
Enhancing the Antiviral Potency of Nucleobases for Potential Broad-Spectrum Antiviral Therapies

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Viruses, and Compounds
2.2. Cell Viability Assay
2.3. Virus-Based Assays and Viral Titer Analysis
2.4. Analysis of T-1105 Conversion and Endogenous Levels of Nucleotides and PRPP
2.5. Viral RNA Isolation for Next-Generation Sequencing
2.6. Next-Generation Sequencing by Click Chemistry
2.7. Data Analysis
3. Results
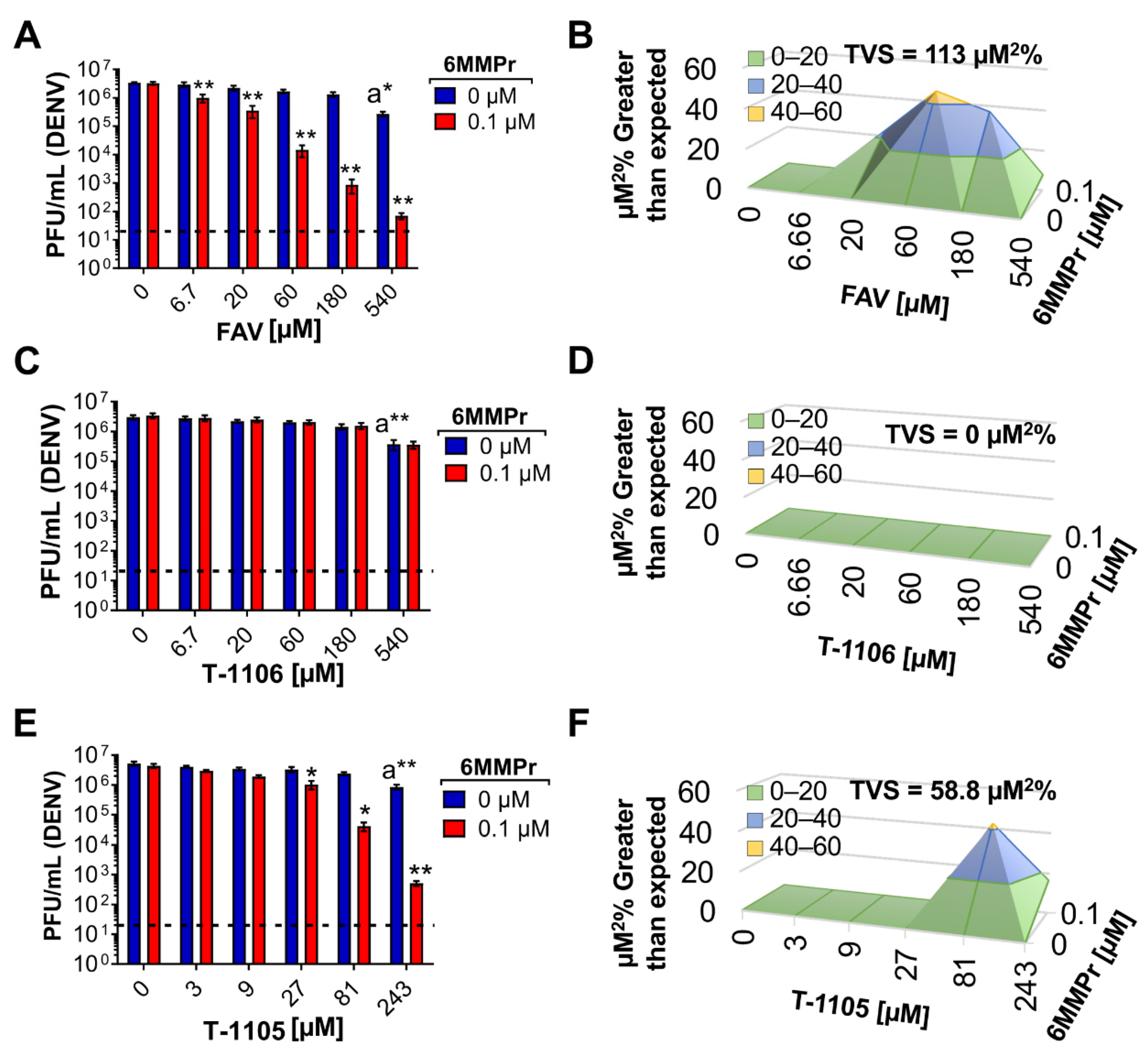
3.1. 6MMPr in Combination with T-1105 or Favipiravir Results in Synergistic Increases in Antiviral Activity against Dengue Virus (DENV)
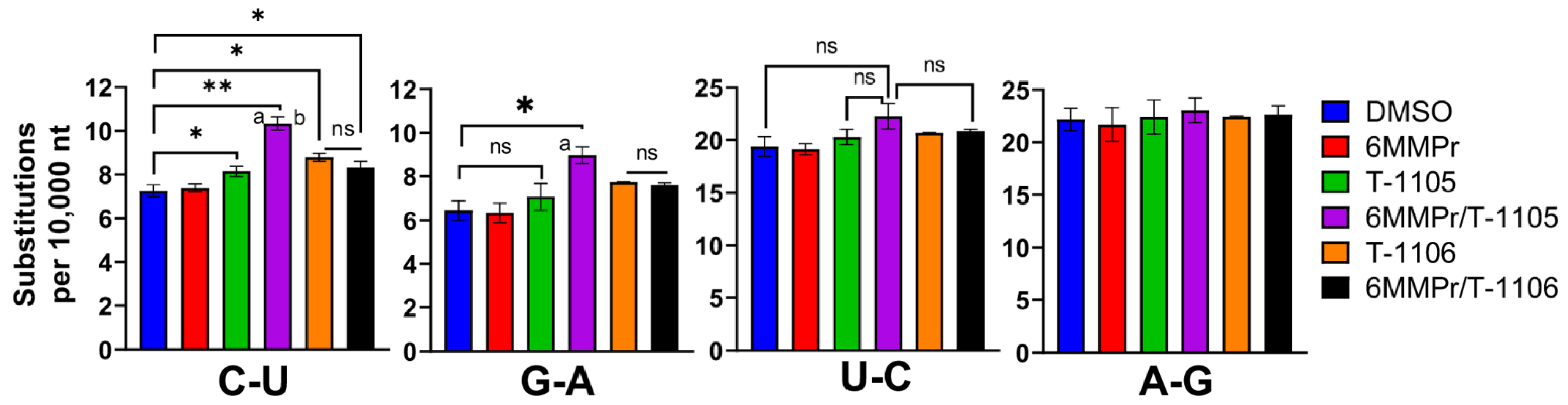
3.2. 6MMPr Co-Treatment Enhanced the Viral Mutation Frequency Observed with T-1105 but Not T-1106
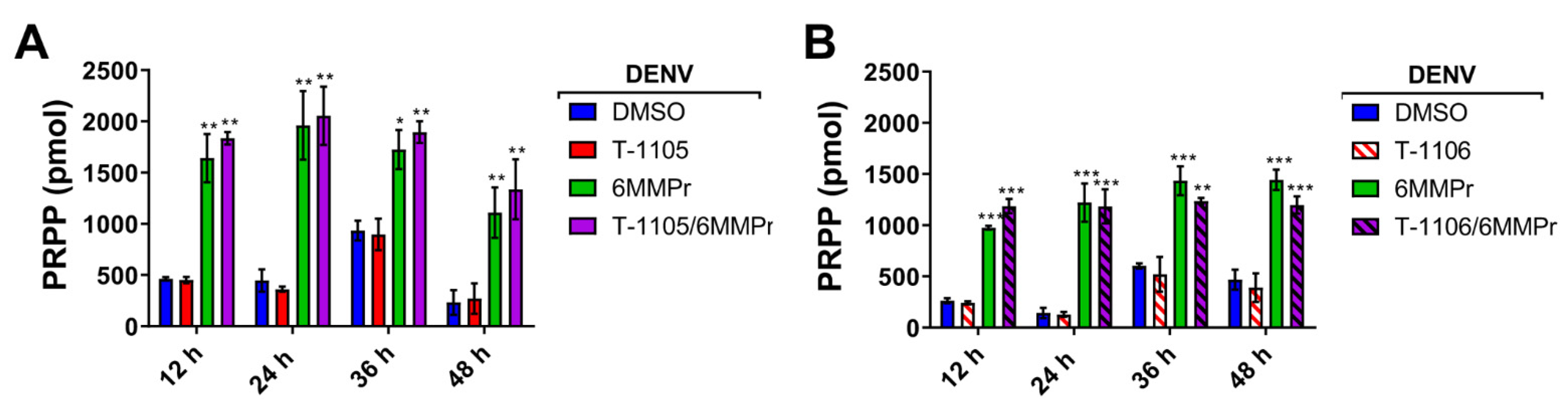
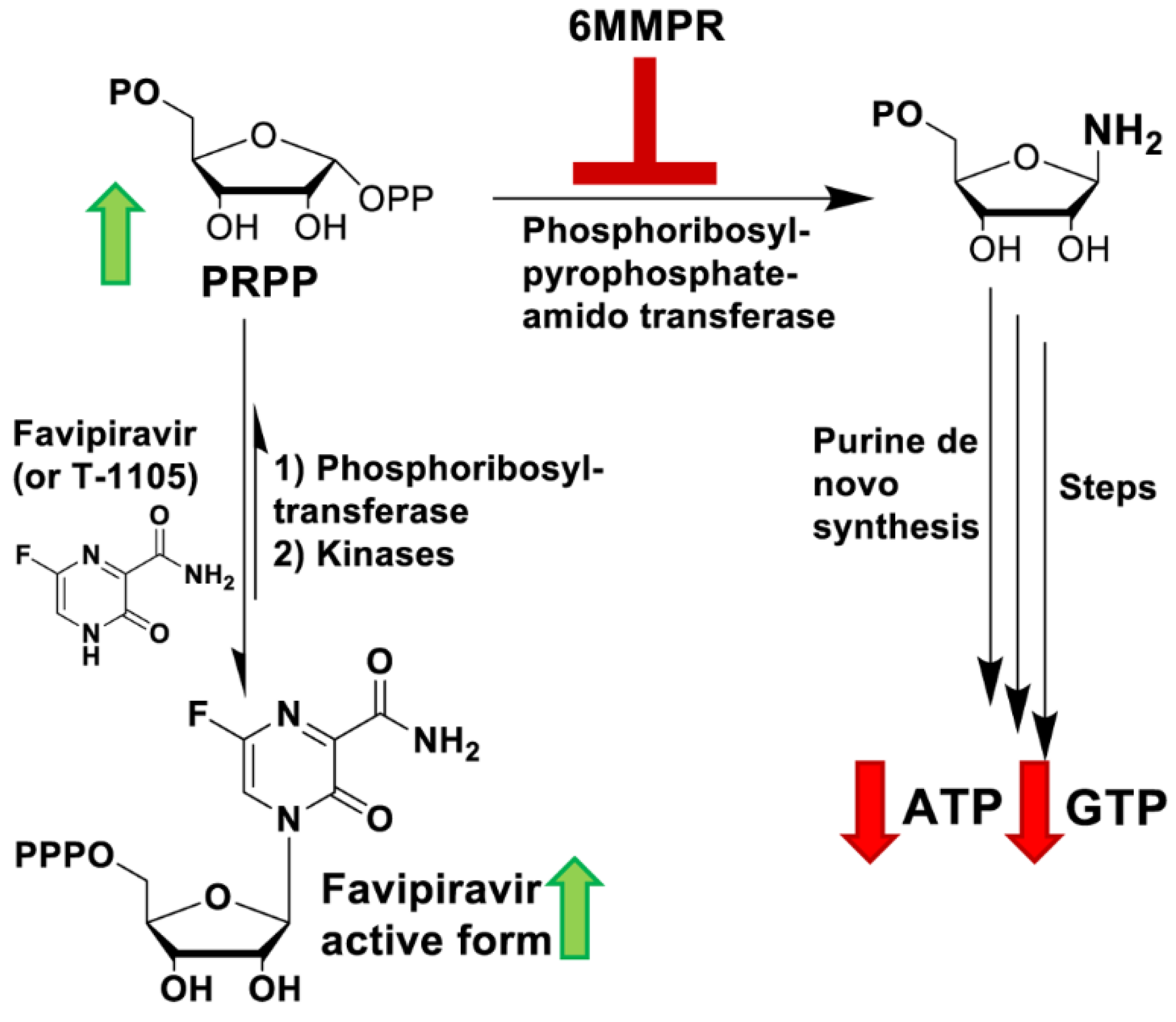
3.3. 6MMPr Increases Intracellular Levels of T-1105-RMP, -RDP, -RTP, and PRPP While Reducing Endogenous ATP and GTP Levels
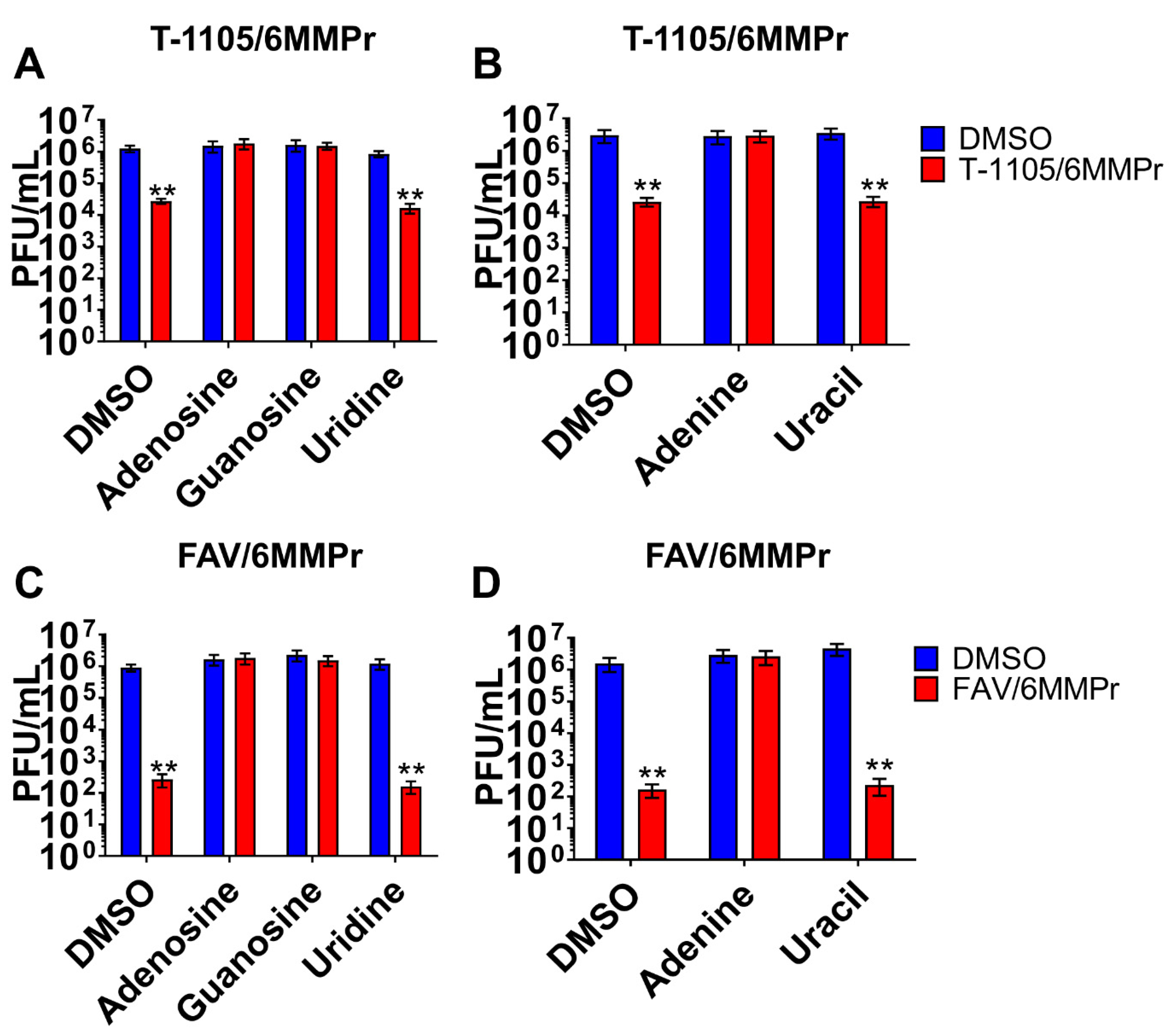
3.4. Supplementation with Exogenous Purines Abolished the Nucleobase/6MMPr Antiviral Activity
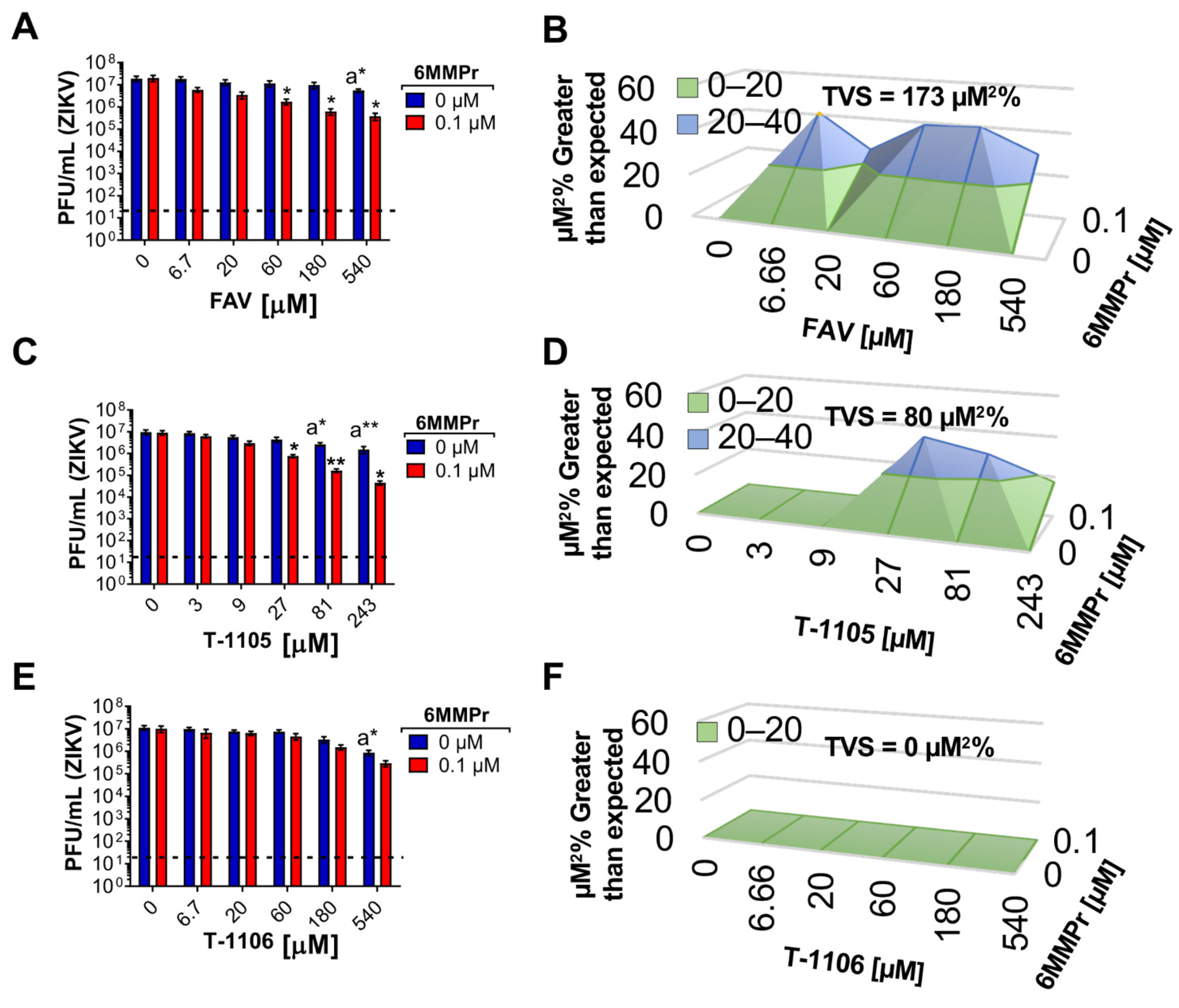
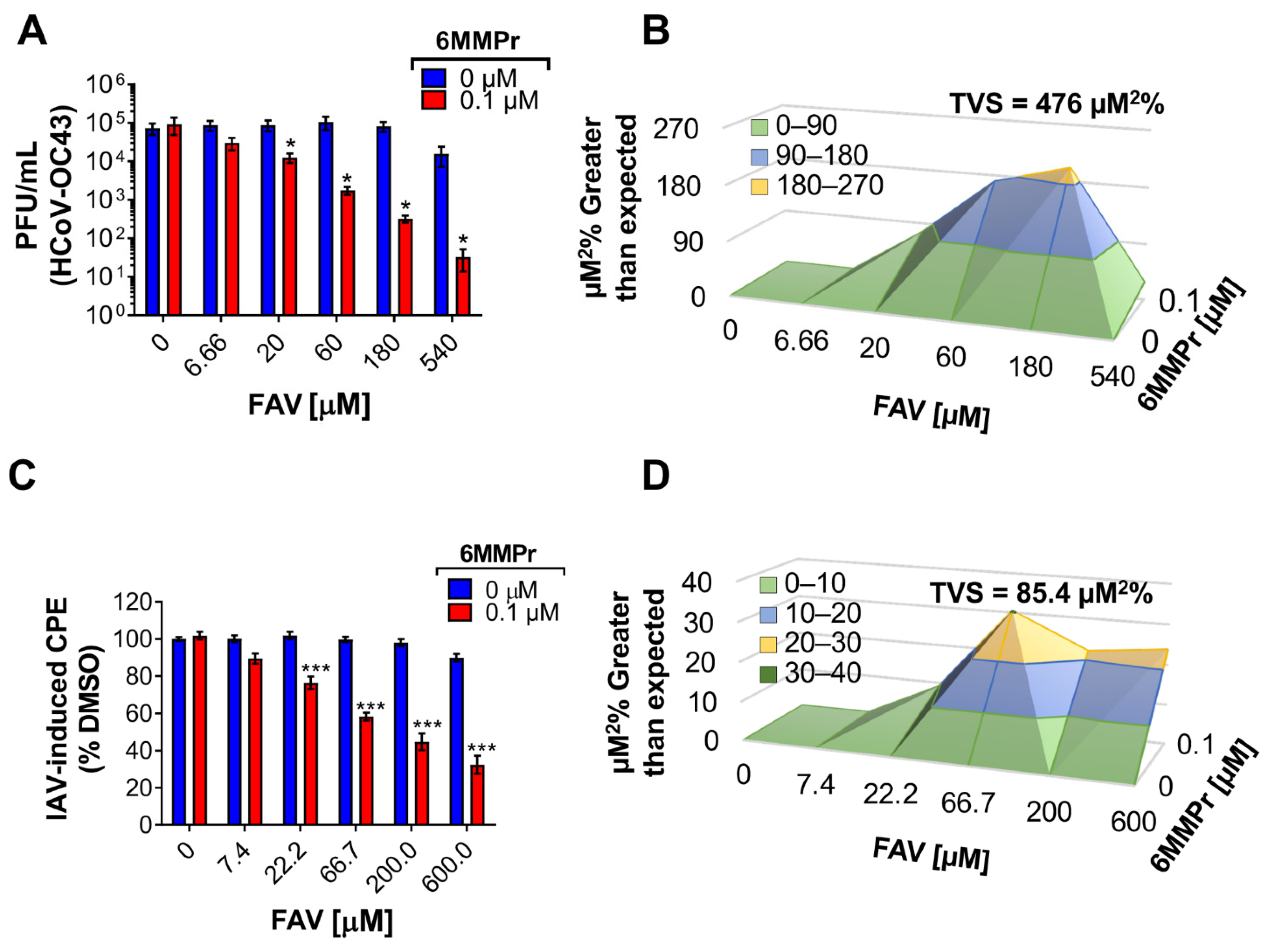
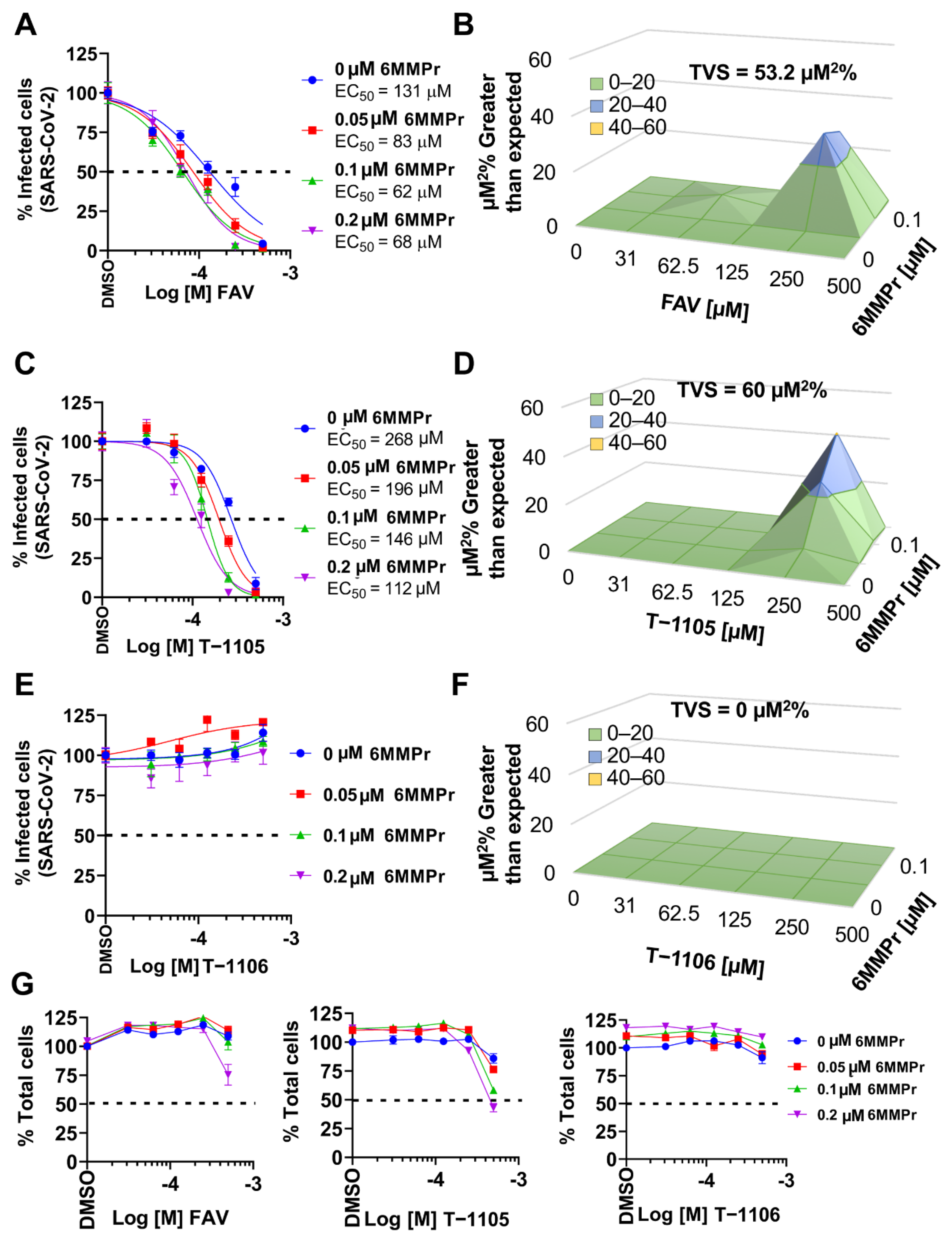
3.5. 6MMPr Synergizes the Antiviral Effect of T-1105 and Favipiravir against Zika Virus and Respiratory RNA Viruses including SARS-CoV-2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delang, L.; Abdelnabi, R.; Neyts, J. Favipiravir as a potential countermeasure against neglected and emerging RNA viruses. Antivir. Res. 2018, 153, 85–94. [Google Scholar] [CrossRef]
- Shiraki, K.; Daikoku, T. Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacol. Ther. 2020, 209, 107512. [Google Scholar] [CrossRef]
- Pilkington, V.; Pepperrell, T.; Hill, A. A review of the safety of favipiravir-a potential treatment in the COVID-19 pandemic? J. Virus Erad. 2020, 6, 45–51. [Google Scholar] [CrossRef]
- Driouich, J.S.; Cochin, M.; Lingas, G.; Moureau, G.; Touret, F.; Petit, P.R.; Piorkowski, G.; Barthélémy, K.; Laprie, C.; Coutard, B.; et al. Favipiravir antiviral efficacy against SARS-CoV-2 in a hamster model. Nat. Commun. 2021, 12, 1735. [Google Scholar] [CrossRef] [PubMed]
- Guedj, J.; Piorkowski, G.; Jacquot, F.; Madelain, V.; Nguyen, T.H.T.; Rodallec, A.; Gunther, S.; Carbonnelle, C.; Mentré, F.; Raoul, H.; et al. Antiviral efficacy of favipiravir against Ebola virus: A translational study in cynomolgus macaques. PLoS Med. 2018, 15, e1002535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irie, K.; Nakagawa, A.; Fujita, H.; Tamura, R.; Eto, M.; Ikesue, H.; Muroi, N.; Tomii, K.; Hashida, T. Pharmacokinetics of Favipiravir in Critically Ill Patients With COVID-19. Clin. Transl. Sci. 2020, 13, 880–885. [Google Scholar] [CrossRef]
- Doi, Y.; Hibino, M.; Hase, R.; Yamamoto, M.; Kasamatsu, Y.; Hirose, M.; Mutoh, Y.; Homma, Y.; Terada, M.; Ogawa, T.; et al. A Prospective, Randomized, Open-Label Trial of Early versus Late Favipiravir Therapy in Hospitalized Patients with COVID-19. Antimicrob. Agents Chemother. 2020, 64, e01897-20. [Google Scholar] [CrossRef]
- Ivashchenko, A.A.; Dmitriev, K.A.; Vostokova, N.V.; Azarova, V.N.; Blinow, A.A.; Egorova, A.N.; Gordeev, I.G.; Ilin, A.P.; Karapetian, R.N.; Kravchenko, D.V.; et al. AVIFAVIR for Treatment of Patients with Moderate Coronavirus Disease 2019 (COVID-19): Interim Results of a Phase II/III Multicenter Randomized Clinical Trial. Clin. Infect. Dis. 2021, 73, 531–534. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Guedj, J.; Anglaret, X.; Laouénan, C.; Madelain, V.; Taburet, A.M.; Baize, S.; Sissoko, D.; Pastorino, B.; Rodallec, A.; et al. Favipiravir pharmacokinetics in Ebola-Infected patients of the JIKI trial reveals concentrations lower than targeted. PLoS Negl. Trop. Dis. 2017, 11, e0005389. [Google Scholar] [CrossRef] [Green Version]
- Shannon, A.; Selisko, B.; Le, N.T.; Huchting, J.; Touret, F.; Piorkowski, G.; Fattorini, V.; Ferron, F.; Decroly, E.; Meier, C.; et al. Rapid incorporation of Favipiravir by the fast and permissive viral RNA polymerase complex results in SARS-CoV-2 lethal mutagenesis. Nat. Commun. 2020, 11, 4682. [Google Scholar] [CrossRef] [PubMed]
- Arias, A.; Thorne, L.; Goodfellow, I. Favipiravir elicits antiviral mutagenesis during virus replication in vivo. eLife 2014, 3, e03679. [Google Scholar] [CrossRef] [PubMed]
- Perales, C.; Gallego, I.; de Ávila, A.I.; Soria, M.E.; Gregori, J.; Quer, J.; Domingo, E. The increasing impact of lethal mutagenesis of viruses. Future Med. Chem. 2019, 11, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Patterson, S.E.; Bonnac, L.F.; Geraghty, R.J. Nucleobases and corresponding nucleosides display potent antiviral activities against dengue virus possibly through viral lethal mutagenesis. PLoS Negl. Trop. Dis. 2018, 12, e0006421. [Google Scholar] [CrossRef]
- Sangawa, H.; Komeno, T.; Nishikawa, H.; Yoshida, A.; Takahashi, K.; Nomura, N.; Furuta, Y. Mechanism of action of T-705 ribosyl triphosphate against influenza virus RNA polymerase. Antimicrob. Agents Chemother. 2013, 57, 5202–5208. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Smith, L.K.; Rajwanshi, V.K.; Kim, B.; Deval, J. The ambiguous base-pairing and high substrate efficiency of T-705 (Favipiravir) Ribofuranosyl 5’-triphosphate towards influenza A virus polymerase. PLoS ONE 2013, 8, e68347. [Google Scholar] [CrossRef] [PubMed]
- Elena, S.F.; Sanjuán, R. Adaptive Value of High Mutation Rates of RNA Viruses: Separating Causes from Consequences. J. Virol. 2005, 79, 11555–11558. [Google Scholar] [CrossRef] [Green Version]
- Kautz, T.F.; Forrester, N.L. RNA Virus Fidelity Mutants: A Useful Tool for Evolutionary Biology or a Complex Challenge? Viruses 2018, 10, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eigen, M. Error catastrophe and antiviral strategy. Proc. Natl. Acad. Sci. USA 2002, 99, 13374–13376. [Google Scholar] [CrossRef] [Green Version]
- Domingo, E.; Perales, C.; Agudo, R.; Arias, A.; Escarmís, C.; Ferrer-Orta, C.; Verdaguer, N. Mutation, Quasispecies, and Lethal Mutagenesis. In The Picornaviruses; Wiley: Hoboken, NJ, USA, 2010; pp. 195–211. [Google Scholar]
- Bull, J.J.; Sanjuán, R.; Wilke, C.O. Theory of Lethal Mutagenesis for Viruses. J. Virol. 2007, 81, 2930–2939. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.M. Chapter 25, Nucleotide Biosynthesis. In Biochemistry, 5th ed.; Tymoczko, J.L., Lubert, S., Eds.; W. H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Naesens, L.; Guddat, L.W.; Keough, D.T.; Van Kuilenburg, A.B.P.; Meijer, J.; Vande Voorde, J.; Balzarini, J. Role of human hypoxanthine guanine phosphoribosyltransferase in activation of the antiviral agent T-705 (favipiravir). Mol. Pharmacol. 2013, 84, 615–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huchting, J.; Vanderlinden, E.; Van Berwaer, R.; Meier, C.; Naesens, L. Cell line-dependent activation and antiviral activity of T-1105, the non-fluorinated analogue of T-705 (favipiravir). Antivir. Res. 2019, 167, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, U.; Raju, R.; Udwadia, Z.F. Favipiravir: A new and emerging antiviral option in COVID-19. Med. J. Armed. Forces India 2020, 76, 370–376. [Google Scholar] [CrossRef]
- Yeo, K.L.; Chen, Y.L.; Xu, H.Y.; Dong, H.; Wang, Q.Y.; Yokokawa, F.; Shi, P.Y. Synergistic suppression of dengue virus replication using a combination of nucleoside analogs and nucleoside synthesis inhibitors. Antimicrob. Agents Chemother. 2015, 59, 2086–2093. [Google Scholar] [CrossRef] [Green Version]
- Westover, J.B.; Sefing, E.J.; Bailey, K.W.; Van Wettere, A.J.; Jung, K.H.; Dagley, A.; Wandersee, L.; Downs, B.; Smee, D.F.; Furuta, Y.; et al. Low-dose ribavirin potentiates the antiviral activity of favipiravir against hemorrhagic fever viruses. Antivir. Res. 2016, 126, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernachio, J.H.; Bleiman, B.; Bryant, K.D.; Chamberlain, S.; Hunley, D.; Hutchins, J.; Ames, B.; Gorovits, E.; Ganguly, B.; Hall, A.; et al. INX-08189, a phosphoramidate prodrug of 6-O-methyl-2′-C-methyl guanosine, is a potent inhibitor of hepatitis C virus replication with excellent pharmacokinetic and pharmacodynamic properties. Antimicrob. Agents Chemother. 2011, 55, 1843–1851. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, A.; Patnayak, D.P.; Chander, Y.; Parsad, M.; Goyal, S.M. Survival of two avian respiratory viruses on porous and nonporous surfaces. Avian Dis. 2006, 50, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Rawson, J.M.; Roth, M.E.; Xie, J.; Daly, M.B.; Clouser, C.L.; Landman, S.R.; Reilly, C.S.; Bonnac, L.; Kim, B.; Patterson, S.E.; et al. Synergistic reduction of HIV-1 infectivity by 5-azacytidine and inhibitors of ribonucleotide reductase. Bioorg. Med. Chem. 2016, 24, 2410–2422. [Google Scholar] [CrossRef]
- Cohen, S.; Megherbi, M.; Jordheim, L.P.; Lefebvre, I.; Perigaud, C.; Dumontet, C.; Guitton, J. Simultaneous analysis of eight nucleoside triphosphates in cell lines by liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 3831–3840. [Google Scholar] [CrossRef]
- Jaworski, E.; Routh, A. Parallel ClickSeq and Nanopore sequencing elucidates the rapid evolution of defective-interfering RNAs in Flock House virus. PLoS Pathog. 2017, 13, e1006365. [Google Scholar] [CrossRef] [Green Version]
- Routh, A.; Head, S.R.; Ordoukhanian, P.; Johnson, J.E. ClickSeq: Fragmentation-Free Next-Generation Sequencing via Click Ligation of Adaptors to Stochastically Terminated 3’-Azido cDNAs. J. Mol. Biol. 2015, 427, 2610–2616. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Routh, A. Mapping RNA-capsid interactions and RNA secondary structure within virus particles using next-generation sequencing. Nucleic Acids Res. 2020, 48, e12. [Google Scholar] [CrossRef] [Green Version]
- Jaworski, E.; Routh, A. ClickSeq: Replacing Fragmentat.tion and Enzymatic Ligation with Click-Chemistry to Prevent Sequence Chimeras. Methods Mol. Biol. 2018, 1712, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smee, D.F.; Prichard, M.N. Comparison of three dimensional synergistic analyses of percentage versus logarithmic data in antiviral studies. Antivir. Res. 2017, 145, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Goldhill, D.H.; Langat, P.; Xie, H.; Galiano, M.; Miah, S.; Kellam, P.; Zambon, M.; Lackenby, A.; Barclay, W.S. Determining the Mutation Bias of Favipiravir in Influenza Virus Using Next-Generation Sequencing. J. Virol. 2019, 93, e01217–e01218. [Google Scholar] [CrossRef] [Green Version]
- Baranovich, T.; Wong, S.S.; Armstrong, J.; Marjuki, H.; Webby, R.J.; Webster, R.G.; Govorkova, E.A. T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J. Virol. 2013, 87, 3741–3751. [Google Scholar] [CrossRef] [Green Version]
- De Ávila, A.I.; Gallego, I.; Soria, M.E.; Gregori, J.; Quer, J.; Ignacio Esteban, J.; Rice, C.M.; Domingo, E.; Perales, C. Lethal mutagenesis of hepatitis C virus induced by favipiravir. PLoS ONE 2016, 11, e0164691. [Google Scholar] [CrossRef] [Green Version]
- Kamal, M.A.; Christopherson, R.I. Accumulation of 5-phosphoribosyl-1-pyrophosphate in human CCRF-CEM leukaemia cells treated with antifolates. Int. J. Biochem. Cell Biol. 2004, 36, 545–551. [Google Scholar] [CrossRef]
- Shi, R.Z.; Lyons, S.D.; Christopherson, R.I. Metabolic effects of thiopurine derivatives against human CCRF-CEM leukaemia cells. Int. J. Biochem. Cell Biol. 1998, 30, 885–895. [Google Scholar] [CrossRef]
- Fotoohi, A.K.; Wrabel, A.; Moshfegh, A.; Peterson, C.; Albertioni, F. Molecular mechanisms underlying the enhanced sensitivity of thiopurine-resistant T-lymphoblastic cell lines to methyl mercaptopurineriboside. Biochem. Pharmacol. 2006, 72, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Bradburne, A.F.; Bynoe, M.L.; Tyrrell, D.A. Effects of a "new" human respiratory virus in volunteers. Br. Med. J. 1967, 3, 767–769. [Google Scholar] [CrossRef] [Green Version]
- Reed, S.E. The behaviour of recent isolates of human respiratory coronavirus in vitro and in volunteers: Evidence of heterogeneity among 229E-related strains. J. Med. Virol. 1984, 13, 179–192. [Google Scholar] [CrossRef]
- Furuta, Y.; Takahashi, K.; Shiraki, K.; Sakamoto, K.; Smee, D.F.; Barnard, D.L.; Gowen, B.B.; Julander, J.G.; Morrey, J.D. T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antivir. Res. 2009, 82, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Udwadia, Z.F.; Singh, P.; Barkate, H.; Patil, S.; Rangwala, S.; Pendse, A.; Kadam, J.; Wu, W.; Caracta, C.F.; Tandon, M. Efficacy and safety of favipiravir, an oral RNA-dependent RNA polymerase inhibitor, in mild-to-moderate COVID-19: A randomized, comparative, open-label, multicenter, phase 3 clinical trial. Int. J. Infect. Dis. 2021, 103, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Takeda, M.; Matsuyama, S. The anti-influenza virus drug favipiravir has little effect on replication of SARS-CoV-2 in cultured cells. Antimicrob. Agents Chemother. 2021, 65, e00020–e00021. [Google Scholar] [CrossRef]
- Vogt, M.H.J.; Stet, E.H.; De Abreu, R.A.; Bökkerink, J.P.M.; Lambooy, L.H.J.; Trijbels, F.J.M. The importance of methylthio-IMP for methylmercaptopurine ribonucleoside (Me-MPR) cytotoxicity in Molt F4 human malignant T-lymphoblasts. BBA-Mol. Basis Dis. 1993, 1181, 189–194. [Google Scholar] [CrossRef]
- Stet, E.H.; De Abreu, R.A.; Bökkerink, J.P.M.; Lambooy, L.H.J.; Vogels-Mentink, T.M.; Keizer-Garritsen, J.J.; Trijbels, F.J.M. Reversal of methylmercaptopurine ribonucleoside cytotoxicity by purine ribonucleosides and adenine. Biochem. Pharmacol. 1995, 49, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.L.; Zaluzec, E.J.; Wery, J.P.; Niu, L.; Switzer, R.L.; Zalkin, H.; Satow, Y. Structure of the allosteric regulatory enzyme of purine biosynthesis. Science 1994, 264, 1427–1433. [Google Scholar] [CrossRef]
- Jin, Z.; Tucker, K.; Lin, X.; Kao, C.C.; Shaw, K.; Tan, H.; Symons, J.; Behera, I.; Rajwanshi, V.K.; Dyatkina, N.; et al. Biochemical evaluation of the inhibition properties of favipiravir and 2=-C-methyl-cytidine triphosphates against human and mouse norovirus RNA polymerases. Antimicrob. Agents Chemother. 2015, 59, 7504–7516. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Peng, R.; Yuan, B.; Wang, M.; Zhao, J.; Fu, L.; Qi, J.; Shi, Y. Structural Basis of SARS-CoV-2 Polymerase Inhibition by Favipiravir. Innovation 2021, 2, 10080. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Romero, E.; De Oya, N.J.; Domingo, E.; Saiza, J.C. Extinction of west nile virus by favipiravir through lethal mutagenesis. Antimicrob. Agents Chemother. 2017, 61, e01400-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Avila, A.I.; Moreno, E.; Perales, C.; Domingo, E. Favipiravir can evoke lethal mutagenesis and extinction of foot-and-mouth disease virus. Virus Res. 2017, 233, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Rozen-Gagnon, K.; Stapleford, K.A.; Mongelli, V.; Blanc, H.; Failloux, A.B.; Saleh, M.C.; Vignuzzi, M. Alphavirus Mutator Variants Present Host-Specific Defects and Attenuation in Mammalian and Insect Models. PLoS Pathog. 2014, 10, e1003877. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Kirkegaard, K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 2005, 1, 0102–0110. [Google Scholar] [CrossRef]
- Domingo, E. Viruses at the edge of adaptation. Virology 2000, 270, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhong, W.; Salam, A.; Tarning, J.; Zhan, Q.; Huang, J.A.; Weng, H.; Bai, C.; Ren, Y.; Yamada, K.; et al. Phase 2a, open-label, dose-escalating, multi-center pharmacokinetic study of favipiravir (T-705) in combination with oseltamivir in patients with severe influenza. EBioMedicine 2020, 62, 103125. [Google Scholar] [CrossRef]
- Dabbous, H.M.; El-Sayed, M.H.; El Assal, G.; Elghazaly, H.; Ebeid, F.F.S.; Sherief, A.F.; Elgaafary, M.; Fawzy, E.; Hassany, S.M.; Riad, A.R.; et al. Safety and efficacy of favipiravir versus hydroxychloroquine in management of COVID-19: A randomised controlled trial. Sci. Rep. 2021, 11, 7282. [Google Scholar] [CrossRef]
- McKimm-Breschkin, J.L.; Jiang, S.; Hui, D.S.; Beigel, J.H.; Govorkova, E.A.; Lee, N. Prevention and treatment of respiratory viral infections: Presentations on antivirals, traditional therapies and host-directed interventions at the 5th ISIRV Antiviral Group conference. Antivir. Res. 2018, 149, 118–142. [Google Scholar] [CrossRef]
- Ison, M.G.; Scheetz, M.H. Understanding the pharmacokinetics of Favipiravir: Implications for treatment of influenza and COVID-19. EBioMedicine 2021, 63, 103204. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.R.; Coenen, M.J.H.; Vermeulen, S.H.; Derijks, L.J.J.; van Marrewijk, C.J.; Klungel, O.H.; Scheffer, H.; Franke, B.; Guchelaar, H.J.; de Jong, D.J.; et al. Early Assessment of Thiopurine Metabolites Identifies Patients at Risk of Thiopurine-induced Leukopenia in Inflammatory Bowel Disease. J. Crohn’s Colitis 2017, 11, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soto-Acosta, R.; Edwards, T.C.; Dreis, C.D.; Krishna, V.D.; Cheeran, M.C.-J.; Qiu, L.; Xie, J.; Bonnac, L.F.; Geraghty, R.J. Enhancing the Antiviral Potency of Nucleobases for Potential Broad-Spectrum Antiviral Therapies. Viruses 2021, 13, 2508. https://doi.org/10.3390/v13122508
Soto-Acosta R, Edwards TC, Dreis CD, Krishna VD, Cheeran MC-J, Qiu L, Xie J, Bonnac LF, Geraghty RJ. Enhancing the Antiviral Potency of Nucleobases for Potential Broad-Spectrum Antiviral Therapies. Viruses. 2021; 13(12):2508. https://doi.org/10.3390/v13122508
Chicago/Turabian StyleSoto-Acosta, Ruben, Tiffany C. Edwards, Christine D. Dreis, Venkatramana D. Krishna, Maxim C-J. Cheeran, Li Qiu, Jiashu Xie, Laurent F. Bonnac, and Robert J. Geraghty. 2021. "Enhancing the Antiviral Potency of Nucleobases for Potential Broad-Spectrum Antiviral Therapies" Viruses 13, no. 12: 2508. https://doi.org/10.3390/v13122508