
Hepatitis C Virus Core Protein Down-Regulates Expression of Src-Homology 2 Domain Containing Protein Tyrosine Phosphatase by Modulating Promoter DNA Methylation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Cell Culture
2.3. Cell Lysis and Western Blot
2.4. RT-qPCR
2.5. Flow Cytometry
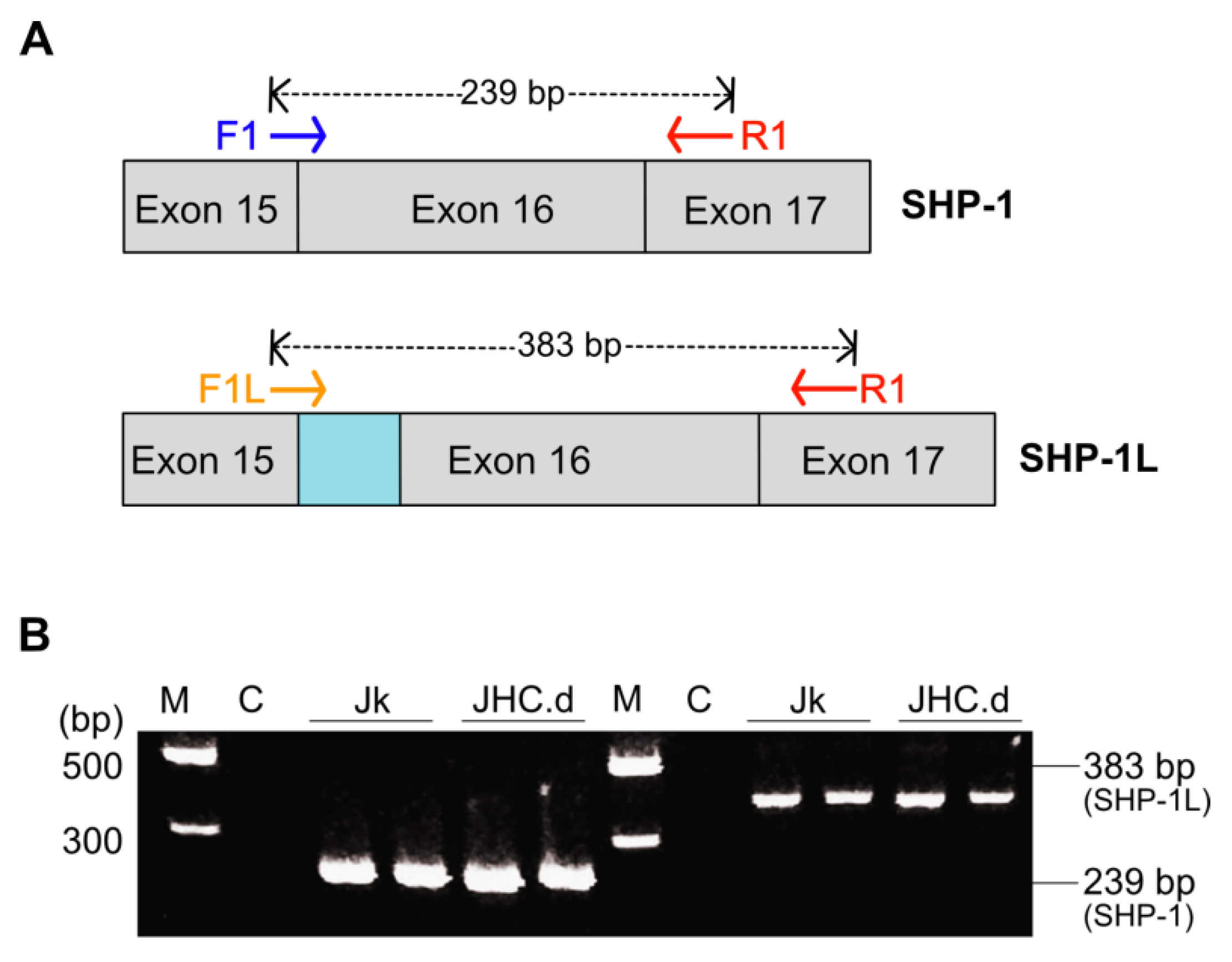
2.6. Detection of Spliced SHP-1 mRNA
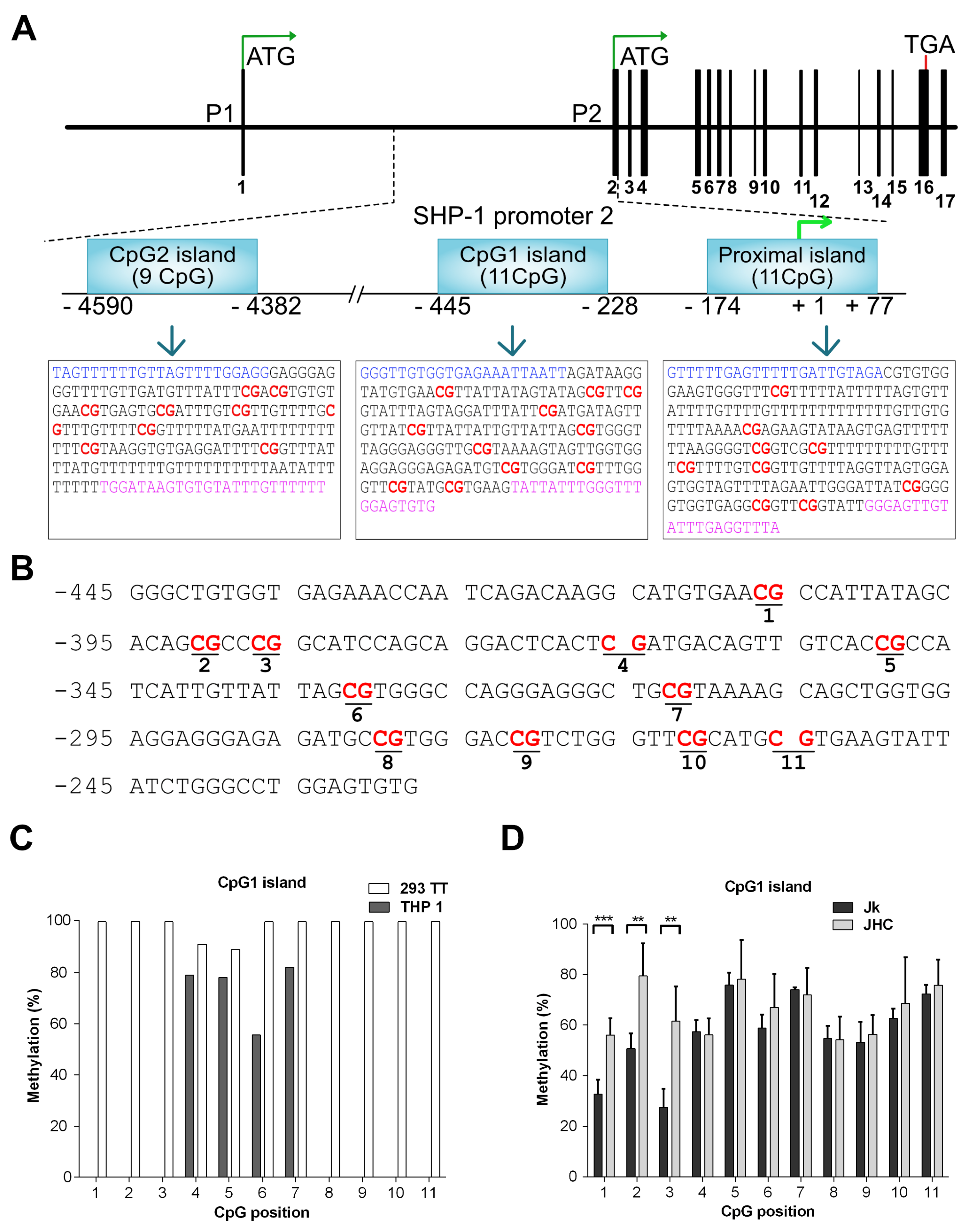
2.7. Promoter CpG Island Identification and Primer Design
2.8. Bisulfite Sequencing
2.9. 5-Aza-2′-deoxycytidine (5-Aza) Treatment
2.10. Statistical Analysis
3. Results
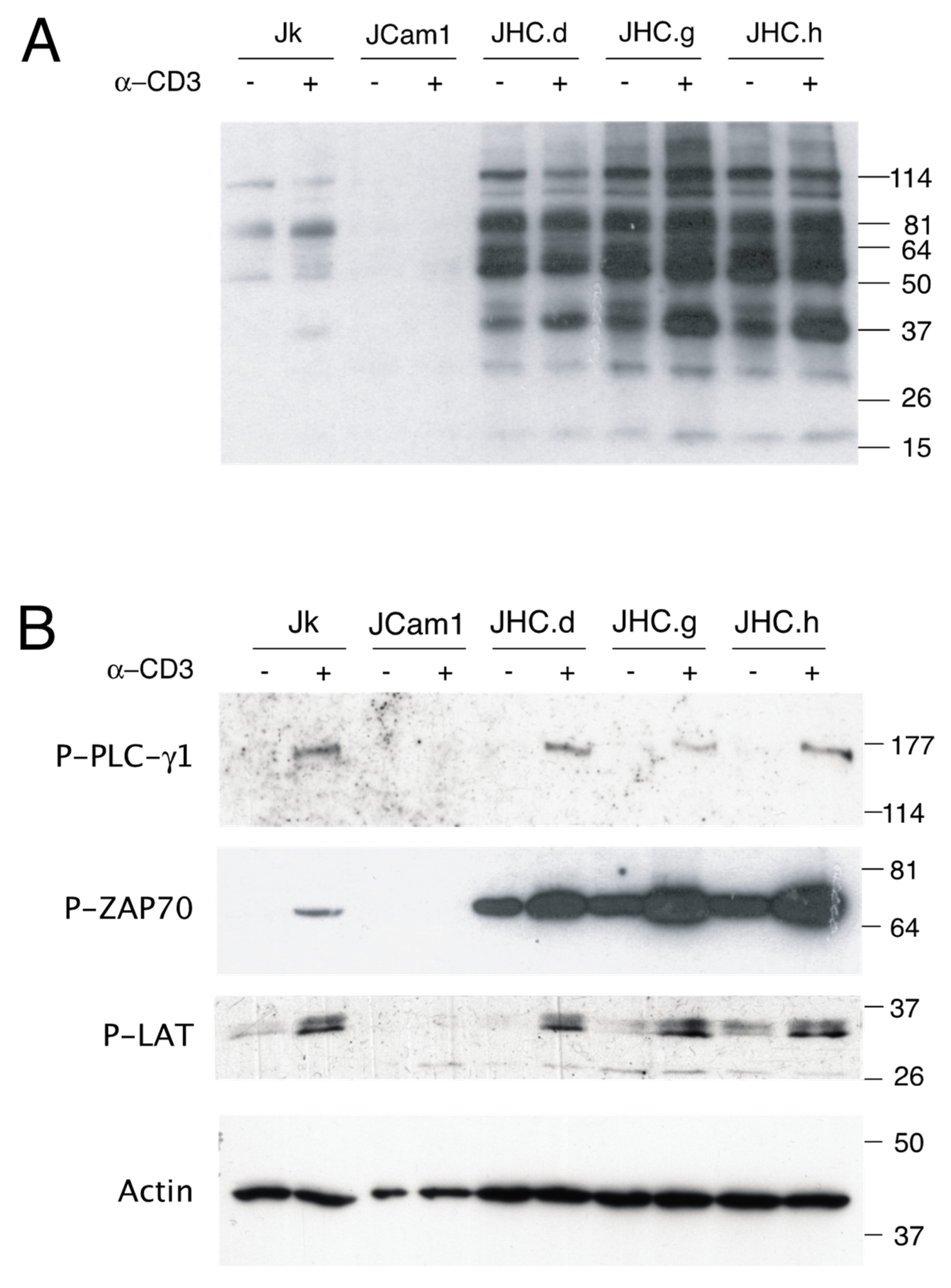
3.1. Altered Tyrosine Phosphorylation of TCR-Related Signaling Molecules in the C Protein Expressing T Cells
3.2. Specific Down-Regulation of the SHP-1 Protein Expression in the C Protein Expressing Cells
3.3. SHP-1 mRNA Splicing Is Not Altered in the C Protein Expressing Cells
3.4. Reduced SHP-1 Expression Correlates with Its P2 Promoter Hypermethylation
3.5. Responsiveness of the SHP-1 Gene Expression to 5-Aza-2’-deoxycytidine (5-Aza) Treatment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El–Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Dash, S.; Aydin, Y.; Widmer, K.E.; Nayak, L. Hepatocellular Carcinoma Mechanisms Associated with Chronic HCV Infection and the Impact of Direct-Acting Antiviral Treatment. J. Hepatocell. Carcinoma 2020, 7, 45–76. [Google Scholar] [CrossRef] [Green Version]
- Pol, S.; Vallet-Pichard, A.; Hermine, O. Extrahepatic cancers and chronic HCV infection. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-T.; Sheu, J.-C.; Lin, J.-T.; Wang, T.-H.; Chen, D.S. Detection of Replicative Form of Hepatitis C Virus RNA in Peripheral Blood Mononuclear Cells. J. Infect. Dis. 1992, 166, 1167–1169. [Google Scholar] [CrossRef]
- Hsieh, T.-T.; Yao, D.-S.; Sheen, I.-S.; Liaw, Y.-F.; Pao, C.C. Hepatitis C Virus in Peripheral Blood Mononuclear Cells. Am. J. Clin. Pathol. 1992, 98, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Huang, J.Y.; Wang, C.-H.; Tahara, S.M.; Zhou, L.; Kondo, Y.; Schechter, J.; Su, L.; Lai, M.M.C.; Wakita, T.; et al. Hepatitis C virus has a genetically determined lymphotropism through co-receptor B7.2. Nat. Commun. 2017, 8, 13882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y.K.; Igarashi, H.; Kanematu, T.; Fujiwara, K.; Wong, D.C.; Purcell, R.H.; Yoshikura, H. Sequence analysis of the hepatitis C virus genome recovered from serum, liver, and peripheral blood mononuclear cells of infected chimpanzees. J. Virol. 1997, 71, 5769–5773. [Google Scholar] [CrossRef] [Green Version]
- Blackard, J.T.; Kemmer, N.; Sherman, K.E. Extrahepatic replication of HCV: Insights into clinical manifestations and biological consequences. Hepatology 2006, 44, 15–22. [Google Scholar] [CrossRef]
- Yan, F.M.; Chen, A.S.; Hao, F.; Zhao, X.P.; Gu, C.H.; Bin Zhao, L.; Yang, D.L.; Hao, L.J. Hepatitis C virus may infect extrahepatic tissues in patients with hepatitis C. World J. Gastroenterol. 2000, 6, 805–811. [Google Scholar] [CrossRef]
- Gokhale, N.S.; Vazquez, C.; Horner, S.M. Hepatitis C virus: Strategies to evade antiviral responses. Futur. Virol. 2014, 9, 1061–1075. [Google Scholar] [CrossRef] [Green Version]
- Grakoui, A.; Shoukry, N.H.; Woollard, D.J.; Han, J.-H.; Hanson, H.L.; Ghrayeb, J.; Murthy, K.K.; Rice, C.M.; Walker, C.M. HCV Persistence and Immune Evasion in the Absence of Memory T Cell Help. Science 2003, 302, 659–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.R.; Marousis, C.G.; Davis, G.L.; Rice, C.M.; Wong, J.; Houghton, M.; Lau, J.Y. The role of hepatitis C virus-specific cytotoxic T lymphocytes in chronic hepatitis C. J. Immunol. 1997, 158, 1473–1481. [Google Scholar]
- Rehermann, B.; Chang, K.M.; McHutchinson, J.; Kokka, R.; Houghton, M.; Rice, C.M.; Chisari, F.V. Differential cytotoxic T-lymphocyte responsiveness to the hepatitis B and C viruses in chronically infected patients. J. Virol. 1996, 70, 7092–7102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, J.; Okamoto, T.; Yamashita, K.; Sakai, Y.; Yamamoto, M.; Horii, T.; Standley, D.M.; Moriishi, K.; Moriya, K.; Koike, K.; et al. Characterization of SPP inhibitors suppressing propagation of HCV and protozoa. Proc. Natl. Acad. Sci. USA 2017, 114, E10782–E10791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergqvist, A.; Rice, C.M. Transcriptional Activation of the Interleukin-2 Promoter by Hepatitis C Virus Core Protein. J. Virol. 2001, 75, 772–781. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.M.; You, L.R.; Hwang, L.H.; Lee, Y.H. Direct interaction of hepatitis C virus core protein with the cellular lymphotoxin-beta receptor modulates the signal pathway of the lymphotoxin-beta receptor. J. Virol. 1997, 71, 9417–9426. [Google Scholar] [CrossRef] [Green Version]
- Sundström, S.; Ota, S.; Dimberg, L.Y.; Masucci, M.; Bergqvist, A. Hepatitis C Virus Core Protein Induces an Anergic State Characterized by Decreased Interleukin-2 Production and Perturbation of Mitogen-Activated Protein Kinase Responses. J. Virol. 2005, 79, 2230–2239. [Google Scholar] [CrossRef] [Green Version]
- Bergqvist, A.; Sundström, S.; Dimberg, L.Y.; Gylfe, E.; Masucci, M. The Hepatitis C Virus Core Protein Modulates T Cell Responses by Inducing Spontaneous and Altering T-cell Receptor-triggered Ca2+ Oscillations. J. Biol. Chem. 2003, 278, 18877–18883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez-Villar, M.; Muñoz-Suano, A.; Anaya-Baz, B.; Aguilar, S.; Novalbos, J.P.; Giron, J.A.; Rodríguez-Iglesias, M.; Garcia-Cozar, F. Hepatitis C virus core protein up-regulates anergy-related genes and a new set of genes, which affects T cell homeostasis. J. Leukoc. Biol. 2007, 82, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8+ T Cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Macian, F.; Garcia-Cozar, F.; Im, S.-H.; Horton, H.F.; Byrne, M.C.; Rao, A. Transcriptional Mechanisms Underlying Lymphocyte Tolerance. Cell 2002, 109, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Ostman, A.; Böhmer, F.D. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases. Trends Cell Biol. 2001, 11, 258–266. [Google Scholar] [CrossRef]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein Tyrosine Phosphatases in the Human Genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Mustelin, T.; Vang, T.; Bottini, N. Protein tyrosine phosphatases and the immune response. Nat. Rev. Immunol. 2005, 5, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Tarlinton, D. CD45: Direct and indirect government of immune regulation. Immunol. Lett. 2004, 94, 167–174. [Google Scholar] [CrossRef]
- Kishihara, K.; Penninger, J.; Wallace, V.A.; Kündig, T.M.; Kawai, K.; Wakeham, A.; Timms, E.; Pfeffer, K.; Ohashi, P.S.; Thomas, M.L.; et al. Normal B lymphocyte development but impaired T cell maturation in CD45-Exon6 protein tyrosine phosphatase-deficient mice. Cell 1993, 74, 143–156. [Google Scholar] [CrossRef]
- McNeill, L.; Salmond, R.J.; Cooper, J.C.; Carret, C.K.; Cassady-Cain, R.L.; Roche-Molina, M.; Tandon, P.; Holmes, N.; Alexander, D.R. The Differential Regulation of Lck Kinase Phosphorylation Sites by CD45 is Critical for T Cell Receptor Signaling Responses. Immunity 2007, 27, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, R.J.; Bowne, D.B.; Flores, E.; Thomas, M.L. Characterization of hematopoietic intracellular protein tyrosine phosphatases: Description of a phosphatase containing an SH2 domain and another enriched in proline-, glutamic acid-, serine-, and threonine-rich sequences. Mol. Cell Biol. 1992, 12, 2396–2405. [Google Scholar]
- Yi, T.L.; Cleveland, J.L.; Ihle, J.N. Protein tyrosine phosphatase containing SH2 domains: Characterization, preferential expression in hematopoietic cells, and localization to human chromosome 12p12-p13. Mol. Cell Biol. 1992, 12, 836–846. [Google Scholar]
- Zhang, J.; Somani, A.-K.; Siminovitch, K.A. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin. Immunol. 2000, 12, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Berrada, K.; Yang, W.; Tabrizi, M.; Platanias, L.C.; Yi, T. Direct association with and dephosphorylation of Jak2 kinase by the SH2-domain-containing protein tyrosine phosphatase SHP-1. Mol. Cell. Biol. 1996, 16, 6985–6992. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Sun, M.; Liu, L.; Zhou, G.W. The function of the protein tyrosine phosphatase SHP-1 in cancer. Gene 2003, 306, 1–12. [Google Scholar] [CrossRef]
- Östman, A.; Hellberg, C.; Böhmer, F.D. Protein-tyrosine phosphatases and cancer. Nat. Rev. Cancer 2006, 6, 307–320. [Google Scholar] [CrossRef]
- Zhang, Q.; Raghunath, P.N.; Vonderheid, E.; Ødum, N.; Wasik, M.A. Lack of Phosphotyrosine Phosphatase SHP-1 Expression in Malignant T-Cell Lymphoma Cells Results from Methylation of the SHP-1 Promoter. Am. J. Pathol. 2000, 157, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Witzig, T.E.; Hu, G.; Offer, S.M.; Wellik, L.E.; Han, J.J.; Stenson, M.J.; Dogan, A.; Diasio, R.B.; Gupta, M. Epigenetic mechanisms of protein tyrosine phosphatase 6 suppression in diffuse large B-cell lymphoma: Implications for epigenetic therapy. Leukemia 2013, 28, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sooman, L.; Ekman, S.; Tsakonas, G.; Jaiswal, A.; Navani, S.; Edqvist, P.-H.; Pontén, F.; Bergström, S.; Johansson, M.; Wu, X.; et al. PTPN6 expression is epigenetically regulated and influences survival and response to chemotherapy in high-grade gliomas. Tumor Biol. 2014, 35, 4479–4488. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, L.; Liu, X.; Nie, Z.; Wang, X.; Pan, Y.; Luo, J. Research on the epigenetic regulation mechanism of thePTPN6gene in advanced chronic myeloid leukaemia. Br. J. Haematol. 2017, 178, 728–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Kydd, A.R.; Nakase, K.; Noonan, K.M.; Murakami, A.; Tao, H.; Dwyer, M.; Xu, C.; Zhu, Q.; Marasco, W.A. Negative regulation of the SH2-homology–containing protein-tyrosine phosphatase-1 (SHP-1) P2 promoter by the HTLV-1 Tax oncoprotein. Blood 2007, 110, 2110–2120. [Google Scholar] [CrossRef]
- Nakase, K.; Cheng, J.; Zhu, Q.; Marasco, W.A. Mechanisms of SHP-1 P2 promoter regulation in hematopoietic cells and its silencing in HTLV-1-transformed T cells. J. Leukoc. Biol. 2008, 85, 165–174. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.Y.; Marzec, M.; Raghunath, P.N.; Nagasawa, T.; Wasik, M.A. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 6948–6953. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Amin, H.M.; Frantz, C.; Franko, B.; Lee, J.; Lin, Q.; Lai, R. Restoration of shp1 expression by 5-AZA-2′-deoxycytidine is associated with downregulation of JAK3/STAT3 signaling in ALK-positive anaplastic large cell lymphoma. Leukemia 2006, 20, 1602–1609. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.-Z.; Ding, K.; Wang, Z.-R.; Ding, C.-H.; Lei, S.-J.; Liu, J.-P.; Yin, C.; Hu, P.-F.; Ding, J.; Chen, W.-S.; et al. SHP-1 Acts as a Tumor Suppressor in Hepatocarcinogenesis and HCC Progression. Cancer Res. 2018, 78, 4680–4691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, C.B.; Pastrana, D.V.; Lowy, D.R.; Schiller, J.T. Efficient Intracellular Assembly of Papillomaviral Vectors. J. Virol. 2004, 78, 751–757. [Google Scholar] [CrossRef] [Green Version]
- 2019-Novel Coronavirus (2019-nCoV) Real-Time rRT-PCR Panel. Primers and Probes. Available online: https://www.cdc.gov/coronavirus/2019-ncov/downloads/rt-pcr-panel-primer-probes.pdf (accessed on 10 November 2021).
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Patterson, K.; Molloy, L.; Qu, W.; Clark, S. DNA Methylation: Bisulphite Modification and Analysis. J. Vis. Exp. 2011, 56, e3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melki, J.R.; Vincent, P.C.; Clark, S.J. Concurrent DNA hypermethylation of multiple genes in acute myeloid leukemia. Cancer Res. 1999, 59, 3730–3740. [Google Scholar] [PubMed]
- Parrish, R.R.; Day, J.J.; Lubin, F.D. Direct Bisulfite Sequencing for Examination of DNA Methylation with Gene and Nucleotide Resolution from Brain Tissues. Curr. Protoc. Neurosci. 2012, 60, 7–24. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhang, Y.; Fei, J.; Chang, X.; Fan, W.; Qian, X.; Zhang, T.; Lu, D. Rapid quantification of DNA methylation by measuring relative peak heights in direct bisulfite-PCR sequencing traces. Lab. Investig. 2009, 90, 282–290. [Google Scholar] [CrossRef]
- Pao, L.I.; Badour, K.; Siminovitch, K.A.; Neel, B.G. Nonreceptor Protein-Tyrosine Phosphatases in Immune Cell Signaling. Annu. Rev. Immunol. 2007, 25, 473–523. [Google Scholar] [CrossRef]
- Shin, E.-C.; Sung, P.S.; Park, S.-H. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat. Rev. Immunol. 2016, 16, 509–523. [Google Scholar] [CrossRef]
- Ma, X.-Z.; Jin, T.; Sakac, D.; Fahim, S.; Zhang, X.; Katsman, Y.; Bali, M.; Branch, D.R. Abnormal splicing of SHP-1 protein tyrosine phosphatase in human T cells: Implications for lymphomagenesis. Exp. Hematol. 2003, 31, 131–142. [Google Scholar] [CrossRef]
- Jin, Y.J.; Yu, C.L.; Burakoff, S.J. Human 70-kDa SHP-1L differs from 68-kDa SHP-1 in its C-terminal structure and catalytic activity. J. Biol. Chem. 1999, 274, 28301–28307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, U. SHP-1 and SHP-2 in T cells: Two phosphatases functioning at many levels. Immunol. Rev. 2009, 228, 342–359. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-H.; Lim, J.S.; Lim, S.-Y.; Tiwari, I.; Jang, K.L. Hepatitis C virus Core protein stimulates cell growth by down-regulating p16 expression via DNA methylation. Cancer Lett. 2011, 310, 61–68. [Google Scholar] [CrossRef]
- Ripoli, M.; Barbano, R.; Balsamo, T.; Piccoli, C.; Brunetti, V.; Coco, M.; Mazzoccoli, G.; Vinciguerra, M.; Pazienza, V. Hypermethylated levels of E-cadherin promoter in Huh-7 cells expressing the HCV core protein. Virus Res. 2011, 160, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Chen, R.; Li, Z.; Liu, Y.; Cheng, D.; Zhou, Q.; Zhou, J.; Lin, Q. Hepatitis C virus core upregulates the methylation status of the RASSF1A promoter through regulation of SMYD3 in hilar cholangiocarcinoma cells. Acta Biochim. Biophys. Sin. 2011, 43, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Kadlecek, T.; Au-Yeung, B.; Goodfellow, H.E.S.; Hsu, L.-Y.; Freedman, T.; Weiss, A. ZAP-70: An Essential Kinase in T-cell Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002279. [Google Scholar] [CrossRef] [Green Version]
- Mège, D.; Di Bartolo, V.; Germain, V.; Tuosto, L.; Michel, F.; Acuto, O. Mutation of tyrosines 492/493 in the kinase domain of ZAP-70 affects multiple T-cell receptor signaling pathways. J. Biol. Chem. 1996, 271, 32644–32652. [Google Scholar] [CrossRef] [Green Version]
- Plas, D.R.; Johnson, R.; Pingel, J.T.; Matthews, R.J.; Dalton, M.; Roy, G.; Chan, A.C.; Thomas, M.L.; Mellors, J.W.; Rinaldo, C.R.; et al. Direct Regulation of ZAP-70 by SHP-1 in T Cell Antigen Receptor Signaling. Science 1996, 272, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, U.; Ravichandran, K.S.; Pei, D.; Walsh, C.T.; Burakoff, S.J.; Neel, B.G. Lck-dependent tyrosyl phosphorylation of the phosphotyrosine phosphatase SH-PTP1 in murine T cells. Mol. Cell Biol. 1994, 14, 1824–1834. [Google Scholar]
- Oka, T.; Ouchida, M.; Koyama, M.; Ogama, Y.; Takada, S.; Nakatani, Y.; Tanaka, T.; Yoshino, T.; Hayashi, K.; Ohara, N.; et al. Gene silencing of the tyrosine phosphatase SHP1 gene by aberrant methylation in leukemias/lymphomas. Cancer Res. 2002, 62, 6390–6394. [Google Scholar] [PubMed]
- Wlodarski, P.; Zhang, Q.; Liu, X.; Kasprzycka, M.; Marzec, M.; Wasik, M.A. PU.1 Activates Transcription of SHP-1 Gene in Hematopoietic Cells. J. Biol. Chem. 2007, 282, 6316–6323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amara, K.; Trimeche, M.; Ziadi, S.; Laatiri, A.; Hachana, M.R.; Korbi, S. Prognostic significance of aberrant promoter hypermethylation of CpG islands in patients with diffuse large B-cell lymphomas. Ann. Oncol. 2008, 19, 1774–1786. [Google Scholar] [CrossRef]
- Koyama, M.; Oka, T.; Ouchida, M.; Nakatani, Y.; Nishiuchi, R.; Yoshino, T.; Hayashi, K.; Akagi, T.; Seino, Y. Activated Proliferation of B-Cell Lymphomas/Leukemias with the SHP1 Gene Silencing by Aberrant CpG Methylation. Lab. Investig. 2003, 83, 1849–1858. [Google Scholar] [CrossRef] [Green Version]
- Arora, P.; Kim, E.-O.; Jung, J.K.; Jang, K.L. Hepatitis C virus core protein downregulates E-cadherin expression via activation of DNA methyltransferase 1 and 3b. Cancer Lett. 2008, 261, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Quan, H.; Zhou, F.; Nie, D.; Chen, Q.; Cai, X.; Shan, X.; Zhou, Z.; Chen, K.; Huang, A.; Li, S.; et al. Hepatitis C virus core protein epigenetically silences SFRP1 and enhances HCC aggressiveness by inducing epithelial–mesenchymal transition. Oncogene 2013, 33, 2826–2835. [Google Scholar] [CrossRef] [Green Version]
- Kwak, J.; Shim, J.H.; Tiwari, I.; Jang, K.L. Hepatitis C virus core protein inhibits E6AP expression via DNA methylation to escape from ubiquitin-dependent proteasomal degradation. Cancer Lett. 2016, 380, 59–68. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devi, P.; Ota, S.; Punga, T.; Bergqvist, A. Hepatitis C Virus Core Protein Down-Regulates Expression of Src-Homology 2 Domain Containing Protein Tyrosine Phosphatase by Modulating Promoter DNA Methylation. Viruses 2021, 13, 2514. https://doi.org/10.3390/v13122514
Devi P, Ota S, Punga T, Bergqvist A. Hepatitis C Virus Core Protein Down-Regulates Expression of Src-Homology 2 Domain Containing Protein Tyrosine Phosphatase by Modulating Promoter DNA Methylation. Viruses. 2021; 13(12):2514. https://doi.org/10.3390/v13122514
Chicago/Turabian StyleDevi, Priya, Seisuke Ota, Tanel Punga, and Anders Bergqvist. 2021. "Hepatitis C Virus Core Protein Down-Regulates Expression of Src-Homology 2 Domain Containing Protein Tyrosine Phosphatase by Modulating Promoter DNA Methylation" Viruses 13, no. 12: 2514. https://doi.org/10.3390/v13122514
APA StyleDevi, P., Ota, S., Punga, T., & Bergqvist, A. (2021). Hepatitis C Virus Core Protein Down-Regulates Expression of Src-Homology 2 Domain Containing Protein Tyrosine Phosphatase by Modulating Promoter DNA Methylation. Viruses, 13(12), 2514. https://doi.org/10.3390/v13122514