
INSTIs and NNRTIs Potently Inhibit HIV-1 Polypurine Tract Mutants in a Single Round Infection Assay


 , ,
, ,  and
and
Abstract
:1. Introduction
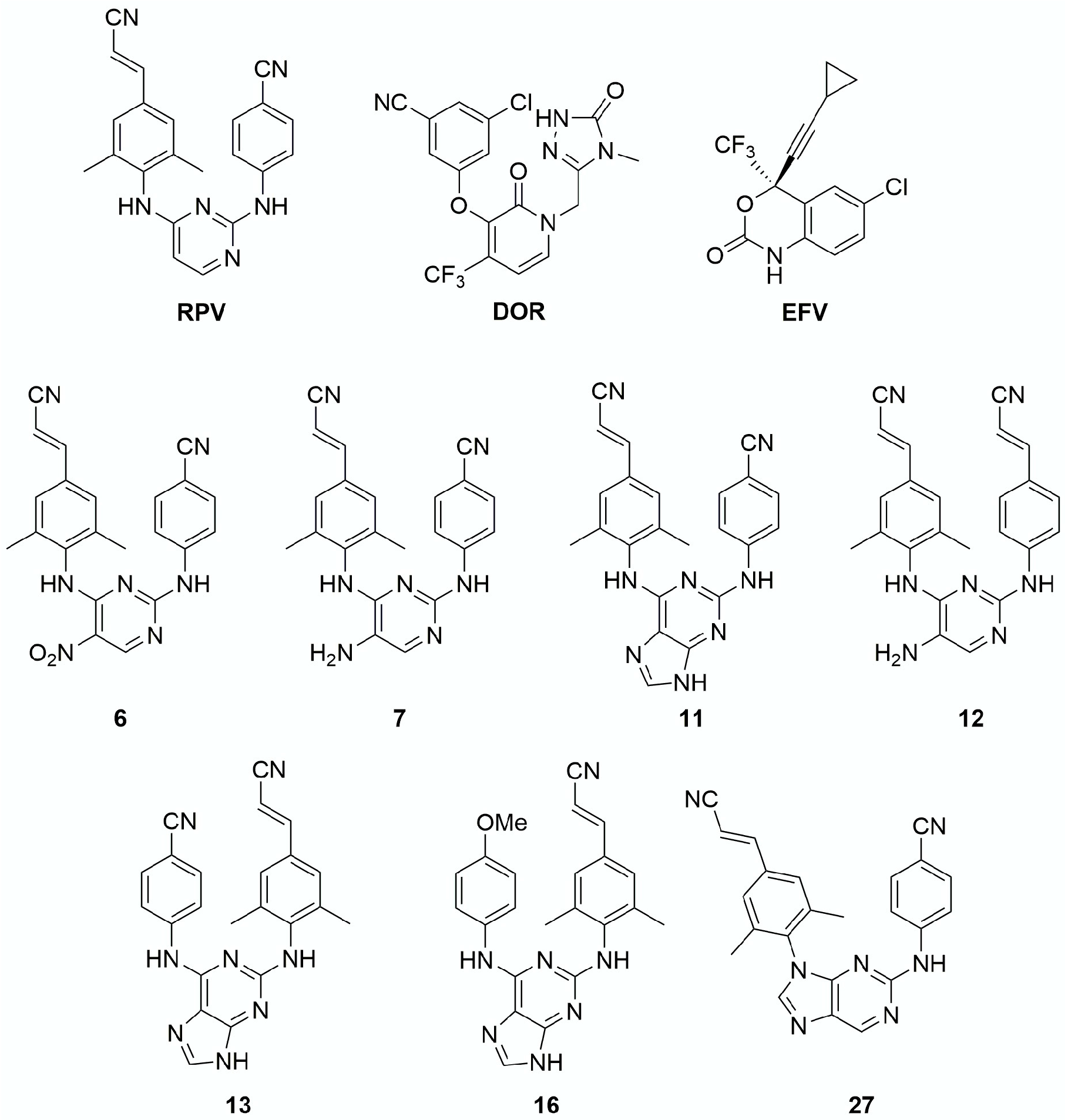
2. Materials and Methods
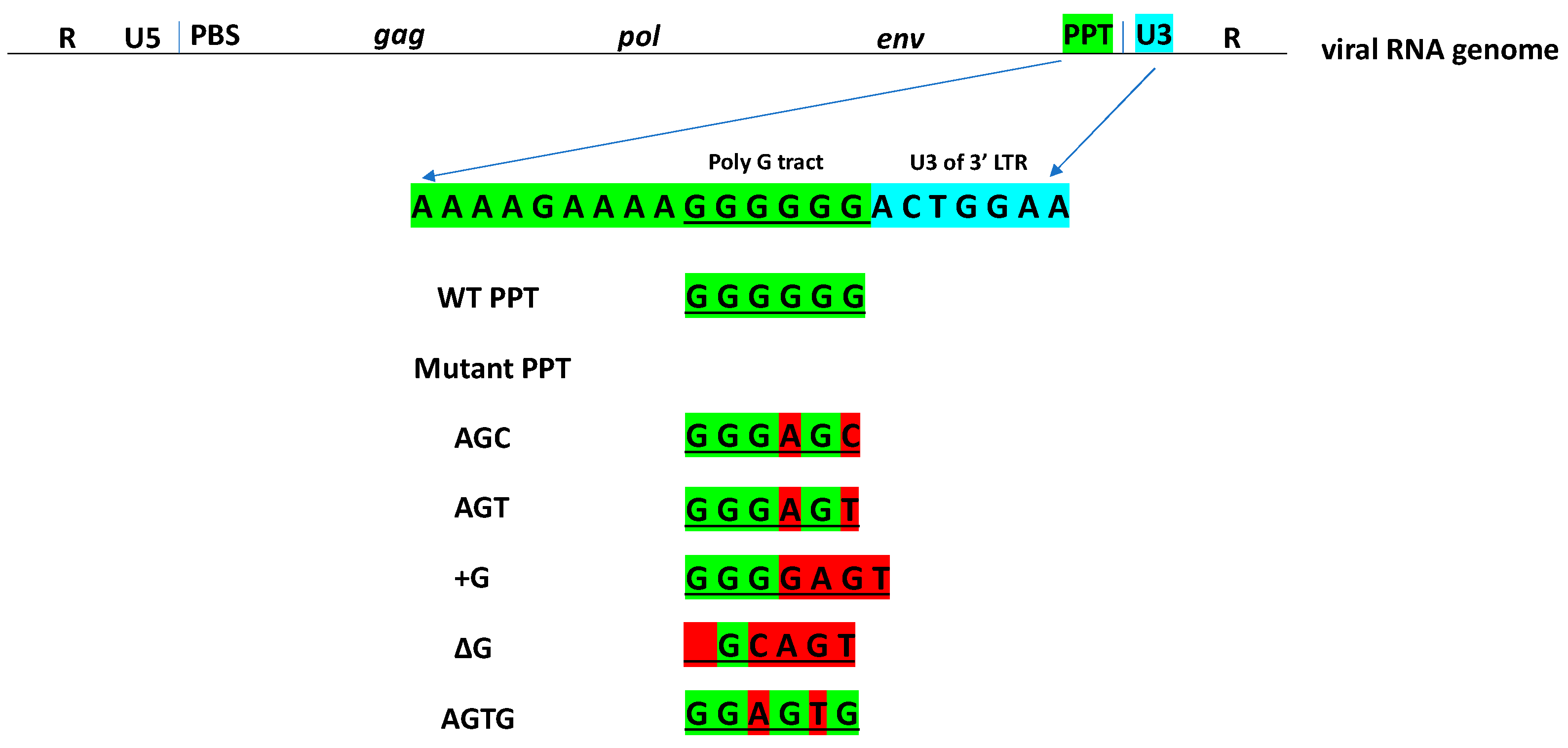
- AGC (GGGAGC), 5′-CCACTTTTTAAAAGAAAAGGGAGCACTGGAAGGGCTAATTC-3′;
- AGT (GGGAGT), 5′-CCACTTTTTAAAAGAAAAGGGAGTACTGGAAGGGCTAATTC-3′;
- +G (GGGGAGT), 5′-CCACTTTTTAAAAGAAAAGGGGAGTACTGGAAGGGCTAATTC-3′;
- +G (GGGGAGT), 5′-CCACTTTTTAAAAGAAAAGGGGAGTACTGGAAGGGCTAATTC-3′;
- ΔG (_GCAGT), 5′-CCACTTTTTAAAAGAAAAGCAGTACTGGAAGGGCTAATTC-3′;
- AGTG (GGAGTG)’ 5′-CCACTTTTTAAAAGAAAAGGAGTGACTGGAAGGGCTAATTC-3′.
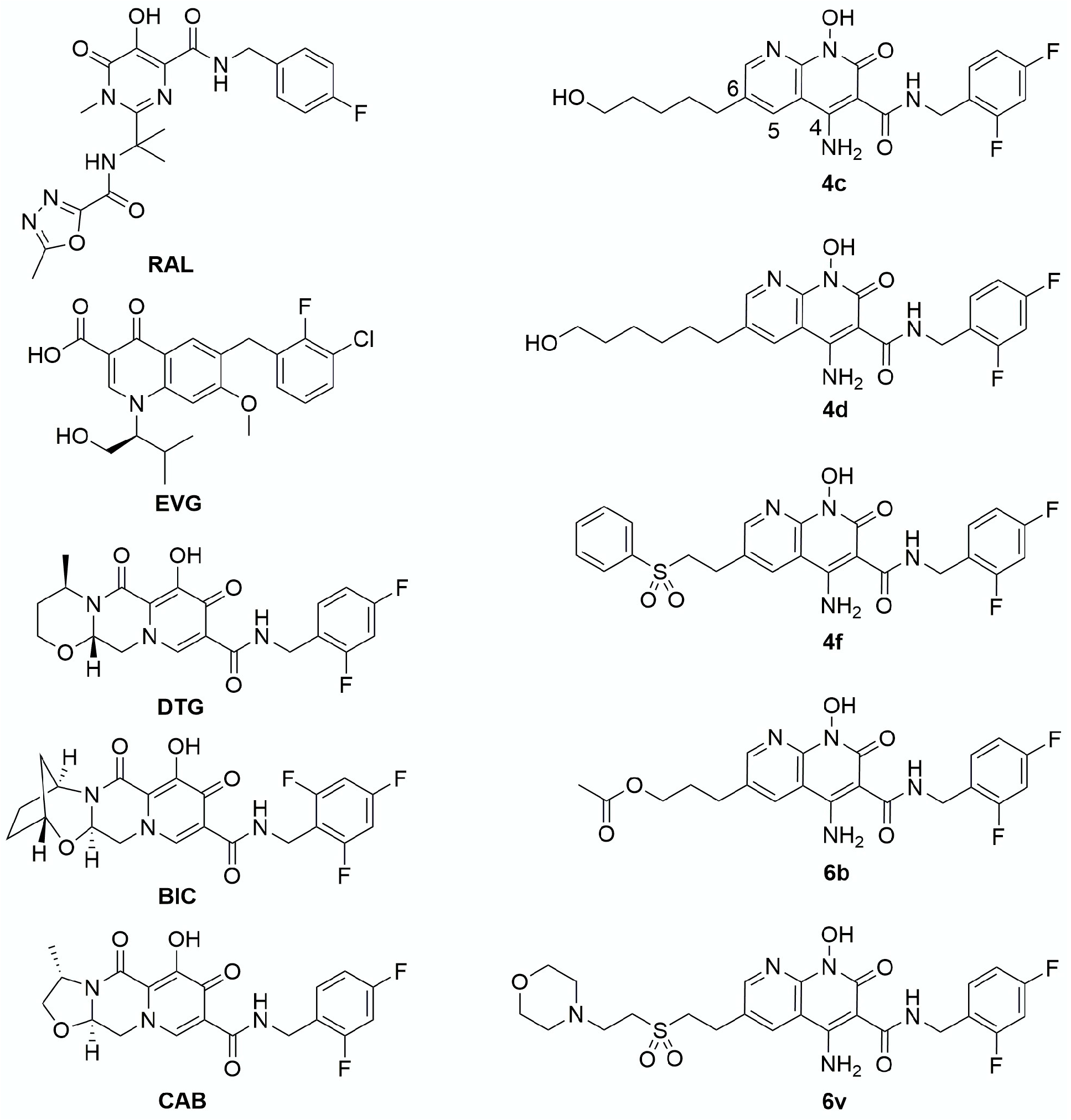
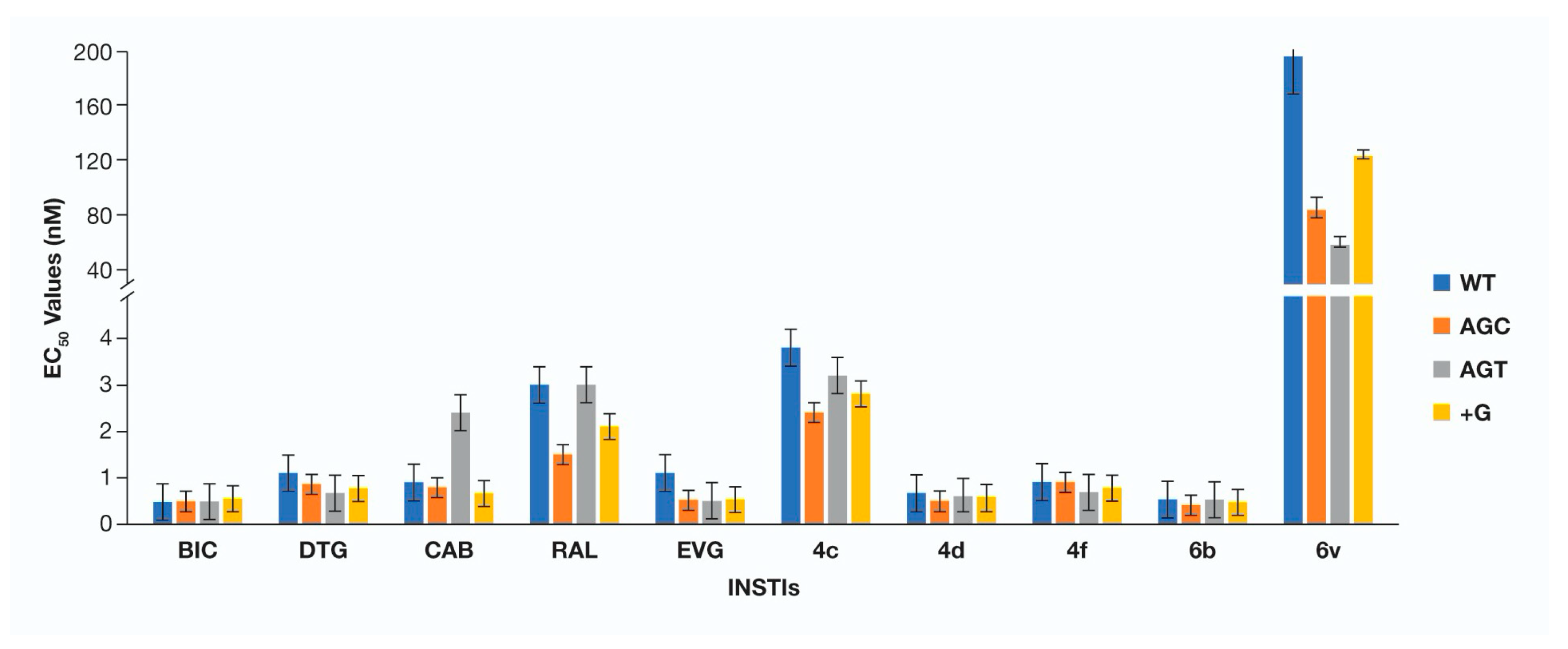
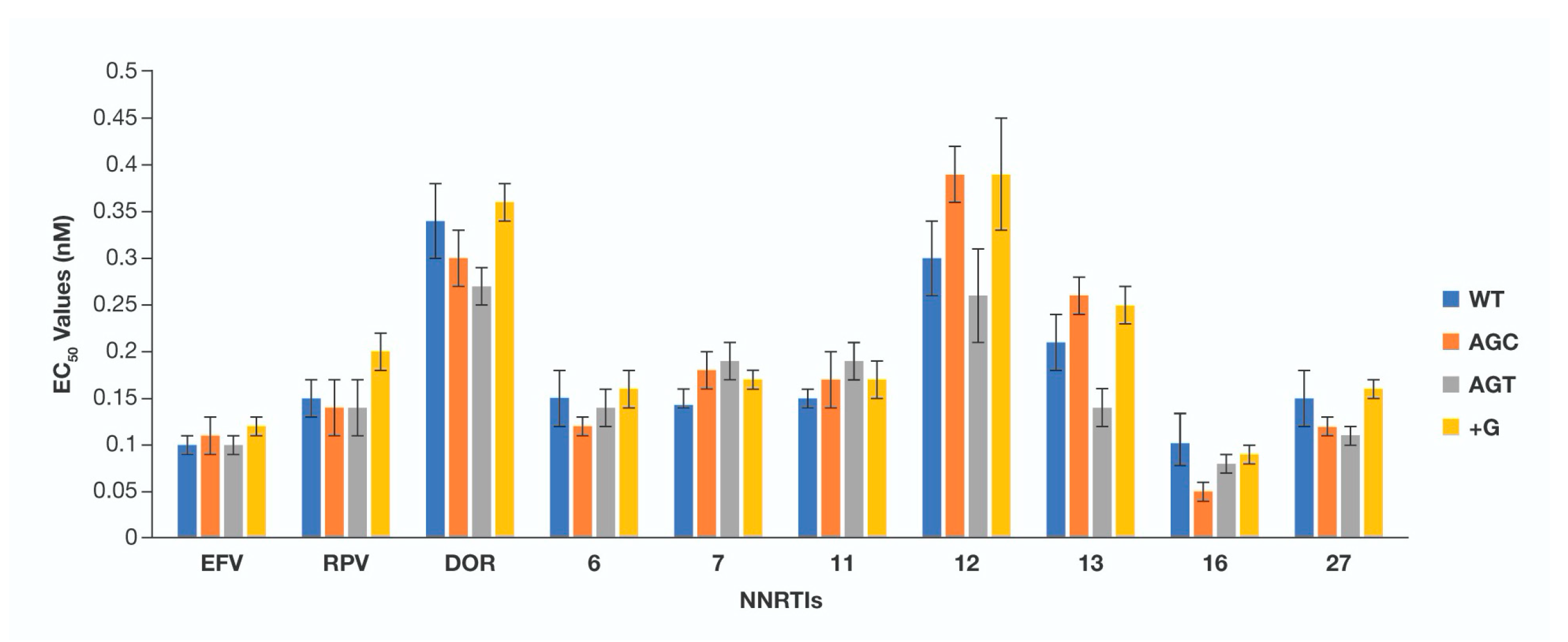
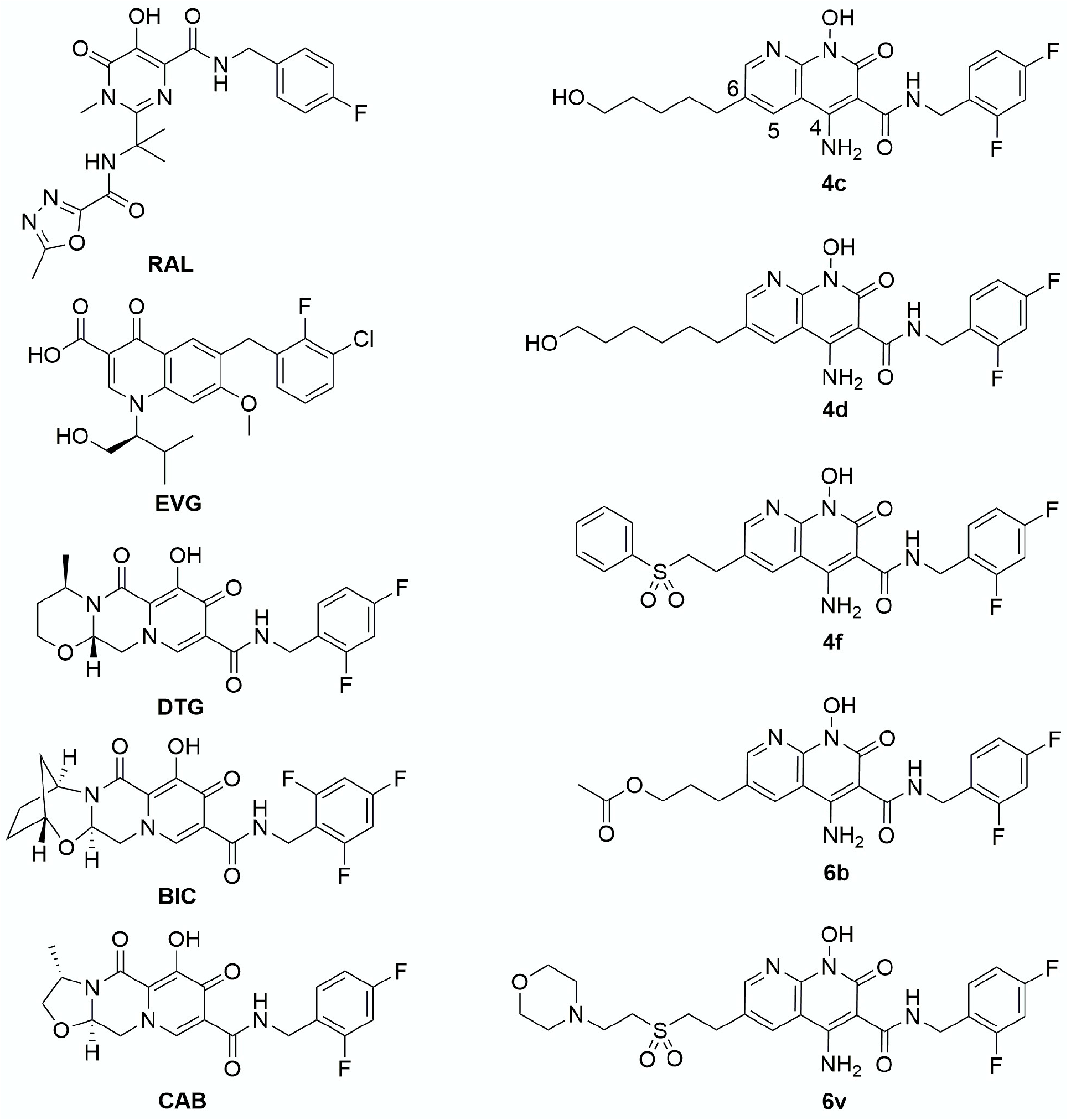
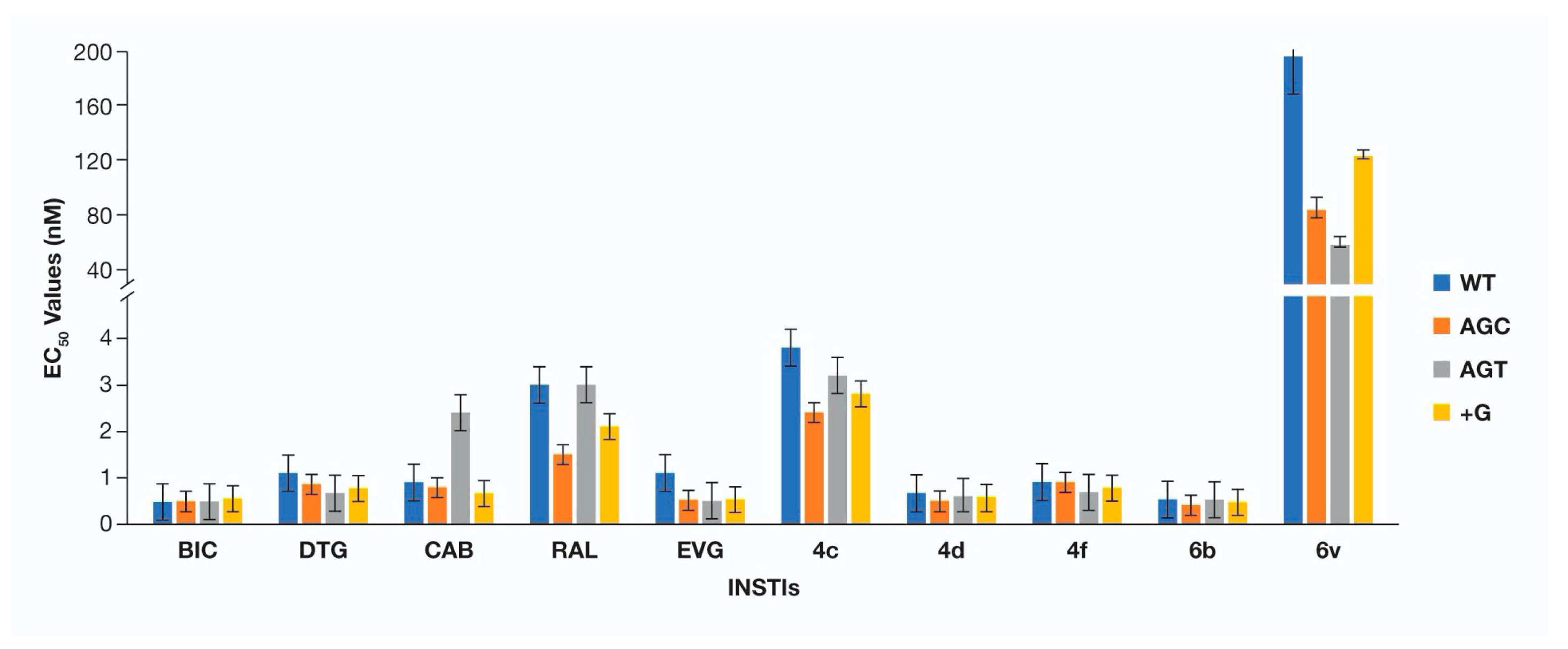
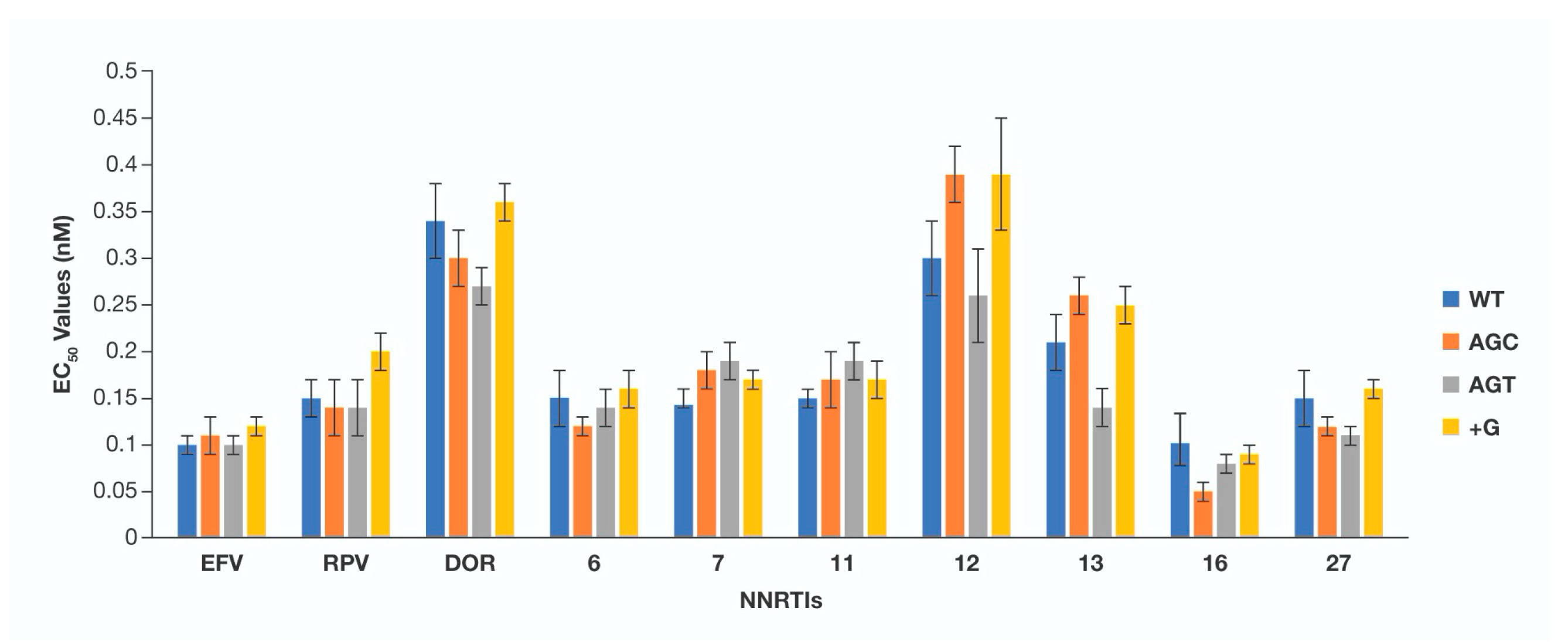
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data availability statement
Acknowledgments
Conflicts of Interest
References
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV; U.S. Department of HHS: Washington, DC, USA, 2021. [Google Scholar]
- Kobayashi, M.; Yoshinaga, T.; Seki, T.; Wakasa-Morimoto, C.; Brown, K.W.; Ferris, R.; Foster, S.A.; Hazen, R.J.; Miki, S.; Suyama-Kagitani, A.; et al. In Vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob. Agents Chemother. 2011, 55, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Tsiang, M.; Jones, G.S.; Goldsmith, J.; Mulato, A.; Hansen, D.; Kan, E.; Tsai, L.; Bam, R.A.; Stepan, G.; Stray, K.M.; et al. Antiviral Activity of Bictegravir (GS-9883), a Novel Potent HIV-1 Integrase Strand Transfer Inhibitor with an Improved Resistance Profile. Antimicrob. Agents Chemother. 2016, 60, 7086–7097. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.J.; Zhao, X.Z.; Burke, T.R., Jr.; Hughes, S.H. Efficacies of Cabotegravir and Bictegravir against drug-resistant HIV-1 integrase mutants. Retrovirology 2018, 15, 37. [Google Scholar] [CrossRef]
- Hare, S.; Smith, S.J.; Metifiot, M.; Jaxa-Chamiec, A.; Pommier, Y.; Hughes, S.H.; Cherepanov, P. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol. Pharmacol. 2011, 80, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Margot, N.A.; Ram, R.R.; White, K.L.; Abram, M.E.; Callebaut, C. Antiviral activity of HIV-1 integrase strand-transfer inhibitors against mutants with integrase resistance-associated mutations and their frequency in treatment-naive individuals. J. Med. Virol. 2019, 91, 2188–2194. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Z.; Smith, S.J.; Metifiot, M.; Marchand, C.; Boyer, P.L.; Pommier, Y.; Hughes, S.H.; Burke, T.R., Jr. 4-amino-1-hydroxy-2-oxo-1,8-naphthyridine-containing compounds having high potency against raltegravir-resistant integrase mutants of HIV-1. J. Med. Chem. 2014, 57, 5190–5202. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Zhao, X.Z.; Passos, D.O.; Lyumkis, D.; Burke, T.R., Jr.; Hughes, S.H. Integrase Strand Transfer Inhibitors Are Effective Anti-HIV Drugs. Viruses 2021, 13, 205. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Grant, P.M.; Tzou, P.L.; Barrow, G.; Harrigan, P.R.; Ioannidis, J.P.A.; Shafer, R.W. A systematic review of the genetic mechanisms of dolutegravir resistance. J. Antimicrob. Chemother. 2019, 74, 3135–3149. [Google Scholar] [CrossRef] [PubMed]
- Castagna, A.; Maggiolo, F.; Penco, G.; Wright, D.; Mills, A.; Grossberg, R.; Molina, J.M.; Chas, J.; Durant, J.; Moreno, S.; et al. Dolutegravir in antiretroviral-experienced patients with raltegravir- and/or elvitegravir-resistant HIV-1: 24-week results of the phase III VIKING-3 study. J. Infect. Dis. 2014, 210, 354–362. [Google Scholar] [CrossRef] [Green Version]
- Eron, J.J.; Clotet, B.; Durant, J.; Katlama, C.; Kumar, P.; Lazzarin, A.; Poizot-Martin, I.; Richmond, G.; Soriano, V.; Ait-Khaled, M.; et al. Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study. J. Infect. Dis. 2013, 207, 740–748. [Google Scholar] [CrossRef]
- Collier, D.A.; Monit, C.; Gupta, R.K. The Impact of HIV-1 Drug Escape on the Global Treatment Landscape. Cell Host Microbe 2019, 26, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.G.; Thomas, R.; Blanco, J.L.; Ibanescu, R.I.; Oliveira, M.; Mesplede, T.; Golubkov, O.; Roger, M.; Garcia, F.; Martinez, E.; et al. Development of a G118R mutation in HIV-1 integrase following a switch to dolutegravir monotherapy leading to cross-resistance to integrase inhibitors. J. Antimicrob. Chemother. 2016, 71, 1948–1953. [Google Scholar] [CrossRef]
- Quashie, P.K.; Mesplede, T.; Han, Y.S.; Oliveira, M.; Singhroy, D.N.; Fujiwara, T.; Underwood, M.R.; Wainberg, M.A. Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J. Virol. 2012, 86, 2696–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quashie, P.K.; Oliviera, M.; Veres, T.; Osman, N.; Han, Y.S.; Hassounah, S.; Lie, Y.; Huang, W.; Mesplede, T.; Wainberg, M.A. Differential effects of the G118R, H51Y, and E138K resistance substitutions in different subtypes of HIV integrase. J. Virol. 2015, 89, 3163–3175. [Google Scholar] [CrossRef] [Green Version]
- Yoshinaga, T.; Kobayashi, M.; Seki, T.; Miki, S.; Wakasa-Morimoto, C.; Suyama-Kagitani, A.; Kawauchi-Miki, S.; Taishi, T.; Kawasuji, T.; Johns, B.A.; et al. Antiviral characteristics of GSK1265744, an HIV integrase inhibitor dosed orally or by long-acting injection. Antimicrob. Agents Chemother. 2015, 59, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshinaga, T.; Seki, T.; Miki, S.; Miyamoto, T.; Suyama-Kagitani, A.; Kawauchi-Miki, S.; Kobayashi, M.; Sato, A.; Stewart, E.; Underwood, M.; et al. Novel secondary mutations C56S and G149A confer resistance to HIV-1 integrase strand transfer inhibitors. Antivir. Res. 2018, 152, 1–9. [Google Scholar] [CrossRef]
- Oliveira, M.; Ibanescu, R.I.; Anstett, K.; Mesplede, T.; Routy, J.P.; Robbins, M.A.; Brenner, B.G.; The Montreal Primary HIV (PHI) Cohort Study Group. Selective resistance profiles emerging in patient-derived clinical isolates with cabotegravir, bictegravir, dolutegravir, and elvitegravir. Retrovirology 2018, 15, 56. [Google Scholar] [CrossRef] [Green Version]
- Margolis, D.A.; Brinson, C.C.; Smith, G.H.R.; de Vente, J.; Hagins, D.P.; Eron, J.J.; Griffith, S.K.; Clair, M.H.S.; Stevens, M.C.; Williams, P.E.; et al. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral-naive adults with HIV-1 infection (LATTE): A randomised, phase 2b, dose-ranging trial. Lancet Infect. Dis. 2015, 15, 1145–1155. [Google Scholar] [CrossRef]
- Malet, I.; Subra, F.; Charpentier, C.; Collin, G.; Descamps, D.; Calvez, V.; Marcelin, A.G.; Delelis, O. Mutations Located outside the Integrase Gene Can Confer Resistance to HIV-1 Integrase Strand Transfer Inhibitors. mBio 2017, 8, e00922-17. [Google Scholar] [CrossRef] [Green Version]
- Wijting, I.E.A.; Lungu, C.; Rijnders, B.J.A.; van der Ende, M.E.; Pham, H.T.; Mesplede, T.; Pas, S.D.; Voermans, J.J.C.; Schuurman, R.; van de Vijver, D.; et al. HIV-1 Resistance Dynamics in Patients With Virologic Failure to Dolutegravir Maintenance Monotherapy. J. Infect. Dis. 2018, 218, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Hikichi, Y.; Van Duyne, R.; Pham, P.; Groebner, J.L.; Wiegand, A.; Mellors, J.W.; Kearney, M.F.; Freed, E.O. Mechanistic Analysis of the Broad Antiretroviral Resistance Conferred by HIV-1 Envelope Glycoprotein Mutations. mBio 2021, 12, e03134-20. [Google Scholar] [CrossRef]
- Van Duyne, R.; Kuo, L.S.; Pham, P.; Fujii, K.; Freed, E.O. Mutations in the HIV-1 envelope glycoprotein can broadly rescue blocks at multiple steps in the virus replication cycle. Proc. Natl. Acad. Sci. USA 2019, 116, 9040–9049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Sluis-Cremer, N. Mutations in the HIV-1 3’-Polypurine Tract and Integrase Strand Transfer Inhibitor Resistance. Antimicrob. Agents Chemother. 2021, 65, 65. [Google Scholar] [CrossRef] [PubMed]
- Ferris, A.L.; Wu, X.; Hughes, C.M.; Stewart, C.; Smith, S.J.; Milne, T.A.; Wang, G.G.; Shun, M.C.; Allis, C.D.; Engelman, A.; et al. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. Natl. Acad. Sci. USA 2010, 107, 3135–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.J.; Hughes, S.H. Rapid screening of HIV reverse transcriptase and integrase inhibitors. J. Vis. Exp. 2014, 86, e51400. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.J.; Zhao, X.Z.; Passos, D.O.; Pye, V.E.; Cherepanov, P.; Lyumkis, D.; Burke, T.R., Jr.; Hughes, S.H. HIV-1 Integrase Inhibitors with Modifications That Affect Their Potencies against Drug Resistant Integrase Mutants. ACS Infect. Dis. 2021, 7, 1469–1482. [Google Scholar] [CrossRef]
- Smith, S.J.; Zhao, X.Z.; Passos, D.O.; Lyumkis, D.; Burke, T.R., Jr.; Hughes, S.H. HIV-1 Integrase Inhibitors That Are Active against Drug-Resistant Integrase Mutants. Antimicrob. Agents Chemother. 2020, 64, e00611-20. [Google Scholar] [CrossRef]
- Zhao, X.Z.; Smith, S.J.; Maskell, D.P.; Metifiot, M.; Pye, V.E.; Fesen, K.; Marchand, C.; Pommier, Y.; Cherepanov, P.; Hughes, S.H.; et al. HIV-1 Integrase Strand Transfer Inhibitors with Reduced Susceptibility to Drug Resistant Mutant Integrases. ACS Chem. Biol. 2016, 11, 1074–1081. [Google Scholar] [CrossRef]
- Zhao, X.Z.; Smith, S.J.; Maskell, D.P.; Metifiot, M.; Pye, V.E.; Fesen, K.; Marchand, C.; Pommier, Y.; Cherepanov, P.; Hughes, S.H.; et al. Structure-Guided Optimization of HIV Integrase Strand Transfer Inhibitors. J. Med. Chem. 2017, 60, 7315–7332. [Google Scholar] [CrossRef]
- Smith, S.J.; Pauly, G.T.; Akram, A.; Melody, K.; Ambrose, Z.; Schneider, J.P.; Hughes, S.H. Rilpivirine and Doravirine Have Complementary Efficacies Against NNRTI-Resistant HIV-1 Mutants. J. Acquir. Immune Defic. Syndr. 2016, 72, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.J.; Pauly, G.T.; Akram, A.; Melody, K.; Rai, G.; Maloney, D.J.; Ambrose, Z.; Thomas, C.J.; Schneider, J.T.; Hughes, S.H. Rilpivirine analogs potently inhibit drug-resistant HIV-1 mutants. Retrovirology 2016, 13, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.J.; Pauly, G.T.; Hewlett, K.; Schneider, J.P.; Hughes, S.H. Structure-based non-nucleoside inhibitor design: Developing inhibitors that are effective against resistant mutants. Chem. Biol. Drug Des. 2021, 97, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Zhao, X.Z.; Burke, T.R., Jr.; Hughes, S.H. HIV-1 Integrase Inhibitors That Are Broadly Effective against Drug-Resistant Mutants. Antimicrob. Agents Chemother. 2018, 62, 62. [Google Scholar] [CrossRef] [Green Version]
- Sluis-Cremer, N.; Tachedjian, G. Mechanisms of inhibition of HIV replication by non-nucleoside reverse transcriptase inhibitors. Virus Res. 2008, 134, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betancor, G.; Alvarez, M.; Marcelli, B.; Andres, C.; Martinez, M.A.; Menendez-Arias, L. Effects of HIV-1 reverse transcriptase connection subdomain mutations on polypurine tract removal and initiation of (+)-strand DNA synthesis. Nucleic Acids Res. 2015, 43, 2259–2270. [Google Scholar] [CrossRef] [Green Version]
- Biondi, M.J.; Beilhartz, G.L.; McCormick, S.; Gotte, M. N348I in HIV-1 reverse transcriptase can counteract the nevirapine-mediated bias toward RNase H cleavage during plus-strand initiation. J. Biol. Chem. 2010, 285, 26966–26975. [Google Scholar] [CrossRef] [Green Version]
- Grobler, J.A.; Dornadula, G.; Rice, M.R.; Simcoe, A.L.; Hazuda, D.J.; Miller, M.D. HIV-1 reverse transcriptase plus-strand initiation exhibits preferential sensitivity to non-nucleoside reverse transcriptase inhibitors in vitro. J. Biol. Chem. 2007, 282, 8005–8010. [Google Scholar] [CrossRef]
- Hare, S.; Maertens, G.N.; Cherepanov, P. 3’-processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J. 2012, 31, 3020–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, T.; Mizuuchi, K. Retroviral DNA integration: Structure of an integration intermediate. Cell 1988, 54, 497–504. [Google Scholar] [CrossRef]
- Miller, M.D.; Farnet, C.M.; Bushman, F.D. Human immunodeficiency virus type 1 preintegration complexes: Studies of organization and composition. J. Virol. 1997, 71, 5382–5390. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.O.; Bowerman, B.; Varmus, H.E.; Bishop, J.M. Retroviral integration: Structure of the initial covalent product and its precursor, and a role for the viral IN protein. Proc. Natl. Acad. Sci. USA 1989, 86, 2525–2529. [Google Scholar] [CrossRef] [Green Version]
- Engelman, A.; Mizuuchi, K.; Craigie, R. HIV-1 DNA integration: Mechanism of viral DNA cleavage and DNA strand transfer. Cell 1991, 67, 1211–1221. [Google Scholar] [CrossRef]
- Hu, W.S.; Hughes, S.H. HIV-1 reverse transcription. Cold Spring Harb. Perspect. Med. 2012, 2, a006882. [Google Scholar] [CrossRef] [Green Version]
- Varadarajan, J.; McWilliams, M.J.; Hughes, S.H. Treatment with suboptimal doses of raltegravir leads to aberrant HIV-1 integrations. Proc. Natl. Acad. Sci. USA 2013, 110, 14747–14752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Chang, K.W.; Hughes, S.H. Mutations in the U5 sequences adjacent to the primer binding site do not affect tRNA cleavage by rous sarcoma virus RNase H but do cause aberrant integrations in vivo. J. Virol. 2006, 80, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Hang, J.Q.; Li, Y.; Yang, Y.; Cammack, N.; Mirzadegan, T.; Klumpp, K. Substrate-dependent inhibition or stimulation of HIV RNase H activity by non-nucleoside reverse transcriptase inhibitors (NNRTIs). Biochem. Biophys. Res. Commun. 2007, 352, 341–350. [Google Scholar] [CrossRef]
- Radzio, J.; Sluis-Cremer, N. Efavirenz accelerates HIV-1 reverse transcriptase ribonuclease H cleavage, leading to diminished zidovudine excision. Mol. Pharmacol. 2008, 73, 601–606. [Google Scholar] [CrossRef]
- Shaw-Reid, C.A.; Feuston, B.; Munshi, V.; Getty, K.; Krueger, J.; Hazuda, D.J.; Parniak, M.A.; Miller, M.D.; Lewis, D. Dissecting the effects of DNA polymerase and ribonuclease H inhibitor combinations on HIV-1 reverse-transcriptase activities. Biochemistry 2005, 44, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PPT Mutant | Single Round Infectivity (% of WT Activity) |
|---|---|
| AGC | 5.0 ± 1.2 |
| AGT | 7.0 ± 1.4 |
| +G | 8.0 ± 2.5 |
| ΔG | 0 * |
| AGTG | 0 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, S.J.; Ferris, A.; Zhao, X.; Pauly, G.; Schneider, J.P.; Burke, T.R., Jr.; Hughes, S.H. INSTIs and NNRTIs Potently Inhibit HIV-1 Polypurine Tract Mutants in a Single Round Infection Assay. Viruses 2021, 13, 2501. https://doi.org/10.3390/v13122501
Smith SJ, Ferris A, Zhao X, Pauly G, Schneider JP, Burke TR Jr., Hughes SH. INSTIs and NNRTIs Potently Inhibit HIV-1 Polypurine Tract Mutants in a Single Round Infection Assay. Viruses. 2021; 13(12):2501. https://doi.org/10.3390/v13122501
Chicago/Turabian StyleSmith, Steven J., Andrea Ferris, Xuezhi Zhao, Gary Pauly, Joel P. Schneider, Terrence R. Burke, Jr., and Stephen H. Hughes. 2021. "INSTIs and NNRTIs Potently Inhibit HIV-1 Polypurine Tract Mutants in a Single Round Infection Assay" Viruses 13, no. 12: 2501. https://doi.org/10.3390/v13122501
APA StyleSmith, S. J., Ferris, A., Zhao, X., Pauly, G., Schneider, J. P., Burke, T. R., Jr., & Hughes, S. H. (2021). INSTIs and NNRTIs Potently Inhibit HIV-1 Polypurine Tract Mutants in a Single Round Infection Assay. Viruses, 13(12), 2501. https://doi.org/10.3390/v13122501