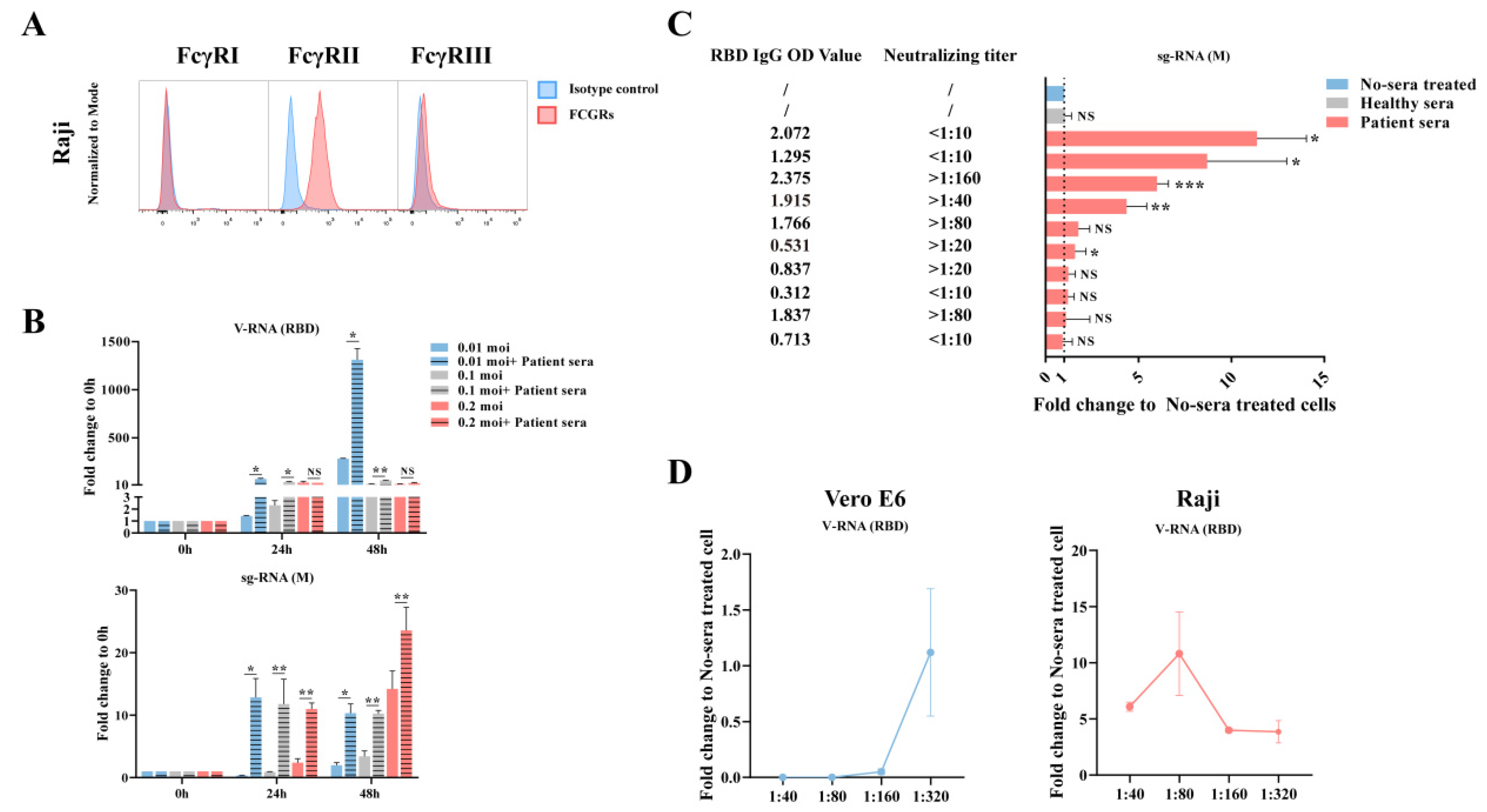
Using software Hisat2 v2.1.0, raw reads was mapped to genome that combined with human GRCH38.913 and Sars-CoV2 (MN996528.1). After transforming the format and sorting in Samtools v1.10-24, the BAM file was passed to StringTie v2.1.0 for transcriptome assembly and quantitation. Read count table of transcriptome generated by prepDE.py, a tool in StringTie, was used for gene differential expression analysis in R v4.1.0 with package DESeq2 v1.32.0. Compared to the mock group, the gene with Log2 Fold Change >2 and p-value < 0.05 was retained. Furthermore, the genes whose expression increased with degree of ADE were passed to the online tool Metascape for enrichment analysis.
2.8. Micro-Neutralization Assay
For detection of the neutralization antibody titer of convalescent sera, SARS-CoV-2 were diluted to 4000 TCID50/mL and incubated with equal-volume diluted patient sera (diluted from 1:10 to 1:1280, two-fold serial dilution) at 37 °C for 30 min. This was added to the mixture of Vero E6 cells seeded in 96-well plates and infected at 37 °C for 1 h. The supernatant was removed and cells washed with PBS. DMEM medium containing 2% FBS were added to the cells. Cell plates were fixed at 24 h post infection. Stained cells with in house made SARS-CoV NP pAb (1:500) and Cy3-anti-Rabbit IgG (H+L) were from Proteintech (SA00009-2) to detect viral NP.
2.9. Serological Test
An in-house anti-SARS-CoV-2 IgG ELISA kit was developed using recombinant RBD of the SARS-CoV-2 isolate WIV04 (MN996528.1). The RBD proteins were expressed in HEK293-17 cell lines. For IgG analysis, MaxiSorp Nunc-immuno 96-well ELISA plates were coated (100 ng per well) overnight at room temperature with RBD protein. Plasma from different donors were used at a dilution of 1:20 for 1 h at 37 °C. A HRP-conjugated anti-human IgG monoclonal antibody (Kyab Biotech Co. Ltd., Wuhan, China) was used at a dilution of 1:40,000. The OD value (450–630 nm) was calculated.
2.10. Statistical Analysis
Data analyses were performed using GraphPad Prism 7.0 software. Data are shown as mean ± SD. Data were analyzed with the Shapiro–Wilk normality test and confirmed with the Gaussian distribution. Statistical analysis was performed using the Student’s t-test with two tailed, 95% confidence. p values less than 0.05 were considered statistically significant.
4. Discussion
Here, we tested the ADE effect of convalescent serum samples using in vitro immune cells, aiming for a better understanding of possible SARS-CoV-2 viral antibody induced pathology in vivo. Our data indicated that ADE could occur to FcγR-expression cells such as B cells, monocytes, or macrophages. Although the ADE effect was not correlated to the dose of antibody in a particular patient, it was found to be highest within a narrow range of preexisting titer in the serum that was known to induce ADE. Finally, the ADE effect includes not only an enhancement of viral replication, but also an excessive immune response in these immune cells.
The role of SARS-CoV-2 antibodies in the severity of diseases is controversial. Based on the observations that severe patients tend to have higher antibody titers, there have been two hypotheses: one possibility is that the extensive viral replication and hyper-inflammation in severe patients induced overproduction of antibodies, while another possibility is that the high levels of antibodies worsen disease severity via ADE. The hyper-inflammation, however, was a result of ADE [
3]. In this study, we showed the in vitro evidence of ADE in multiple peripheral blood immune cells, supporting the second hypothesis that non-neutralizing antibodies could worsen the disease severity by enhancing viral infection or enhancing excessive immune activation in certain immune cells, suggesting they contributed to SARS-CoV-2 induced immunopathology in severe patients.
The ADE effect has been well characterized in cat coronavirus FIPV infection. FIPV infected macrophages and monocytes via APN receptor, and this infection or infection induced host responses were greatly enhanced by pre-existing native antibodies in cats [
6,
9,
10]. Likewise, a list of in vitro experiments also proved that SARS-CoV-1 or MERS-CoV induced ADE, although there is a lack of in vivo evidence [
11,
12]. Therefore, it has been suggested that strong antibody titers are more closely linked to severe COVID-19, which could be important in the lower respiratory tract that contributes to lung pathology [
3]. Our data proved the theory that more viral infection and more excessive immune responses in B cells, macrophages, and monocytes, the three cell types that are heavily recruited in lung under severe condition, and thus would increase the severity in lung. Moreover, a medium level of antibody can more easily induce ADE, a phenomenon that is also observed in dengue disease [
4], suggesting that the patients with antibody titers at or near the peak enhancement titer may place these individuals at greater risk of severe disease than if they only developed a small amount of antibody. Finally, the ADE effect may further dampen our immune defense mechanism by causing the dysfunction of B cells or macrophages, which eventually leads to impaired adaptive immunity.
Our study also has some limitations. First, this in vitro study provides insight into the pathogenesis of COVID-19 enhanced by antibodies. However, we were unable to provide in vivo data in the pathogenesis of a more severe clinical outcome in patients or in experimental animals because the effector functions of antibodies are altered by species–species interactions between antibodies and immune cells [
16]. Second, our study may not apply to predict potential ADE effect upon vaccination. It was revealed that SARS-CoV-2 natural infection induced a list of “bad antibodies” that would not show up after vaccination, for example, autoantibodies or lower levels of fucosylation of SARS-CoV-2-specific antibodies [
18]. The IgG types may also vary between infection and vaccination.
Collectively, we showed in vitro evidence of convalescent serum-dependent enhancement of either SARS-CoV-2 infection or viral induced excessive immune responses in immune cells. Our study provides insights into the understanding of an association of high viral antibody titer and severe lung pathology in severe patients with COVID-19.


 ,
, 
{kind=link}
{kind=link}
{kind=link}