
Could Interleukin-33 (IL-33) Govern the Outcome of an Equine Influenza Virus Infection? Learning from Other Species


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
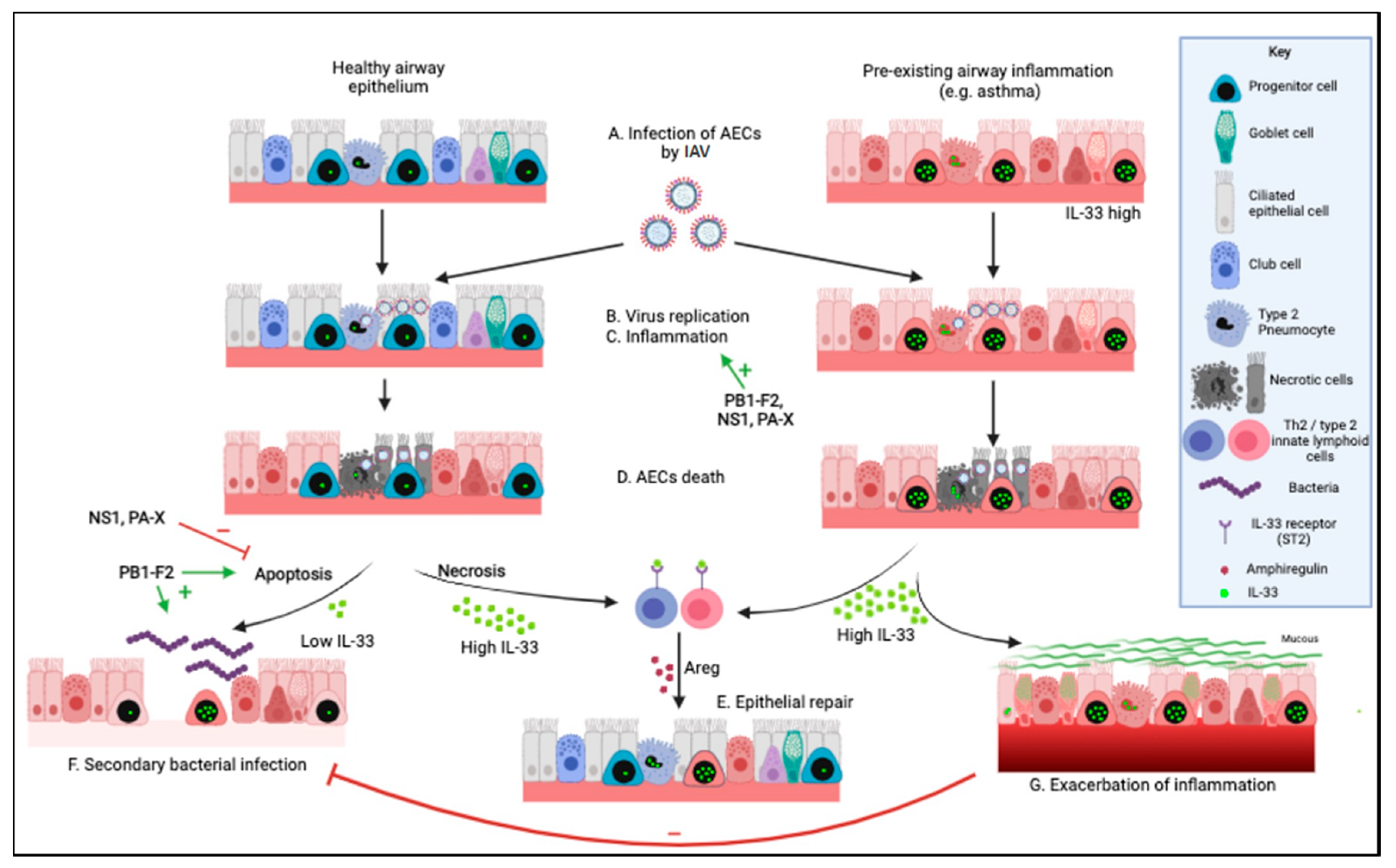
2. Determinants of Lung Damage and Pulmonary Complications during an EIV Infection
2.1. Viral Determinants of Airway Epithelial Cell Death
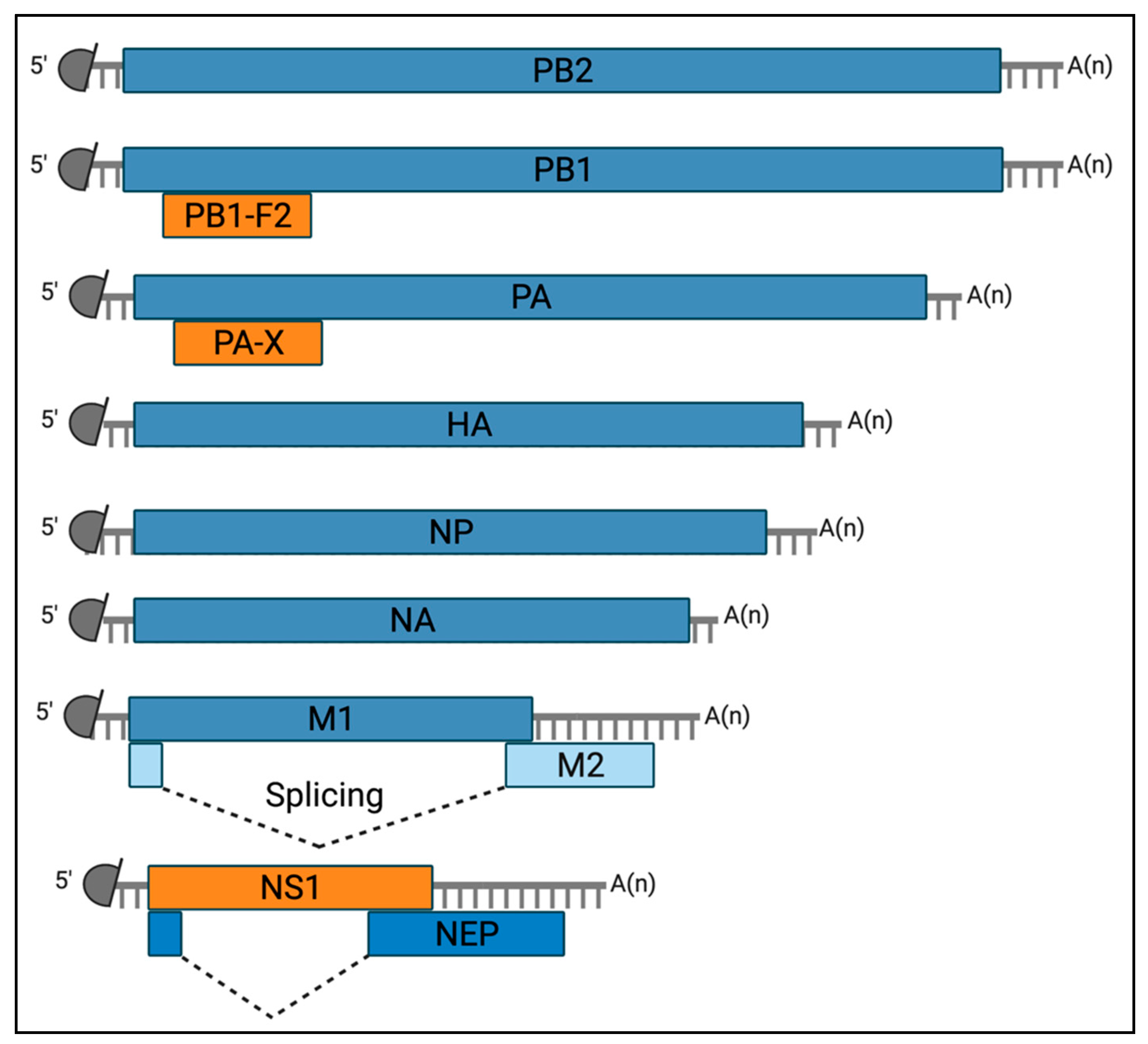
2.1.1. NS1
2.1.2. PA-X
2.1.3. PB1-F2
2.2. Immune-Mediated Damage to the Respiratory Epithelium during EIV Infection
2.3. Secondary Bacterial Pneumonia Post EIV Infection
3. IL-33 during an IAV Infection
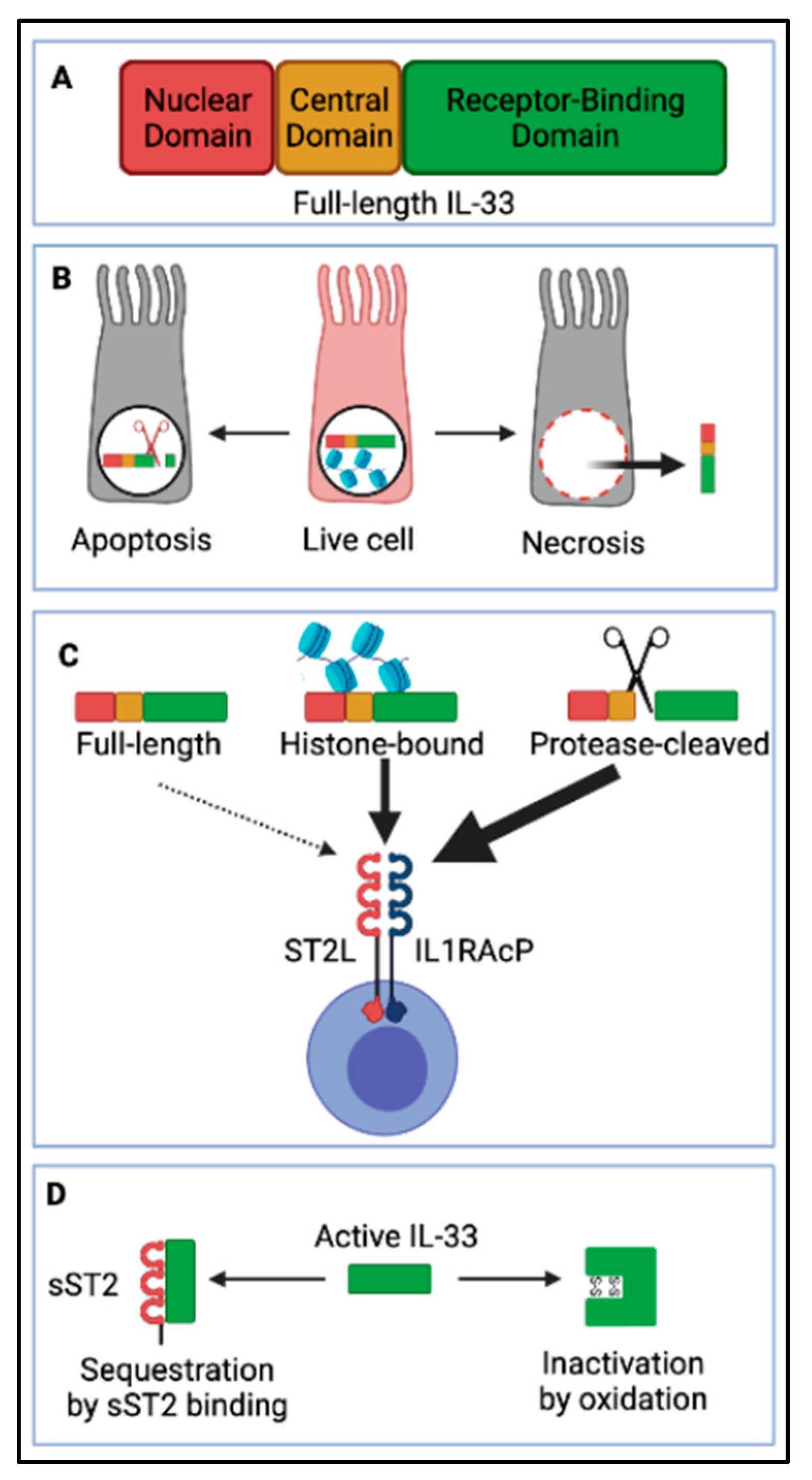
3.1. IL-33 Biology
3.2. IL-33, a Multifunctional Cytokine during IAV Infection
3.2.1. IL-33 Regulates Immune Responses during IAV Infection
3.2.2. The Balance IL-33/Type I IFN Regulates IAV Disease Severity in Asthmatics
3.2.3. IL-33 Promotes Anti-Bacterial Host Defense during IAV Infection
3.3. IL-33 Is an Important Adjuvant for IAV Vaccines
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Timoney, P.J. Equine Influenza. Comp. Immunol. Microbiol. Infect. Dis. 1996, 19, 205–211. [Google Scholar] [CrossRef]
- Van Maanen, C.; Cullinane, A. Equine Influenza Virus Infections: An Update. Vet. Q. 2002, 24, 79–94. [Google Scholar] [CrossRef]
- Yamanaka, T.; Niwa, H.; Tsujimura, K.; Kondo, T.; Matsumura, T. Epidemic of Equine Influenza among Vaccinated Racehorses in Japan in 2007. J. Vet. Med. Sci. 2008, 70, 623–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxburgh, L.; Berg, M.; Klingeborn, B.; Emmoth, E.; Linné, T. Evolution of H3N8 Equine Influenza Virus from 1963 to 1991. Virus Res. 1994, 34, 153–165. [Google Scholar] [CrossRef]
- Berg, M.; Desselberger, U.; Abusugra, I.A.; Klingeborn, B.; Linné, T. Genetic Drift of Equine 2 Influenza a Virus (H3N8), 1963–1988: Analysis by Oligonucleotide Mapping. Vet. Microbiol. 1990, 22, 225–236. [Google Scholar] [CrossRef]
- Kitchen, R.H.; Kehler, W.H.; Henthorne, J.C. The 1963 Equine Influenza Epizootic. J. Am. Vet. Med. Assoc. 1963, 143, 1108–1110. [Google Scholar]
- Waddell, G.H.; Teigland, M.B.; Sigel, M.M. A new influenza virus associated with equine respiratory disease. J. Am. Vet. Med. Assoc. 1963, 143, 587–590. [Google Scholar] [PubMed]
- Muranaka, M.; Yamanaka, T.; Katayama, Y.; Niwa, H.; Oku, K.; Matsumura, T.; Oyamada, T. Time-Related Pathological Changes in Horses Experimentally Inoculated with Equine Influenza A Virus. J. Equine Sci. 2012, 23, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Sutton, G.A.; Viel, L.; Carman, P.S.; Boag, B.L. Study of the Duration and Distribution of Equine Influenza Virus Subtype 2 (H3N8) Antigens in Experimentally Infected Ponies In Vivo. Can. J. Vet. Res. 1997, 61, 113–120. [Google Scholar]
- Murcia, P.R.; Wood, J.L.N.; Holmes, E.C. Genome-Scale Evolution and Phylodynamics of Equine H3N8 Influenza A Virus. J. Virol. 2011, 85, 5312–5322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fougerolle, S.; Legrand, L.; Lecouturier, F.; Sailleau, C.; Paillot, R.; Hans, A.; Pronost, S. Genetic Evolution of Equine Influenza Virus Strains (H3N8) Isolated in France from 1967 to 2015 and the Implications of Several Potential Pathogenic Factors. Virology 2017, 505, 210–217. [Google Scholar] [CrossRef]
- Legrand, L.J.; Pitel, P.-H.Y.; Cullinane, A.A.; Fortier, G.D.; Pronost, S.L. Genetic Evolution of Equine Influenza Strains Isolated in France from 2005 to 2010: Equine Influenza Genetic Characterisation. Equine Vet. J. 2015, 47, 207–211. [Google Scholar] [CrossRef]
- Rash, A.; Woodward, A.; Bryant, N.; McCauley, J.; Elton, D. An Efficient Genome Sequencing Method for Equine Influenza [H3N8] Virus Reveals a New Polymorphism in the PA-X Protein. Virol. J. 2014, 11, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rash, A.; Morton, R.; Woodward, A.; Maes, O.; McCauley, J.; Bryant, N.; Elton, D. Evolution and Divergence of H3N8 Equine Influenza Viruses Circulating in the United Kingdom from 2013 to 2015. Pathogens 2017, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- World Organisation for Animal Health. OIE Expert Surveillance Panel on Equine Influenza Vaccine Composition. Conclusions and Recommendations. 2021. Available online: Https://Www.Oie.Int/En/Disease/Equine-Influenza-2/ (accessed on 7 November 2021).
- Nemoto, M.; Ohta, M.; Yamanaka, T.; Kambayashi, Y.; Bannai, H.; Tsujimura, K.; Yamayoshi, S.; Kawaoka, Y.; Cullinane, A. Antigenic Differences between Equine Influenza Virus Vaccine Strains and Florida Sublineage Clade 1 Strains Isolated in Europe in 2019. Vet. J. 2021, 272, 105674. [Google Scholar] [CrossRef]
- Oladunni, F.S.; Oseni, S.O.; Martinez-Sobrido, L.; Chambers, T.M. Equine Influenza Virus and Vaccines. Viruses 2021, 13, 1657. [Google Scholar] [CrossRef] [PubMed]
- Binns, M.M.; Daly, J.M.; Chirnside, E.D.; Mumford, J.A.; Wood, J.M.; Richards, C.M.; Daniels, R.S. Genetic and Antigenic Analysis of an Equine Influenza H 3 Isolate from the 1989 Epidemic. Arch. Virol. 1993, 130, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Livesay, G.; O’Neill, T.; Hannant, D.; Yadav, M.; Mumford, J. The Outbreak of Equine Influenza (H3N8) in the United Kingdom in 1989: Diagnostic Use of an Antigen Capture ELISA. Vet. Rec. 1993, 133, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Boon, A.C.M.; Finkelstein, D.; Zheng, M.; Liao, G.; Allard, J.; Klumpp, K.; Webster, R.; Peltz, G.; Webby, R.J. H5N1 Influenza Virus Pathogenesis in Genetically Diverse Mice Is Mediated at the Level of Viral Load. mBio 2011, 2, e00171-11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.D.; Chau, T.N.B.; Hoang, D.M.; Van Vinh Chau, N.; Khanh, T.H.; Dong, V.C.; et al. Fatal Outcome of Human Influenza A (H5N1) Is Associated with High Viral Load and Hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef]
- Granados, A.; Peci, A.; McGeer, A.; Gubbay, J.B. Influenza and Rhinovirus Viral Load and Disease Severity in Upper Respiratory Tract Infections. J. Clin. Virol. 2017, 86, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Marathe, B.M.; Wong, S.-S.; Vogel, P.; Garcia-Alcalde, F.; Webster, R.G.; Webby, R.J.; Najera, I.; Govorkova, E.A. Combinations of Oseltamivir and T-705 Extend the Treatment Window for Highly Pathogenic Influenza A(H5N1) Virus Infection in Mice. Sci. Rep. 2016, 6, 26742. [Google Scholar] [CrossRef] [Green Version]
- Toapanta, F.R.; Ross, T.M. Impaired Immune Responses in the Lungs of Aged Mice Following Influenza Infection. Respir. Res. 2009, 10, 112. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.A.; Tyrell, D.J.; Kulkarni, U.A.; Wood, S.; Leng, L.; Zemans, R.L.; Bucala, R.; Goldstein, D.R. Macrophage Migration Inhibitory Factor Enhances Influenza-Associated Mortality in Mice. JCI Insight 2019, 4, e128034. [Google Scholar] [CrossRef]
- Gao, R.; Bhatnagar, J.; Blau, D.M.; Greer, P.; Rollin, D.C.; Denison, A.M.; Deleon-Carnes, M.; Shieh, W.-J.; Sambhara, S.; Tumpey, T.M.; et al. Cytokine and Chemokine Profiles in Lung Tissues from Fatal Cases of 2009 Pandemic Influenza A (H1N1). Am. J. Pathol. 2013, 183, 1258–1268. [Google Scholar] [CrossRef]
- Laghlali, G.; Lawlor, K.E.; Tate, M.D. Die Another Way: Interplay between Influenza A Virus, Inflammation and Cell Death. Viruses 2020, 12, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkin-Smith, G.K.; Duan, M.; Chen, W.; Poon, I.K.H. The Induction and Consequences of Influenza A Virus-Induced Cell Death. Cell Death Dis. 2018, 9, 1002. [Google Scholar] [CrossRef]
- Downey, J.; Pernet, E.; Coulombe, F.; Divangahi, M. Dissecting Host Cell Death Programs in the Pathogenesis of Influenza. Microbes Infect. 2018, 20, 560–569. [Google Scholar] [CrossRef]
- Fujikura, D.; Miyazaki, T. Programmed Cell Death in the Pathogenesis of Influenza. Int. J. Mol. Sci. 2018, 19, 2065. [Google Scholar] [CrossRef] [Green Version]
- Cayrol, C.; Girard, J.-P. Interleukin-33 (IL-33): A Nuclear Cytokine from the IL-1 Family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Becker, C.; Ridge, K.M.; Budinger, G.R.S. Influenza Virus-Induced Lung Injury: Pathogenesis and Implications for Treatment. Eur. Respir. J. 2015, 45, 1463–1478. [Google Scholar] [CrossRef] [Green Version]
- Wattrang, E.; Jessett, D.M.; Yates, P.; Fuxler, L.; Hannant, D. Experimental Infection of Ponies with Equine Influenza A2 (H3N8) Virus Strains of Different Pathogenicity Elicits Varying Interferon and Interleukin-6 Responses. Viral Immunol. 2003, 16, 57–67. [Google Scholar] [CrossRef]
- Le Goffic, R.; Arshad, M.I.; Rauch, M.; L’Helgoualc’h, A.; Delmas, B.; Piquet-Pellorce, C.; Samson, M. Infection with Influenza Virus Induces IL-33 in Murine Lungs. Am. J. Respir. Cell Mol. Biol. 2011, 4, 1125–1132. [Google Scholar] [CrossRef]
- Ravanetti, L.; Dijkhuis, A.; Dekker, T.; Sabogal Pineros, Y.S.; Ravi, A.; Dierdorp, B.S.; Erjefält, J.S.; Mori, M.; Pavlidis, S.; Adcock, I.M.; et al. IL-33 Drives Influenza-Induced Asthma Exacerbations by Halting Innate and Adaptive Antiviral Immunity. J. Allergy Clin. Immunol. 2019, 143, 1355–1370.e16. [Google Scholar] [CrossRef]
- Robinson, K.M.; Ramanan, K.; Clay, M.E.; McHugh, K.J.; Rich, H.E.; Alcorn, J.F. Novel Protective Mechanism for Interleukin-33 at the Mucosal Barrier during Influenza-Associated Bacterial Superinfection. Mucosal Immunol. 2018, 11, 199–208. [Google Scholar] [CrossRef]
- Stubbs, T.M.; te Velthuis, A.J. The RNA-Dependent RNA Polymerase of the Influenza A Virus. Future Virol. 2014, 9, 863–876. [Google Scholar] [CrossRef] [Green Version]
- Szewczyk, B.; Bieńkowska-Szewczyk, K.; Król, E. Introduction to Molecular Biology of Influenza a Viruses. Acta Biochim. Pol. 2014, 61, 397–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.-A.; Wills, N.M.; Xiao, Y.-L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An Overlapping Protein-Coding Region in Influenza A Virus Segment 3 Modulates the Host Response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Chauché, C.; Nogales, A.; Zhu, H.; Goldfarb, D.; Ahmad Shanizza, A.I.; Gu, Q.; Parrish, C.R.; Martínez-Sobrido, L.; Marshall, J.F.; Murcia, P.R. Mammalian Adaptation of an Avian Influenza A Virus Involves Stepwise Changes in NS1. J. Virol. 2018, 92, e01875-17. [Google Scholar] [CrossRef] [Green Version]
- Feng, K.H.; Sun, M.; Iketani, S.; Holmes, E.C.; Parrish, C.R. Comparing the Functions of Equine and Canine Influenza H3N8 Virus PA-X Proteins: Suppression of Reporter Gene Expression and Modulation of Global Host Gene Expression. Virology 2016, 496, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Ayllon, J.; García-Sastre, A. The NS1 Protein: A Multitasking Virulence Factor. In Influenza Pathogenesis and Control—Volume II; Oldstone, M.B.A., Compans, R.W., Eds.; Current Topics in Microbiology and Immunology; Springer International Publishing: Cham, Switzerland, 2014; Volume 386, pp. 73–107. [Google Scholar] [CrossRef]
- Nogales, A.; Martinez-Sobrido, L.; Topham, D.; DeDiego, M. Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins. Viruses 2018, 10, 708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alymova, I.V.; Green, A.M.; van de Velde, N.; McAuley, J.L.; Boyd, K.L.; Ghoneim, H.E.; McCullers, J.A. Immunopathogenic and Antibacterial Effects of H3N2 Influenza A Virus PB1-F2 Map to Amino Acid Residues 62, 75, 79, and 82. J. Virol. 2011, 85, 12324–12333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alymova, I.V.; Samarasinghe, A.; Vogel, P.; Green, A.M.; Weinlich, R.; McCullers, J.A. A Novel Cytotoxic Sequence Contributes to Influenza A Viral Protein PB1-F2 Pathogenicity and Predisposition to Secondary Bacterial Infection. J. Virol. 2014, 88, 503–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnham, A.J.; Miller, J.R.; Singh, I.; Billings, E.A.; Rush, M.A.; Air, G.M.; Bour, S. Novel Isoforms of Influenza Virus PA-X and PB1-F2 Indicated by Automatic Annotation. Virus Res. 2021, 304, 198545. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I Interferons in Infectious Disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A Virus Protein PB1-F2 Translocates into Mitochondria via Tom40 Channels and Impairs Innate Immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Mo, Y.; Wang, X.; Gu, M.; Hu, Z.; Zhong, L.; Wu, Q.; Hao, X.; Hu, S.; Liu, W.; et al. PA-X Decreases the Pathogenicity of Highly Pathogenic H5N1 Influenza A Virus in Avian Species by Inhibiting Virus Replication and Host Response. J. Virol. 2015, 89, 4126–4142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marc, D. Influenza Virus Non-Structural Protein NS1: Interferon Antagonism and Beyond. J. Gen. Virol. 2014, 95, 2594–2611. [Google Scholar] [CrossRef] [PubMed]
- García-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A Virus Lacking the NS1 Gene Replicates in Interferon-Deficient Systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef] [Green Version]
- Kochs, G.; García-Sastre, A.; Martínez-Sobrido, L. Multiple Anti-Interferon Actions of the Influenza A Virus NS1 Protein. J. Virol. 2007, 81, 7011–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egorov, A.; Brandt, S.; Sereinig, S.; Romanova, J.; Ferko, B.; Katinger, D.; Grassauer, A.; Alexandrova, G.; Katinger, H.; Muster, T. Transfectant Influenza A Viruses with Long Deletions in the NS1 Protein Grow Efficiently in Vero Cells. J. Virol. 1998, 72, 6437–6441. [Google Scholar] [CrossRef] [Green Version]
- Geiss, G.K.; Salvatore, M.; Tumpey, T.M.; Carter, V.S.; Wang, X.; Basler, C.F.; Taubenberger, J.K.; Bumgarner, R.E.; Palese, P.; Katze, M.G.; et al. Cellular Transcriptional Profiling in Influenza A Virus-Infected Lung Epithelial Cells: The Role of the Nonstructural NS1 Protein in the Evasion of the Host Innate Defense and Its Potential Contribution to Pandemic Influenza. Proc. Natl. Acad. Sci. USA 2002, 99, 10736–10741. [Google Scholar] [CrossRef] [Green Version]
- Twu, K.Y.; Kuo, R.-L.; Marklund, J.; Krug, R.M. The H5N1 Influenza Virus NS Genes Selected after 1998 Enhance Virus Replication in Mammalian Cells. J. Virol. 2007, 81, 8112–8121. [Google Scholar] [CrossRef] [Green Version]
- Hayman, A.; Comely, S.; Lackenby, A.; Hartgroves, L.C.S.; Goodbourn, S.; McCauley, J.W.; Barclay, W.S. NS1 Proteins of Avian Influenza A Viruses Can Act as Antagonists of the Human Alpha/Beta Interferon Response. J. Virol. 2007, 81, 2318–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obenauer, J.C.; Denson, J.; Mehta, P.K.; Su, X.; Mukatira, S.; Finkelstein, D.B.; Xu, X.; Wang, J.; Ma, J.; Fan, Y.; et al. Large-Scale Sequence Analysis of Avian Influenza Isolates. Science 2006, 311, 1576–1580. [Google Scholar] [CrossRef]
- Javier, R.T.; Rice, A.P. Emerging Theme: Cellular PDZ Proteins as Common Targets of Pathogenic Viruses. J. Virol. 2011, 85, 11544–11556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Golebiewski, L.; Dow, E.C.; Krug, R.M.; Javier, R.T.; Rice, A.P. The ESEV PDZ-Binding Motif of the Avian Influenza A Virus NS1 Protein Protects Infected Cells from Apoptosis by Directly Targeting Scribble. J. Virol. 2010, 84, 11164–11174. [Google Scholar] [CrossRef] [Green Version]
- Golebiewski, L.; Liu, H.; Javier, R.T.; Rice, A.P. The Avian Influenza Virus NS1 ESEV PDZ Binding Motif Associates with Dlg1 and Scribble to Disrupt Cellular Tight Junctions. J. Virol. 2011, 85, 10639–10648. [Google Scholar] [CrossRef] [Green Version]
- Barba, M.; Daly, J. The Influenza NS1 Protein: What Do We Know in Equine Influenza Virus Pathogenesis? Pathogens 2016, 5, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrhardt, C.; Marjuki, H.; Wolff, T.; Nurnberg, B.; Planz, O.; Pleschka, S.; Ludwig, S. Bivalent Role of the Phosphatidylinositol-3-Kinase (PI3K) during Influenza Virus Infection and Host Cell Defence. Cell. Microbiol. 2006, 8, 1336–1348. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C.; Wolff, T.; Pleschka, S.; Planz, O.; Beermann, W.; Bode, J.G.; Schmolke, M.; Ludwig, S. Influenza A Virus NS1 Protein Activates the PI3K/Akt Pathway To Mediate Antiapoptotic Signaling Responses. J. Virol. 2007, 81, 3058–3067. [Google Scholar] [CrossRef] [Green Version]
- Hale, B.G.; Jackson, D.; Chen, Y.-H.; Lamb, R.A.; Randall, R.E. Influenza A Virus NS1 Protein Binds P85beta and Activates Phosphatidylinositol-3-Kinase Signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.-K.; Li, Y.; Liu, Q.; Anderson, D.H.; Babiuk, L.A.; Zhou, Y. SH3 Binding Motif 1 in Influenza A Virus NS1 Protein Is Essential for PI3K/Akt Signaling Pathway Activation. J. Virol. 2007, 81, 12730–12739. [Google Scholar] [CrossRef] [Green Version]
- Rosário-Ferreira, N.; Preto, A.J.; Melo, R.; Moreira, I.S.; Brito, R.M.M. The Central Role of Non-Structural Protein 1 (NS1) in Influenza Biology and Infection. Int. J. Mol. Sci. 2020, 21, 1511. [Google Scholar] [CrossRef] [Green Version]
- Nogales, A.; Chauché, C.; DeDiego, M.L.; Topham, D.J.; Parrish, C.R.; Murcia, P.R.; Martínez-Sobrido, L. The K186E Amino Acid Substitution in the Canine Influenza Virus H3N8 NS1 Protein Restores Its Ability To Inhibit Host Gene Expression. J. Virol. 2017, 91, e00877-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caroline, C. Molecular Evolution of Equine Influenza Virus Non-Structural Protein 1. Ph.D. Thesis, University of Glasgow, Glasgow, UK, 2018. [Google Scholar]
- Shi, M.; Jagger, B.W.; Wise, H.M.; Digard, P.; Holmes, E.C.; Taubenberger, J.K. Evolutionary Conservation of the PA-X Open Reading Frame in Segment 3 of Influenza A Virus. J. Virol. 2012, 86, 12411–12413. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; MacDonald, L.A.; Takimoto, T. Influenza A Virus Protein PA-X Contributes to Viral Growth and Suppression of the Host Antiviral and Immune Responses. J. Virol. 2015, 89, 6442–6452. [Google Scholar] [CrossRef] [Green Version]
- Khaperskyy, D.A.; Schmaling, S.; Larkins-Ford, J.; McCormick, C.; Gaglia, M.M. Selective Degradation of Host RNA Polymerase II Transcripts by Influenza A Virus PA-X Host Shutoff Protein. PLoS Pathog. 2016, 12, e1005427. [Google Scholar] [CrossRef] [Green Version]
- Kamal, R.; Alymova, I.; York, I. Evolution and Virulence of Influenza A Virus Protein PB1-F2. Int. J. Mol. Sci. 2017, 19, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conenello, G.M.; Zamarin, D.; Perrone, L.A.; Tumpey, T.; Palese, P. A Single Mutation in the PB1-F2 of H5N1 (HK/97) and 1918 Influenza A Viruses Contributes to Increased Virulence. PLoS Pathog. 2007, 3, e141. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.; Paust, S. Dynamic Natural Killer Cell and T Cell Responses to Influenza Infection. Front. Cell. Infect. Microbiol. 2020, 10, 425. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Maeda, N.; Shibata, K.; Yamada, H.; Kase, T.; Yoshikai, Y. Interleukin-15 Is Critical in the Pathogenesis of Influenza A Virus-Induced Acute Lung Injury. J. Virol. 2010, 84, 5574–5582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Careem, M.F.; Mian, M.F.; Yue, G.; Gillgrass, A.; Chenoweth, M.J.; Barra, N.G.; Chew, M.V.; Chan, T.; Al-Garawi, A.A.; Jordana, M.; et al. Critical Role of Natural Killer Cells in Lung Immunopathology During Influenza Infection in Mice. J. Infect. Dis. 2012, 206, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Weiss, I.D.; Wald, O.; Wald, H.; Beider, K.; Abraham, M.; Galun, E.; Nagler, A.; Peled, A. IFN-γ Treatment at Early Stages of Influenza Virus Infection Protects Mice from Death in a NK Cell-Dependent Manner. J. Interferon Cytokine Res. 2010, 30, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Califano, D.; Furuya, Y.; Roberts, S.; Avram, D.; McKenzie, A.N.J.; Metzger, D.W. IFN-γ Increases Susceptibility to Influenza A Infection through Suppression of Group II Innate Lymphoid Cells. Mucosal Immunol. 2018, 11, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermejo-Martin, J.F.; Ortiz de Lejarazu, R.; Pumarola, T.; Rello, J.; Almansa, R.; Ramírez, P.; Martin-Loeches, I.; Varillas, D.; Gallegos, M.C.; Serón, C.; et al. Th1 and Th17 Hypercytokinemia as Early Host Response Signature in Severe Pandemic Influenza. Crit. Care 2009, 13, R201. [Google Scholar] [CrossRef] [Green Version]
- McKinstry, K.K.; Strutt, T.M.; Buck, A.; Curtis, J.D.; Dibble, J.P.; Huston, G.; Tighe, M.; Hamada, H.; Sell, S.; Dutton, R.W.; et al. IL-10 Deficiency Unleashes an Influenza-Specific Th17 Response and Enhances Survival against High-Dose Challenge. J. Immunol. 2009, 182, 7353–7363. [Google Scholar] [CrossRef] [Green Version]
- Van Reeth, K. Cytokines in the Pathogenesis of Influenza. Vet. Microbiol. 2000, 74, 109–116. [Google Scholar] [CrossRef]
- Prantner, D.; Shirey, K.A.; Lai, W.; Lu, W.; Cole, A.M.; Vogel, S.N.; Garzino-Demo, A. The θ-Defensin Retrocyclin 101 Inhibits TLR4- and TLR2-Dependent Signaling and Protects Mice against Influenza Infection. J. Leukoc. Biol. 2017, 102, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Major, J.; Crotta, S.; Llorian, M.; McCabe, T.M.; Gad, H.H.; Priestnall, S.L.; Hartmann, R.; Wack, A. Type I and III Interferons Disrupt Lung Epithelial Repair during Recovery from Viral Infection. Science 2020, 369, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.J.; Thomas, P.G. New Fronts Emerge in the Influenza Cytokine Storm. Semin. Immunopathol. 2017, 39, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Oslund, K.L.; Baumgarth, N. Influenza-Induced Innate Immunity: Regulators of Viral Replication, Respiratory Tract Pathology & Adaptive Immunity. Future Virol. 2011, 6, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Elton, D.; Bryant, N. Facing the Threat of Equine Influenza: Facing the Threat of Equine Influenza. Equine Vet. J. 2011, 43, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; Crotta, S.; McCabe, T.M.; Wack, A. Pathogenic Potential of Interferon Aβ in Acute Influenza Infection. Nat. Commun. 2014, 5, 3864. [Google Scholar] [CrossRef] [Green Version]
- Högner, K.; Wolff, T.; Pleschka, S.; Plog, S.; Gruber, A.D.; Kalinke, U.; Walmrath, H.-D.; Bodner, J.; Gattenlöhner, S.; Lewe-Schlosser, P.; et al. Macrophage-Expressed IFN-β Contributes to Apoptotic Alveolar Epithelial Cell Injury in Severe Influenza Virus Pneumonia. PLoS Pathog. 2013, 9, e1003188. [Google Scholar] [CrossRef]
- Topham, D.J.; Tripp, R.A.; Doherty, P.C. CD8+ T Cells Clear Influenza Virus by Perforin or Fas-Dependent Processes. J. Immunol. 1997, 159, 5197–5200. [Google Scholar]
- Ishikawa, E.; Nakazawa, M.; Yoshinari, M.; Minami, M. Role of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand in Immune Response to Influenza Virus Infection in Mice. J. Virol. 2005, 79, 7658–7663. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Thomas, P.G. Balancing Immune Protection and Immune Pathology by CD8+ T-Cell Responses to Influenza Infection. Front. Immunol. 2016, 7, 25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Bevan, M.J. CD8+ T Cells: Foot Soldiers of the Immune System. Immunity 2011, 35, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.-L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef] [Green Version]
- La Gruta, N.L.; Turner, S.J. T Cell Mediated Immunity to Influenza: Mechanisms of Viral Control. Trends Immunol. 2014, 35, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Kreijtz, J.H.C.M.; Fouchier, R.A.M.; Rimmelzwaan, G.F. Immune Responses to Influenza Virus Infection. Virus Res. 2011, 162, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Van de Sandt, C.E.; Bárcena, M.; Koster, A.J.; Kasper, J.; Kirkpatrick, C.J.; Scott, D.P.; de Vries, R.D.; Herold, S.; Rimmelzwaan, G.F.; Kuiken, T.; et al. Human CD8 + T Cells Damage Noninfected Epithelial Cells during Influenza Virus Infection In Vitro. Am. J. Respir. Cell Mol. Biol. 2017, 57, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Sareneva, T.; Matikainen, S.; Kurimoto, M.; Julkunen, I. Influenza A Virus-Induced IFN-Alpha/Beta and IL-18 Synergistically Enhance IFN-Gamma Gene Expression in Human T Cells. J. Immunol. 1998, 160, 6032–6038. [Google Scholar]
- Maloney, N.S.; Thackray, L.B.; Goel, G.; Hwang, S.; Duan, E.; Vachharajani, P.; Xavier, R.; Virgin, H.W. Essential Cell-Autonomous Role for Interferon (IFN) Regulatory Factor 1 in IFN- -Mediated Inhibition of Norovirus Replication in Macrophages. J. Virol. 2012, 86, 12655–12664. [Google Scholar] [CrossRef] [Green Version]
- Sarawar, S.R.; Sangster, M.; Coffman, R.L.; Doherty, P.C. Administration of Anti-IFN-Gamma Antibody to Beta 2-Microglobulin-Deficient Mice Delays Influenza Virus Clearance but Does Not Switch the Response to a T Helper Cell 2 Phenotype. J. Immunol. 1994, 153, 1246–1253. [Google Scholar]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The Role of Macrophages in the Resolution of Inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, Y.; Moki, T.; Takizawa, T.; Shiratsuchi, A.; Nakanishi, Y. Evidence for Phagocytosis of Influenza Virus-Infected, Apoptotic Cells by Neutrophils and Macrophages in Mice. J. Immunol. 2007, 178, 2448–2457. [Google Scholar] [CrossRef] [Green Version]
- Perrone, L.A.; Plowden, J.K.; García-Sastre, A.; Katz, J.M.; Tumpey, T.M. H5N1 and 1918 Pandemic Influenza Virus Infection Results in Early and Excessive Infiltration of Macrophages and Neutrophils in the Lungs of Mice. PLoS Pathog. 2008, 4, e1000115. [Google Scholar] [CrossRef] [Green Version]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.-A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive Neutrophils and Neutrophil Extracellular Traps Contribute to Acute Lung Injury of Influenza Pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef]
- Singh, R.K.; Dhama, K.; Karthik, K.; Khandia, R.; Munjal, A.; Khurana, S.K.; Chakraborty, S.; Malik, Y.S.; Virmani, N.; Singh, R.; et al. A Comprehensive Review on Equine Influenza Virus: Etiology, Epidemiology, Pathobiology, Advances in Developing Diagnostics, Vaccines, and Control Strategies. Front. Microbiol. 2018, 9, 1941. [Google Scholar] [CrossRef]
- Landolt, G.A. Equine Influenza Virus. Vet. Clin. N. Am. Equine Pract. 2014, 30, 507–522. [Google Scholar] [CrossRef]
- Na, W.; Yeom, M.; Yuk, H.; Moon, H.; Kang, B.; Song, D. Influenza Virus Vaccine for Neglected Hosts: Horses and Dogs. Clin. Exp. Vaccine Res. 2016, 5, 117. [Google Scholar] [CrossRef] [PubMed]
- Patterson-Kane, J.C.; Carrick, J.B.; Axon, J.E.; Wilkie, I.; Begg, A.P. The Pathology of Bronchointerstitial Pneumonia in Young Foals Associated with the First Outbreak of Equine Influenza in Australia. Equine Vet. J. 2008, 40, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Begg, A.; Reece, R.; Hum, S.; Townsend, W.; Gordon, A.; Carrick, J. Pathological Changes in Horses Dying with Equine Influenza in Australia, 2007: EQUINE INFLUENZA. Aust. Vet. J. 2011, 89, 19–22. [Google Scholar] [CrossRef] [PubMed]
- López, A.; Martinson, S.A. Chapter 9—Respiratory System, Mediastinum, and Pleurae. Pathol. Basis Vet. Dis. 2017, 471–560.e1. [Google Scholar] [CrossRef]
- Sarasola, P.; Taylor, D.; Love, S.; McKellar, Q. Secondary Bacterial Infections Following an Outbreak of Equine Influenza. Vet. Rec. 1992, 131, 441–442. [Google Scholar] [CrossRef]
- Yamanaka, T.; Tsujimura, K.; Kondo, T.; Hobo, S.; Matsumura, T. Efficacy of Oseltamivir Phosphate to Horses Inoculated with Equine Influenza A Virus. J. Vet. Med. Sci. 2006, 68, 923–928. [Google Scholar] [CrossRef] [Green Version]
- McCullers, J.A.; Rehg, J.E. Lethal Synergism between Influenza Virus and Streptococcus Pneumoniae: Characterization of a Mouse Model and the Role of Platelet-Activating Factor Receptor. J. Infect. Dis. 2002, 186, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickerson, C.L.; Jakab, G.J. Pulmonary Antibacterial Defenses during Mild and Severe Influenza Virus Infection. Infect. Immun. 1990, 58, 2809–2814. [Google Scholar] [CrossRef] [Green Version]
- Brundage, J.F. Interactions between Influenza and Bacterial Respiratory Pathogens: Implications for Pandemic Preparedness. Lancet Infect. Dis. 2006, 6, 303–312. [Google Scholar] [CrossRef]
- Didierlaurent, A.; Goulding, J.; Patel, S.; Snelgrove, R.; Low, L.; Bebien, M.; Lawrence, T.; van Rijt, L.S.; Lambrecht, B.N.; Sirard, J.-C.; et al. Sustained Desensitization to Bacterial Toll-like Receptor Ligands after Resolutionof Respiratory Influenza Infection. J. Exp. Med. 2008, 205, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNamee, L.A.; Harmsen, A.G. Both Influenza-Induced Neutrophil Dysfunction and Neutrophil-Independent Mechanisms Contribute to Increased Susceptibility to a Secondary Streptococcus Pneumoniae Infection. Infect. Immun. 2006, 74, 6707–6721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Sluijs, K.F.; van Elden, L.J.R.; Nijhuis, M.; Schuurman, R.; Pater, J.M.; Florquin, S.; Goldman, M.; Jansen, H.M.; Lutter, R.; van der Poll, T. IL-10 Is an Important Mediator of the Enhanced Susceptibility to Pneumococcal Pneumonia after Influenza Infection. J. Immunol. 2004, 172, 7603–7609. [Google Scholar] [CrossRef] [Green Version]
- Dockrell, D.H.; Marriott, H.M.; Prince, L.R.; Ridger, V.C.; Ince, P.G.; Hellewell, P.G.; Whyte, M.K.B. Alveolar Macrophage Apoptosis Contributes to Pneumococcal Clearance in a Resolving Model of Pulmonary Infection. J. Immunol. 2003, 171, 5380–5388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knapp, S.; Leemans, J.C.; Florquin, S.; Branger, J.; Maris, N.A.; Pater, J.; van Rooijen, N.; van der Poll, T. Alveolar Macrophages Have a Protective Antiinflammatory Role during Murine Pneumococcal Pneumonia. Am. J. Respir. Crit. Care Med. 2003, 167, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Metzger, D.W. Inhibition of Pulmonary Antibacterial Defense by Interferon-γ during Recovery from Influenza Infection. Nat. Med. 2008, 14, 558–564. [Google Scholar] [CrossRef]
- Atochina, O.; Harn, D. LNFPIII/LeX-Stimulated Macrophages Activate Natural Killer Cells via CD40-CD40L Interaction. Clin. Vaccine Immunol. 2005, 12, 1041–1049. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.J.; Hoth, J.J.; Stagner, M.K.; Gardner, S.A.; Peyton, J.C.; Cheadle, W.G. CD40-CD154 Interactions between Macrophages and Natural Killer Cells during Sepsis Are Critical for Macrophage Activation and Are Not Interferon Gamma Dependent. Clin. Exp. Immunol. 2004, 137, 469–477. [Google Scholar] [CrossRef]
- Small, C.-L.; Shaler, C.R.; McCormick, S.; Jeyanathan, M.; Damjanovic, D.; Brown, E.G.; Arck, P.; Jordana, M.; Kaushic, C.; Ashkar, A.A.; et al. Influenza Infection Leads to Increased Susceptibility to Subsequent Bacterial Superinfection by Impairing NK Cell Responses in the Lung. J. Immunol. 2010, 184, 2048–2056. [Google Scholar] [CrossRef] [Green Version]
- MacMicking, J.D. Interferon-Inducible Effector Mechanisms in Cell-Autonomous Immunity. Nat. Rev. Immunol. 2012, 12, 367–382. [Google Scholar] [CrossRef]
- Mancuso, G.; Midiri, A.; Biondo, C.; Beninati, C.; Zummo, S.; Galbo, R.; Tomasello, F.; Gambuzza, M.; Macrì, G.; Ruggeri, A.; et al. Type I IFN Signaling Is Crucial for Host Resistance against Different Species of Pathogenic Bacteria. J. Immunol. 2007, 178, 3126–3133. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.; Martin, F.J.; Soong, G.; Harfenist, B.S.; Aguilar, J.L.; Ratner, A.J.; Fitzgerald, K.A.; Schindler, C.; Prince, A. Streptococcus Pneumoniae DNA Initiates Type I Interferon Signaling in the Respiratory Tract. mBio 2011, 2, e00016-11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigent, D.A.; Huff, T.L.; Peterson, J.W.; Stanton, G.J.; Baron, S. Role of Interferon in Streptococcal Infection in the Mouse. Microb. Pathog. 1986, 1, 399–407. [Google Scholar] [CrossRef]
- Martin, F.J.; Gomez, M.I.; Wetzel, D.M.; Memmi, G.; O’Seaghdha, M.; Soong, G.; Schindler, C.; Prince, A. Staphylococcus Aureus Activates Type I IFN Signaling in Mice and Humans through the Xr Repeated Sequences of Protein A. J. Clin. Investig. 2009, 119, 1931–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iverson, A.R.; Boyd, K.L.; McAuley, J.L.; Plano, L.R.; Hart, M.E.; McCullers, J.A. Influenza Virus Primes Mice for Pneumonia From Staphylococcus Aureus. J. Infect. Dis. 2011, 203, 880–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAuley, J.L.; Hornung, F.; Boyd, K.L.; Smith, A.M.; McKeon, R.; Bennink, J.; Yewdell, J.W.; McCullers, J.A. Expression of the 1918 Influenza A Virus PB1-F2 Enhances the Pathogenesis of Viral and Secondary Bacterial Pneumonia. Cell Host Microbe 2007, 2, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Al-Garawi, A.; Fattouh, R.; Botelho, F.; Walker, T.D.; Goncharova, S.; Moore, C.-L.; Mori, M.; Erjefalt, J.S.; Chu, D.K.; Humbles, A.A.; et al. Influenza A Facilitates Sensitization to House Dust Mite in Infant Mice Leading to an Asthma Phenotype in Adulthood. Mucosal Immunol. 2011, 4, 682–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estabragh, Z.R.; Mamas, M.A. The Cardiovascular Manifestations of Influenza: A Systematic Review. Int. J. Cardiol. 2013, 167, 2397–2403. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.R.; Bartlett, N.W.; Hussell, T.; Openshaw, P.; Johnston, S.L. The Microbiology of Asthma. Nat. Rev. Microbiol. 2012, 10, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.W. Influenza-Associated Hospitalizations in the United States. JAMA 2004, 292, 1333. [Google Scholar] [CrossRef]
- Simonsen, L.; Fukuda, K.; Schonberger, L.B.; Cox, N.J. The Impact of Influenza Epidemics on Hospitalizations. J. Infect. Dis. 2000, 181, 831–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taubenberger, J.K.; Morens, D.M. The Pathology of Influenza Virus Infections. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 499–522. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.A.; García-Sastre, A. Influenza A Viruses: New Research Developments. Nat. Rev. Microbiol. 2011, 9, 590–603. [Google Scholar] [CrossRef]
- LeMessurier, K.S.; Iverson, A.R.; Chang, T.-C.; Palipane, M.; Vogel, P.; Rosch, J.W.; Samarasinghe, A.E. Allergic Inflammation Alters the Lung Microbiome and Hinders Synergistic Co-Infection with H1N1 Influenza Virus and Streptococcus Pneumoniae in C57BL/6 Mice. Sci. Rep. 2019, 9, 19360. [Google Scholar] [CrossRef] [PubMed]
- Saravia, J.; You, D.; Shrestha, B.; Jaligama, S.; Siefker, D.; Lee, G.I.; Harding, J.N.; Jones, T.L.; Rovnaghi, C.; Bagga, B.; et al. Respiratory Syncytial Virus Disease Is Mediated by Age-Variable IL-33. PLoS Pathog. 2015, 11, e1005217. [Google Scholar] [CrossRef] [Green Version]
- Préfontaine, D.; Nadigel, J.; Chouiali, F.; Audusseau, S.; Semlali, A.; Chakir, J.; Martin, J.G.; Hamid, Q. Increased IL-33 Expression by Epithelial Cells in Bronchial Asthma. J. Allergy Clin. Immunol. 2010, 125, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Chauché, C.; Pirie, S.; McSorley, H.J.; Schwarze, J. Role of the IL-33 Cytokine in Lung Repair Post Equine Influenza Virus Infection. Equine Vet. J. 2021, 53, 9. [Google Scholar] [CrossRef]
- Pichery, M.; Mirey, E.; Mercier, P.; Lefrancais, E.; Dujardin, A.; Ortega, N.; Girard, J.-P. Endogenous IL-33 Is Highly Expressed in Mouse Epithelial Barrier Tissues, Lymphoid Organs, Brain, Embryos, and Inflamed Tissues: In Situ Analysis Using a Novel Il-33–LacZ Gene Trap Reporter Strain. J. Immunol. 2012, 188, 3488–3495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travers, J.; Rochman, M.; Miracle, C.E.; Habel, J.E.; Brusilovsky, M.; Caldwell, J.M.; Rymer, J.K.; Rothenberg, M.E. Chromatin Regulates IL-33 Release and Extracellular Cytokine Activity. Nat. Commun. 2018, 9, 3244. [Google Scholar] [CrossRef]
- Gatti, F.; Mia, S.; Hammarström, C.; Frerker, N.; Fosby, B.; Wang, J.; Pietka, W.; Sundnes, O.; Hol, J.; Kasprzycka, M.; et al. Nuclear IL-33 Restrains the Early Conversion of Fibroblasts to an Extracellular Matrix-Secreting Phenotype. Sci. Rep. 2021, 11, 108. [Google Scholar] [CrossRef] [PubMed]
- Hatzioannou, A.; Banos, A.; Sakelaropoulos, T.; Fedonidis, C.; Vidali, M.-S.; Köhne, M.; Händler, K.; Boon, L.; Henriques, A.; Koliaraki, V.; et al. An Intrinsic Role of IL-33 in Treg Cell–Mediated Tumor Immunoevasion. Nat. Immunol. 2020, 21, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Mohs, A.; Thomas, M.; Klare, J.; Ross, R.; Schmitz, M.L.; Martin, M.U. The Dual Function Cytokine IL-33 Interacts with the Transcription Factor NF-ΚB To Dampen NF-ΚB–Stimulated Gene Transcription. J. Immunol. 2011, 187, 1609–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, K.; McSorley, H.J. Interleukin-33 in the Developing Lung—Roles in Asthma and Infection. Pediatr. Allergy Immunol. 2019, 30, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Shlomovitz, I.; Erlich, Z.; Speir, M.; Zargarian, S.; Baram, N.; Engler, M.; Edry-Botzer, L.; Munitz, A.; Croker, B.A.; Gerlic, M. Necroptosis Directly Induces the Release of Full-length Biologically Active IL -33 In Vitro and in an Inflammatory Disease Model. FEBS J. 2019, 286, 507–522. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, E.; Asanuma, H.; Momose, H.; Furuhata, K.; Mizukami, T.; Hamaguchi, I. Nasal Alum-Adjuvanted Vaccine Promotes IL-33 Release from Alveolar Epithelial Cells That Elicits IgA Production via Type 2 Immune Responses. PLoS Pathog. 2021, 17, e1009890. [Google Scholar] [CrossRef]
- Roussel, L.; Erard, M.; Cayrol, C.; Girard, J. Molecular Mimicry between IL-33 and KSHV for Attachment to Chromatin through the H2A–H2B Acidic Pocket. EMBO Rep. 2008, 9, 1006–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefrancais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.-P.; Cayrol, C. IL-33 Is Processed into Mature Bioactive Forms by Neutrophil Elastase and Cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef] [Green Version]
- Lüthi, A.U.; Cullen, S.P.; McNeela, E.A.; Duriez, P.J.; Afonina, I.S.; Sheridan, C.; Brumatti, G.; Taylor, R.C.; Kersse, K.; Vandenabeele, P.; et al. Suppression of Interleukin-33 Bioactivity through Proteolysis by Apoptotic Caspases. Immunity 2009, 31, 84–98. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an Interleukin-1-like Cytokine That Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Talabot-Ayer, D.; Lamacchia, C.; Gabay, C.; Palmer, G. Interleukin-33 Is Biologically Active Independently of Caspase-1 Cleavage. J. Biol. Chem. 2009, 284, 19420–19426. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.; Vince, J.E. Pyroptosis versus Necroptosis: Similarities, Differences, and Crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef]
- Oboki, K.; Ohno, T.; Kajiwara, N.; Arae, K.; Morita, H.; Ishii, A.; Nambu, A.; Abe, T.; Kiyonari, H.; Matsumoto, K.; et al. IL-33 Is a Crucial Amplifier of Innate Rather than Acquired Immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 18581–18586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhao, Y. Interleukin-33 and Its Receptor in Pulmonary Inflammatory Diseases. Crit. Rev. Immunol. 2015, 35, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Griesenauer, B.; Paczesny, S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 2017, 8, 475. [Google Scholar] [CrossRef]
- Lohning, M.; Stroehmann, A.; Coyle, A.J.; Grogan, J.L.; Lin, S.; Gutierrez-Ramos, J.-C.; Levinson, D.; Radbruch, A.; Kamradt, T. T1/ST2 Is Preferentially Expressed on Murine Th2 Cells, Independent of Interleukin 4, Interleukin 5, and Interleukin 10, and Important for Th2 Effector Function. Proc. Natl. Acad. Sci. USA 1998, 95, 6930–6935. [Google Scholar] [CrossRef] [Green Version]
- Turnquist, H.R.; Zhao, Z.; Rosborough, B.R.; Liu, Q.; Castellaneta, A.; Isse, K.; Wang, Z.; Lang, M.; Beer Stolz, D.; Zheng, X.X.; et al. IL-33 Expands Suppressive CD11b+ Gr-1int and Regulatory T Cells, Including ST2L + Foxp3 + Cells, and Mediates Regulatory T Cell-Dependent Promotion of Cardiac Allograft Survival. J. Immunol. 2011, 187, 4598–4610. [Google Scholar] [CrossRef] [Green Version]
- Matta, B.M.; Reichenbach, D.K.; Zhang, X.; Mathews, L.; Koehn, B.H.; Dwyer, G.K.; Lott, J.M.; Uhl, F.M.; Pfeifer, D.; Feser, C.J.; et al. Peri-AlloHCT IL-33 Administration Expands Recipient T-Regulatory Cells That Protect Mice against Acute GVHD. Blood 2016, 128, 427–439. [Google Scholar] [CrossRef]
- Schiering, C.; Krausgruber, T.; Chomka, A.; Fröhlich, A.; Adelmann, K.; Wohlfert, E.A.; Pott, J.; Griseri, T.; Bollrath, J.; Hegazy, A.N.; et al. The Alarmin IL-33 Promotes Regulatory T-Cell Function in the Intestine. Nature 2014, 513, 564–568. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Li, G.; Zhu, Y.; Liu, L.; Chen, E.; Turnquist, H.; Zhang, X.; Finn, O.J.; Chen, X.; Lu, B. IL-33 Synergizes with TCR and IL-12 Signaling to Promote the Effector Function of CD8 + T Cells. Eur. J. Immunol. 2011, 41, 3351–3360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonilla, W.V.; Fröhlich, A.; Senn, K.; Kallert, S.; Fernandez, M.; Johnson, S.; Kreutzfeldt, M.; Hegazy, A.N.; Schrick, C.; Fallon, P.G.; et al. The Alarmin Interleukin-33 Drives Protective Antiviral CD8+ T Cell Responses. Science 2012, 335, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate Production of TH2 Cytokines by Adipose Tissue-Associated c-Kit+Sca-1+ Lymphoid Cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.A.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes Represent a New Innate Effector Leukocyte That Mediates Type-2 Immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [Green Version]
- Price, A.E.; Liang, H.-E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically Dispersed Innate IL-13-Expressing Cells in Type 2 Immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11489–11494. [Google Scholar] [CrossRef] [Green Version]
- Mjösberg, J.M.; Trifari, S.; Crellin, N.K.; Peters, C.P.; van Drunen, C.M.; Piet, B.; Fokkens, W.J.; Cupedo, T.; Spits, H. Human IL-25- and IL-33-Responsive Type 2 Innate Lymphoid Cells Are Defined by Expression of CRTH2 and CD161. Nat. Immunol. 2011, 12, 1055–1062. [Google Scholar] [CrossRef]
- Kurowska-Stolarska, M.; Stolarski, B.; Kewin, P.; Murphy, G.; Corrigan, C.J.; Ying, S.; Pitman, N.; Mirchandani, A.; Rana, B.; van Rooijen, N.; et al. IL-33 Amplifies the Polarization of Alternatively Activated Macrophages That Contribute to Airway Inflammation. J. Immunol. 2009, 183, 6469–6477. [Google Scholar] [CrossRef] [Green Version]
- Suzukawa, M.; Iikura, M.; Koketsu, R.; Nagase, H.; Tamura, C.; Komiya, A.; Nakae, S.; Matsushima, K.; Ohta, K.; Yamamoto, K.; et al. An IL-1 Cytokine Member, IL-33, Induces Human Basophil Activation via Its ST2 Receptor. J. Immunol. 2008, 181, 5981–5989. [Google Scholar] [CrossRef]
- Enoksson, M.; Lyberg, K.; Möller-Westerberg, C.; Fallon, P.G.; Nilsson, G.; Lunderius-Andersson, C. Mast Cells as Sensors of Cell Injury through IL-33 Recognition. J. Immunol. 2011, 186, 2523–2528. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Jiang, H.-R.; Kewin, P.; Li, Y.; Mu, R.; Fraser, A.R.; Pitman, N.; Kurowska-Stolarska, M.; McKenzie, A.N.J.; McInnes, I.B.; et al. IL-33 Exacerbates Antigen-Induced Arthritis by Activating Mast Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 10913–10918. [Google Scholar] [CrossRef] [Green Version]
- Morita, H.; Arae, K.; Unno, H.; Miyauchi, K.; Toyama, S.; Nambu, A.; Oboki, K.; Ohno, T.; Motomura, K.; Matsuda, A.; et al. An Interleukin-33-Mast Cell-Interleukin-2 Axis Suppresses Papain-Induced Allergic Inflammation by Promoting Regulatory T Cell Numbers. Immunity 2015, 43, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherry, W.B.; Yoon, J.; Bartemes, K.R.; Iijima, K.; Kita, H. A Novel IL-1 Family Cytokine, IL-33, Potently Activates Human Eosinophils. J. Allergy Clin. Immunol. 2008, 121, 1484–1490. [Google Scholar] [CrossRef] [Green Version]
- Smithgall, M.D.; Comeau, M.R.; Park Yoon, B.-R.; Kaufman, D.; Armitage, R.; Smith, D.E. IL-33 Amplifies Both Th1- and Th2-Type Responses through Its Activity on Human Basophils, Allergen-Reactive Th2 Cells, INKT and NK Cells. Int. Immunol. 2008, 20, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Maywald, R.L.; Doerner, S.K.; Pastorelli, L.; De Salvo, C.; Benton, S.M.; Dawson, E.P.; Lanza, D.G.; Berger, N.A.; Markowitz, S.D.; Lenz, H.-J.; et al. IL-33 Activates Tumor Stroma to Promote Intestinal Polyposis. Proc. Natl. Acad. Sci. USA 2015, 112, E2487–E2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadani, S.P.; Walsh, J.T.; Smirnov, I.; Zheng, J.; Kipnis, J. The Glia-Derived Alarmin IL-33 Orchestrates the Immune Response and Promotes Recovery Following CNS Injury. Neuron 2015, 85, 703–709. [Google Scholar] [CrossRef] [Green Version]
- Mahapatro, M.; Foersch, S.; Hefele, M.; He, G.-W.; Giner-Ventura, E.; Mchedlidze, T.; Kindermann, M.; Vetrano, S.; Danese, S.; Günther, C.; et al. Programming of Intestinal Epithelial Differentiation by IL-33 Derived from Pericryptal Fibroblasts in Response to Systemic Infection. Cell Rep. 2016, 15, 1743–1756. [Google Scholar] [CrossRef] [Green Version]
- Gautier, V.; Cayrol, C.; Farache, D.; Roga, S.; Monsarrat, B.; Burlet-Schiltz, O.; Gonzalez de Peredo, A.; Girard, J.-P. Extracellular IL-33 Cytokine, but Not Endogenous Nuclear IL-33, Regulates Protein Expression in Endothelial Cells. Sci. Rep. 2016, 6, 34255. [Google Scholar] [CrossRef]
- Liu, B.; Tai, Y.; Achanta, S.; Kaelberer, M.M.; Caceres, A.I.; Shao, X.; Fang, J.; Jordt, S.-E. IL-33/ST2 Signaling Excites Sensory Neurons and Mediates Itch Response in a Mouse Model of Poison Ivy Contact Allergy. Proc. Natl. Acad. Sci. USA 2016, 113, E7572–E7579. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.S.; Scott, I.C.; Majithiya, J.B.; Rapley, L.; Kemp, B.P.; England, E.; Rees, D.G.; Overed-Sayer, C.L.; Woods, J.; Bond, N.J.; et al. Oxidation of the Alarmin IL-33 Regulates ST2-Dependent Inflammation. Nat. Commun. 2015, 6, 8327. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.W.; Yoo, H.J.; Park, J.H.; Oh, J.E.; Lee, H.K. Exogenous Interleukin-33 Contributes to Protective Immunity via Cytotoxic T-Cell Priming against Mucosal Influenza Viral Infection. Viruses 2019, 11, 840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, K.A.; Fraser, K.A.; Schenkel, J.M.; Moran, A.; Abt, M.C.; Beura, L.K.; Lucas, P.J.; Artis, D.; Wherry, E.J.; Hogquist, K.; et al. Antigen-Independent Differentiation and Maintenance of Effector-like Resident Memory T Cells in Tissues. J. Immunol. 2012, 188, 4866–4875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slütter, B.; Van Braeckel-Budimir, N.; Abboud, G.; Varga, S.M.; Salek-Ardakani, S.; Harty, J.T. Dynamics of Influenza-Induced Lung-Resident Memory T Cells Underlie Waning Heterosubtypic Immunity. Sci. Immunol. 2017, 2, eaag2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritzl, C.J.; Daniels, M.A.; Teixeiro, E. Interplay of Inflammatory, Antigen and Tissue-Derived Signals in the Development of Resident CD8 Memory T Cells. Front. Immunol. 2021, 12, 636240. [Google Scholar] [CrossRef]
- Bourgeois, E.; Van, L.P.; Samson, M.; Diem, S.; Barra, A.; Roga, S.; Gombert, J.-M.; Schneider, E.; Dy, M.; Gourdy, P.; et al. The Pro-Th2 Cytokine IL-33 Directly Interacts with Invariant NKT and NK Cells to Induce IFN-γ Production. Eur. J. Immunol. 2009, 39, 1046–1055. [Google Scholar] [CrossRef]
- Sattler, S.; Smits, H.H.; Xu, D.; Huang, F.-P. The Evolutionary Role of the IL-33/ST2 System in Host Immune Defence. Arch. Immunol. Ther. Exp. 2013, 61, 107–117. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Kracht, M.; Saul, V.V. The Intricate Interplay between RNA Viruses and NF-ΚB. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2014, 1843, 2754–2764. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, M.; Zheng, H.; Muster, T.; Palese, P.; Beg, A.A.; Garcıía-Sastre, A. Influenza A Virus NS1 Protein Prevents Activation of NF-ΚB and Induction of Alpha/Beta Interferon. J. Virol. 2000, 74, 11566–11573. [Google Scholar] [CrossRef] [Green Version]
- Rückle, A.; Haasbach, E.; Julkunen, I.; Planz, O.; Ehrhardt, C.; Ludwig, S. The NS1 Protein of Influenza A Virus Blocks RIG-I-Mediated Activation of the Noncanonical NF-ΚB Pathway and P52/RelB-Dependent Gene Expression in Lung Epithelial Cells. J. Virol. 2012, 86, 10211–10217. [Google Scholar] [CrossRef] [Green Version]
- Tisoncik, J.R.; Billharz, R.; Burmakina, S.; Belisle, S.E.; Proll, S.C.; Korth, M.J.; García-Sastre, A.; Katze, M.G. The NS1 Protein of Influenza A Virus Suppresses Interferon-Regulated Activation of Antigen-Presentation and Immune-Proteasome Pathways. J. Gen. Virol. 2011, 92, 2093–2104. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.K.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate Lymphoid Cells Promote Lung-Tissue Homeostasis after Infection with Influenza Virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.J.; Dash, P.; Crawford, J.C.; Allen, E.K.; Zamora, A.E.; Boyd, D.F.; Duan, S.; Bajracharya, R.; Awad, W.A.; Apiwattanakul, N.; et al. Lung Γδ T Cells Mediate Protective Responses during Neonatal Influenza Infection That Are Associated with Type 2 Immunity. Immunity 2018, 49, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arpaia, N.; Green, J.A.; Moltedo, B.; Arvey, A.; Hemmers, S.; Yuan, S.; Treuting, P.M.; Rudensky, A.Y. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 2015, 162, 1078–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Grinchuk, V.; Urban, J.F.; Bohl, J.; Sun, R.; Notari, L.; Yan, S.; Ramalingam, T.; Keegan, A.D.; Wynn, T.A.; et al. Macrophages as IL-25/IL-33-Responsive Cells Play an Important Role in the Induction of Type 2 Immunity. PLoS ONE 2013, 8, e59441. [Google Scholar] [CrossRef]
- Melo, E.M.; Oliveira, V.L.S.; Boff, D.; Galvão, I. Pulmonary Macrophages and Their Different Roles in Health and Disease. Int. J. Biochem. Cell Biol. 2021, 141, 106095. [Google Scholar] [CrossRef]
- Allard, B.; Panariti, A.; Martin, J.G. Alveolar Macrophages in the Resolution of Inflammation, Tissue Repair, and Tolerance to Infection. Front. Immunol. 2018, 9, 1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffatt, M.F.; Gut, I.G.; Demenais, F.; Strachan, D.P.; Bouzigon, E.; Heath, S.; von Mutius, E.; Farrall, M.; Lathrop, M.; Cookson, W.O.C.M. A Large-Scale, Consortium-Based Genomewide Association Study of Asthma. N. Engl. J. Med. 2010, 363, 1211–1221.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grotenboer, N.S.; Ketelaar, M.E.; Koppelman, G.H.; Nawijn, M.C. Decoding Asthma: Translating Genetic Variation in IL33 and IL1RL1 into Disease Pathophysiology. J. Allergy Clin. Immunol. 2013, 131, 856–865. [Google Scholar] [CrossRef]
- Savenije, O.E.; Mahachie John, J.M.; Granell, R.; Kerkhof, M.; Dijk, F.N.; de Jongste, J.C.; Smit, H.A.; Brunekreef, B.; Postma, D.S.; Van Steen, K.; et al. Association of IL33–IL-1 Receptor–like 1 (IL1RL1) Pathway Polymorphisms with Wheezing Phenotypes and Asthma in Childhood. J. Allergy Clin. Immunol. 2014, 134, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauché, C.; Vacca, F.; Chia, S.L.; Richards, J.; Gregory, W.F.; Ogunkanbi, A.; Wear, M.; McSorley, H.J. A Truncated Form of HpARI Stabilizes IL-33, Amplifying Responses to the Cytokine. Front. Immunol. 2020, 11, 1363. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Rhee, C.K.; Kang, J.Y.; Byun, J.H.; Choi, J.Y.; Kim, S.J.; Kim, Y.K.; Kwon, S.S.; Lee, S.Y. Blockade of IL-33/ST2 Ameliorates Airway Inflammation in a Murine Model of Allergic Asthma. Exp. Lung Res. 2014, 40, 66–76. [Google Scholar] [CrossRef] [PubMed]
- McSorley, H.J.; Blair, N.F.; Smith, K.A.; McKenzie, A.N.J.; Maizels, R.M. Blockade of IL-33 Release and Suppression of Type 2 Innate Lymphoid Cell Responses by Helminth Secreted Products in Airway Allergy. Mucosal Immunol. 2014, 7, 1068–1078. [Google Scholar] [CrossRef]
- Saglani, S.; Lui, S.; Ullmann, N.; Campbell, G.A.; Sherburn, R.T.; Mathie, S.A.; Denney, L.; Bossley, C.J.; Oates, T.; Walker, S.A.; et al. IL-33 Promotes Airway Remodeling in Pediatric Patients with Severe Steroid-Resistant Asthma. J. Allergy Clin. Immunol. 2013, 132, 676–685. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.-S.; Tsai, M.-C. Asthma Exacerbation in Children: A Practical Review. Pediatr. Neonatol. 2014, 55, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Nimmerjahn, F.; Dudziak, D.; Dirmeier, U.; Hobom, G.; Riedel, A.; Schlee, M.; Staudt, L.M.; Rosenwald, A.; Behrends, U.; Bornkamm, G.W.; et al. Active NF-ΚB Signalling Is a Prerequisite for Influenza Virus Infection. J. Gen. Virol. 2004, 85, 2347–2356. [Google Scholar] [CrossRef]
- Chan, R.W.Y.; Chan, M.C.W.; Nicholls, J.M.; Malik Peiris, J.S. Use of Ex Vivo and in Vitro Cultures of the Human Respiratory Tract to Study the Tropism and Host Responses of Highly Pathogenic Avian Influenza A (H5N1) and Other Influenza Viruses. Virus Res. 2013, 178, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Davoine, F.; Cao, M.; Wu, Y.; Ajamian, F.; Ilarraza, R.; Kokaji, A.I.; Moqbel, R.; Adamko, D.J. Virus-Induced Eosinophil Mediator Release Requires Antigen-Presenting and CD4+ T Cells. J. Allergy Clin. Immunol. 2008, 122, 69–77.e2. [Google Scholar] [CrossRef]
- Beale, J.; Jayaraman, A.; Jackson, D.J.; Macintyre, J.D.R.; Edwards, M.R.; Walton, R.P.; Zhu, J.; Ching, Y.M.; Shamji, B.; Edwards, M.; et al. Rhinovirus-Induced IL-25 in Asthma Exacerbation Drives Type 2 Immunity and Allergic Pulmonary Inflammation. Sci. Transl. Med. 2014, 6, 256ra134. [Google Scholar] [CrossRef] [Green Version]
- Samarasinghe, A.E.; Woolard, S.N.; Boyd, K.L.; Hoselton, S.A.; Schuh, J.M.; McCullers, J.A. The Immune Profile Associated with Acute Allergic Asthma Accelerates Clearance of Influenza Virus. Immunol. Cell Biol. 2014, 92, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Forbes, R.L.; Gibson, P.G.; Murphy, V.E.; Wark, P.A.B. Impaired Type I and III Interferon Response to Rhinovirus Infection during Pregnancy and Asthma. Thorax 2012, 67, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werder, R.B.; Zhang, V.; Lynch, J.P.; Snape, N.; Upham, J.W.; Spann, K.; Phipps, S. Chronic IL-33 Expression Predisposes to Virus-Induced Asthma Exacerbations by Increasing Type 2 Inflammation and Dampening Antiviral Immunity. J. Allergy Clin. Immunol. 2018, 141, 1607–1619.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byers, D.E.; Alexander-Brett, J.; Patel, A.C.; Agapov, E.; Dang-Vu, G.; Jin, X.; Wu, K.; You, Y.; Alevy, Y.; Girard, J.-P.; et al. Long-Term IL-33–Producing Epithelial Progenitor Cells in Chronic Obstructive Lung Disease. J. Clin. Investig. 2013, 123, 3967–3982. [Google Scholar] [CrossRef] [PubMed]
- Duerr, C.U.; McCarthy, C.D.A.; Mindt, B.C.; Rubio, M.; Meli, A.P.; Pothlichet, J.; Eva, M.M.; Gauchat, J.-F.; Qureshi, S.T.; Mazer, B.D.; et al. Type I Interferon Restricts Type 2 Immunopathology through the Regulation of Group 2 Innate Lymphoid Cells. Nat. Immunol. 2016, 17, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Alves-Filho, J.C.; Sônego, F.; Souto, F.O.; Freitas, A.; Verri, W.A.; Auxiliadora-Martins, M.; Basile-Filho, A.; McKenzie, A.N.; Xu, D.; Cunha, F.Q.; et al. Interleukin-33 Attenuates Sepsis by Enhancing Neutrophil Influx to the Site of Infection. Nat. Med. 2010, 16, 708–712. [Google Scholar] [CrossRef]
- Lan, F.; Yuan, B.; Liu, T.; Luo, X.; Huang, P.; Liu, Y.; Dai, L.; Yin, H. Interleukin-33 Facilitates Neutrophil Recruitment and Bacterial Clearance in S. Aureus-Caused Peritonitis. Mol. Immunol. 2016, 72, 74–80. [Google Scholar] [CrossRef]
- Yin, H.; Li, X.; Hu, S.; Liu, T.; Yuan, B.; Ni, Q.; Lan, F.; Luo, X.; Gu, H.; Zheng, F. IL-33 Promotes Staphylococcus Aureus-Infected Wound Healing in Mice. Int. Immunopharmacol. 2013, 17, 432–438. [Google Scholar] [CrossRef]
- Kudva, A.; Scheller, E.V.; Robinson, K.M.; Crowe, C.R.; Choi, S.M.; Slight, S.R.; Khader, S.A.; Dubin, P.J.; Enelow, R.I.; Kolls, J.K.; et al. Influenza A Inhibits Th17-Mediated Host Defense against Bacterial Pneumonia in Mice. J. Immunol. 2011, 186, 1666–1674. [Google Scholar] [CrossRef] [Green Version]
- Damjanovic, D.; Lai, R.; Jeyanathan, M.; Hogaboam, C.M.; Xing, Z. Marked Improvement of Severe Lung Immunopathology by Influenza-Associated Pneumococcal Superinfection Requires the Control of Both Bacterial Replication and Host Immune Responses. Am. J. Pathol. 2013, 183, 868–880. [Google Scholar] [CrossRef]
- Jain, S.; Kamimoto, L.; Bramley, A.M.; Schmitz, A.M.; Benoit, S.R.; Louie, J.; Sugerman, D.E.; Druckenmiller, J.K.; Ritger, K.A.; Chugh, R.; et al. Hospitalized Patients with 2009 H1N1 Influenza in the United States, April–June 2009. N. Engl. J. Med. 2009, 361, 1935–1944. [Google Scholar] [CrossRef] [Green Version]
- Veerapandian, R.; Snyder, J.D.; Samarasinghe, A.E. Influenza in Asthmatics: For Better or for Worse? Front. Immunol. 2018, 9, 1843. [Google Scholar] [CrossRef] [Green Version]
- Gilca, R.; De Serres, G.; Boulianne, N.; Ouhoummane, N.; Papenburg, J.; Douville-Fradet, M.; Fortin, É.; Dionne, M.; Boivin, G.; Skowronski, D.M. Risk Factors for Hospitalization and Severe Outcomes of 2009 Pandemic H1N1 Influenza in Quebec, Canada. Influenza Other Respir. Viruses 2011, 5, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Bramley, A.M.; Dasgupta, S.; Skarbinski, J.; Kamimoto, L.; Fry, A.M.; Finelli, L.; Jain, S.; for the 2009 Pandemic Influenza A (H1N1) Virus Hospitalizations Investigation Team. Intensive Care Unit Patients with 2009 Pandemic Influenza A (H1N1pdm09) Virus Infection—United States, 2009: ICU Patients with Pandemic Influenza. Influenza Other Respir. Viruses 2012, 6, e134–e142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kerkhove, M.D.; Vandemaele, K.A.H.; Shinde, V.; Jaramillo-Gutierrez, G.; Koukounari, A.; Donnelly, C.A.; Carlino, L.O.; Owen, R.; Paterson, B.; Pelletier, L.; et al. Risk Factors for Severe Outcomes Following 2009 Influenza A (H1N1) Infection: A Global Pooled Analysis. PLoS Med. 2011, 8, e1001053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivester, K.M.; Couëtil, L.L.; Moore, G.E. An Observational Study of Environmental Exposures, Airway Cytology, and Performance in Racing Thoroughbreds. J. Vet. Intern. Med. 2018, 32, 1754–1762. [Google Scholar] [CrossRef]
- Kobari, S.; Kusakabe, T.; Momota, M.; Shibahara, T.; Hayashi, T.; Ozasa, K.; Morita, H.; Matsumoto, K.; Saito, H.; Ito, S.; et al. IL-33 Is Essential for Adjuvant Effect of Hydroxypropyl-β-Cyclodexrin on the Protective Intranasal Influenza Vaccination. Front. Immunol. 2020, 11, 360. [Google Scholar] [CrossRef]
- Marichal, T.; Ohata, K.; Bedoret, D.; Mesnil, C.; Sabatel, C.; Kobiyama, K.; Lekeux, P.; Coban, C.; Akira, S.; Ishii, K.J.; et al. DNA Released from Dying Host Cells Mediates Aluminum Adjuvant Activity. Nat. Med. 2011, 17, 996–1002. [Google Scholar] [CrossRef]
- McKee, A.S.; Burchill, M.A.; Munks, M.W.; Jin, L.; Kappler, J.W.; Friedman, R.S.; Jacobelli, J.; Marrack, P. Host DNA Released in Response to Aluminum Adjuvant Enhances MHC Class II-Mediated Antigen Presentation and Prolongs CD4 T-Cell Interactions with Dendritic Cells. Proc. Natl. Acad. Sci. USA 2013, 110, E1122–E1131. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, E.; Ozasa, K.; Temizoz, B.; Ohata, K.; Koo, C.X.; Kanuma, T.; Kusakabe, T.; Kobari, S.; Horie, M.; Morimoto, Y.; et al. Inhaled Fine Particles Induce Alveolar Macrophage Death and Interleukin-1α Release to Promote Inducible Bronchus-Associated Lymphoid Tissue Formation. Immunity 2016, 45, 1299–1310. [Google Scholar] [CrossRef] [Green Version]
- O’Grady, K.; Hearnden, C.C.H.; Bento, D.; Oleszycka, E.; Andersen, P.; Muñoz-Wolf, N.; Lavelle, E.C. IL-33 Is a Negative Regulator of Vaccine-Induced Antigen-Specific Cellular Immunity. J. Immunol. 2019, 202, 1145–1152. [Google Scholar] [CrossRef] [Green Version]
- Kayamuro, H.; Yoshioka, Y.; Abe, Y.; Arita, S.; Katayama, K.; Nomura, T.; Yoshikawa, T.; Kubota-Koketsu, R.; Ikuta, K.; Okamoto, S.; et al. Interleukin-1 Family Cytokines as Mucosal Vaccine Adjuvants for Induction of Protective Immunity against Influenza Virus. J. Virol. 2010, 84, 12703–12712. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.M.; Roy, S.; Califano, D.; McKenzie, A.N.J.; Metzger, D.W.; Furuya, Y. The Interleukin-33–Group 2 Innate Lymphoid Cell Axis Represents a Potential Adjuvant Target To Increase the Cross-Protective Efficacy of Influenza Vaccine. J. Virol. 2021, 95, e00598-21. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozario, C.; Martínez-Sobrido, L.; McSorley, H.J.; Chauché, C. Could Interleukin-33 (IL-33) Govern the Outcome of an Equine Influenza Virus Infection? Learning from Other Species. Viruses 2021, 13, 2519. https://doi.org/10.3390/v13122519
Rozario C, Martínez-Sobrido L, McSorley HJ, Chauché C. Could Interleukin-33 (IL-33) Govern the Outcome of an Equine Influenza Virus Infection? Learning from Other Species. Viruses. 2021; 13(12):2519. https://doi.org/10.3390/v13122519
Chicago/Turabian StyleRozario, Christoforos, Luis Martínez-Sobrido, Henry J. McSorley, and Caroline Chauché. 2021. "Could Interleukin-33 (IL-33) Govern the Outcome of an Equine Influenza Virus Infection? Learning from Other Species" Viruses 13, no. 12: 2519. https://doi.org/10.3390/v13122519