
Return of the Neurotropic Enteroviruses: Co-Opting Cellular Pathways for Infection

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
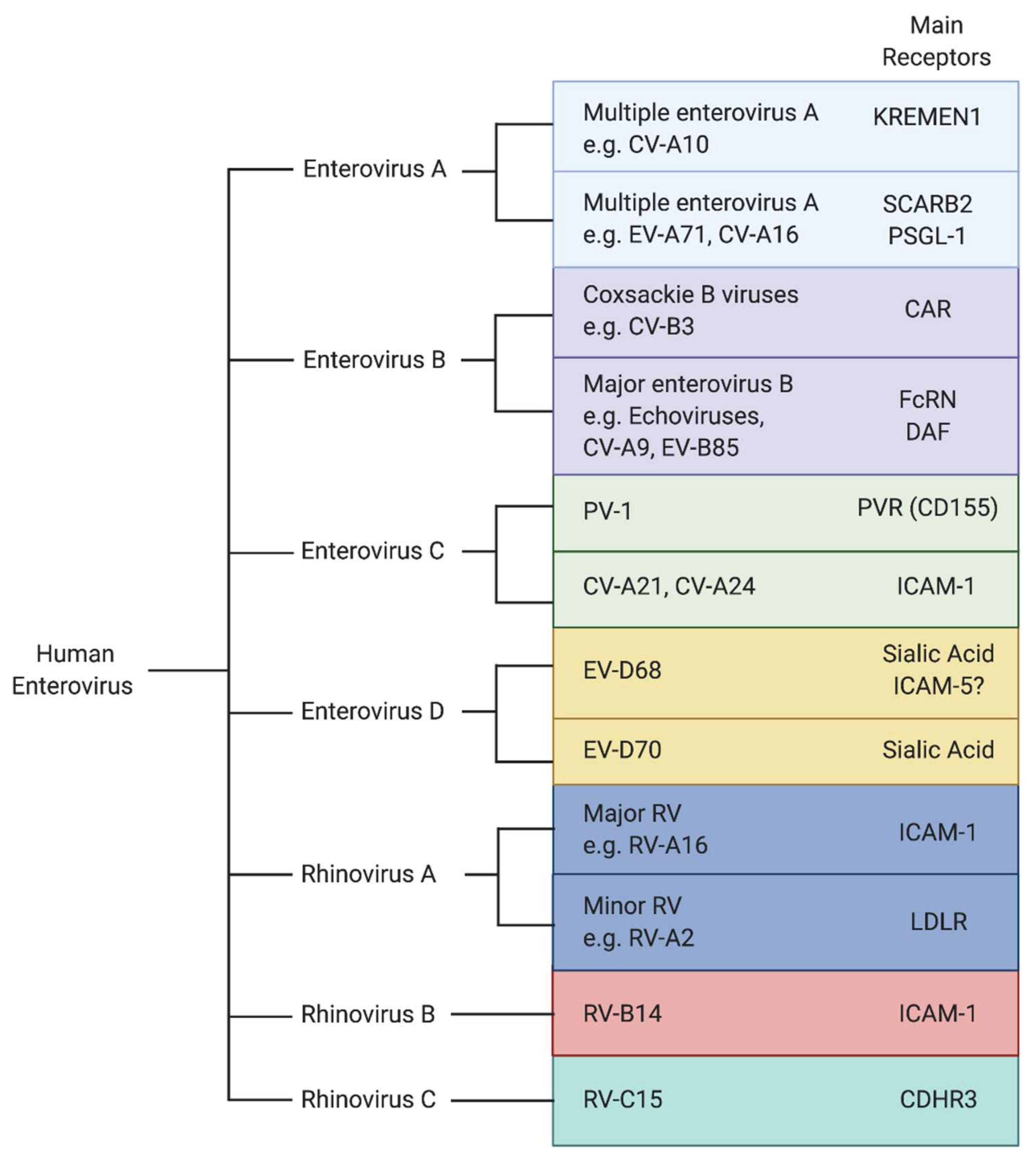
2. The Critical Role of Cellular Receptors in Viral Entry
3. EV-D68 Receptors
4. EV-A71 Receptors
5. Receptors for Other Neurotropic Enteroviruses
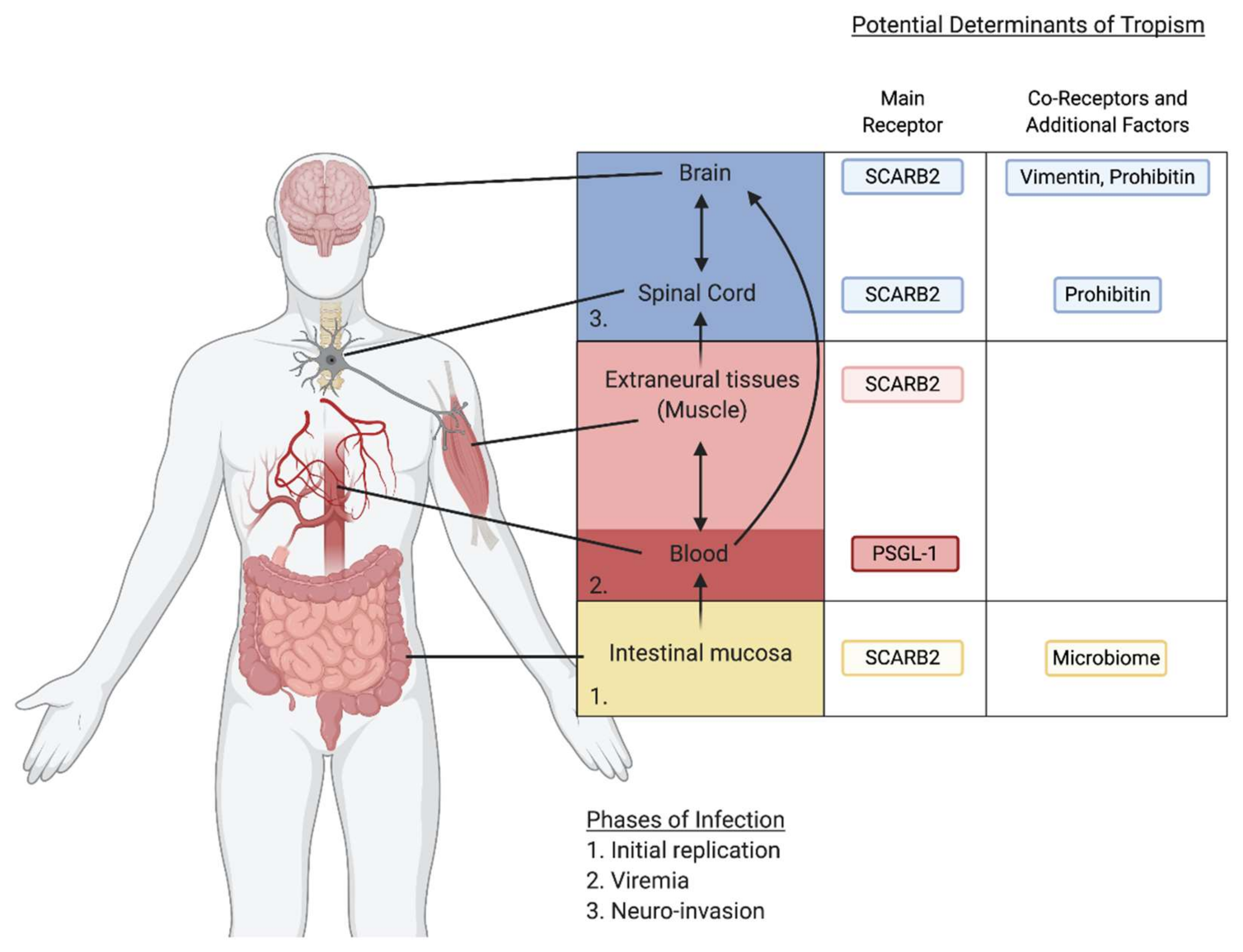
6. Contribution of Viral Receptors to Neurotropism
7. Contribution of Other Host Factors to Neurotropism and Pathogenesis
8. Entry Factors
9. IRES-Mediated Translation
10. Genome Amplification
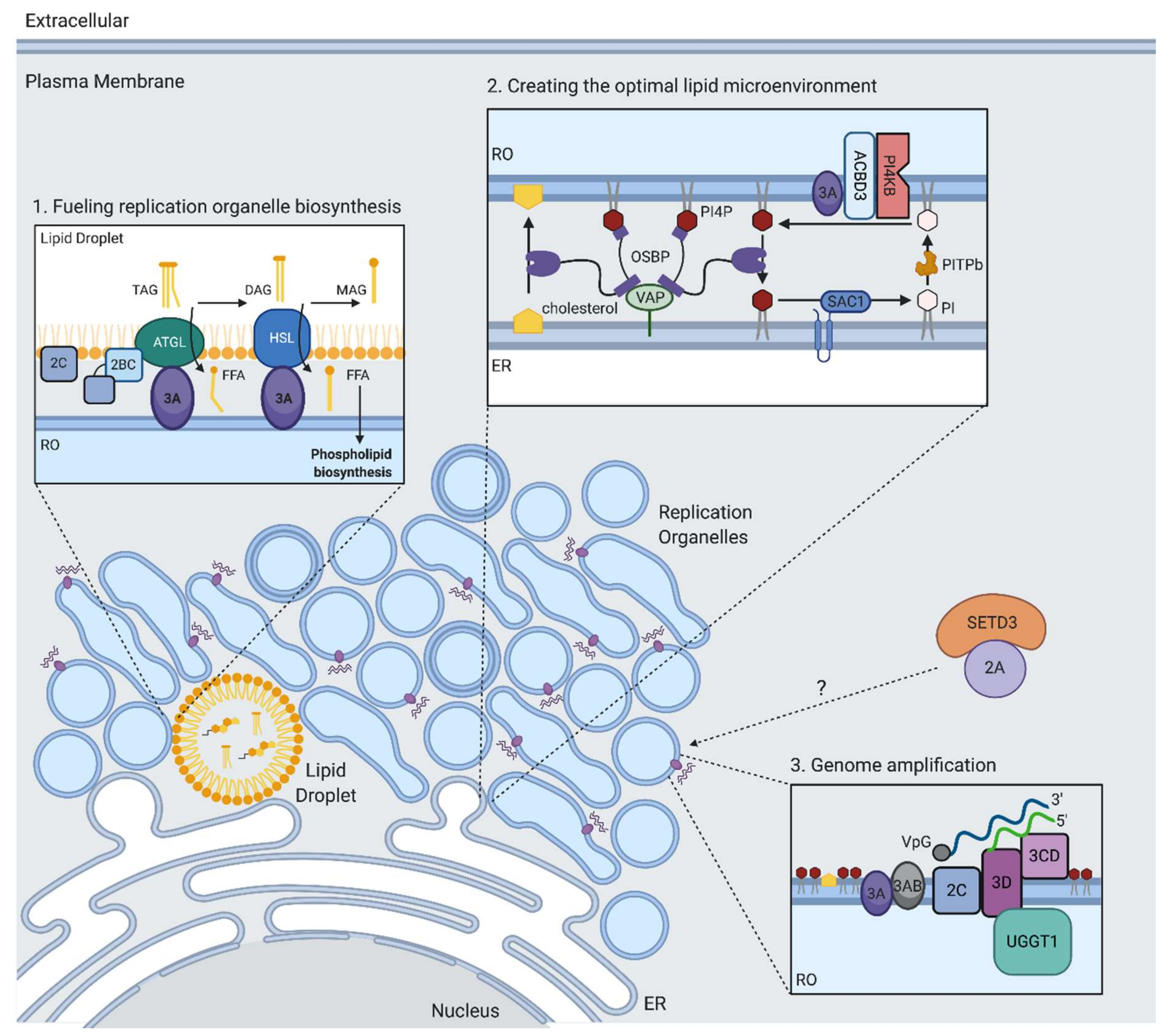
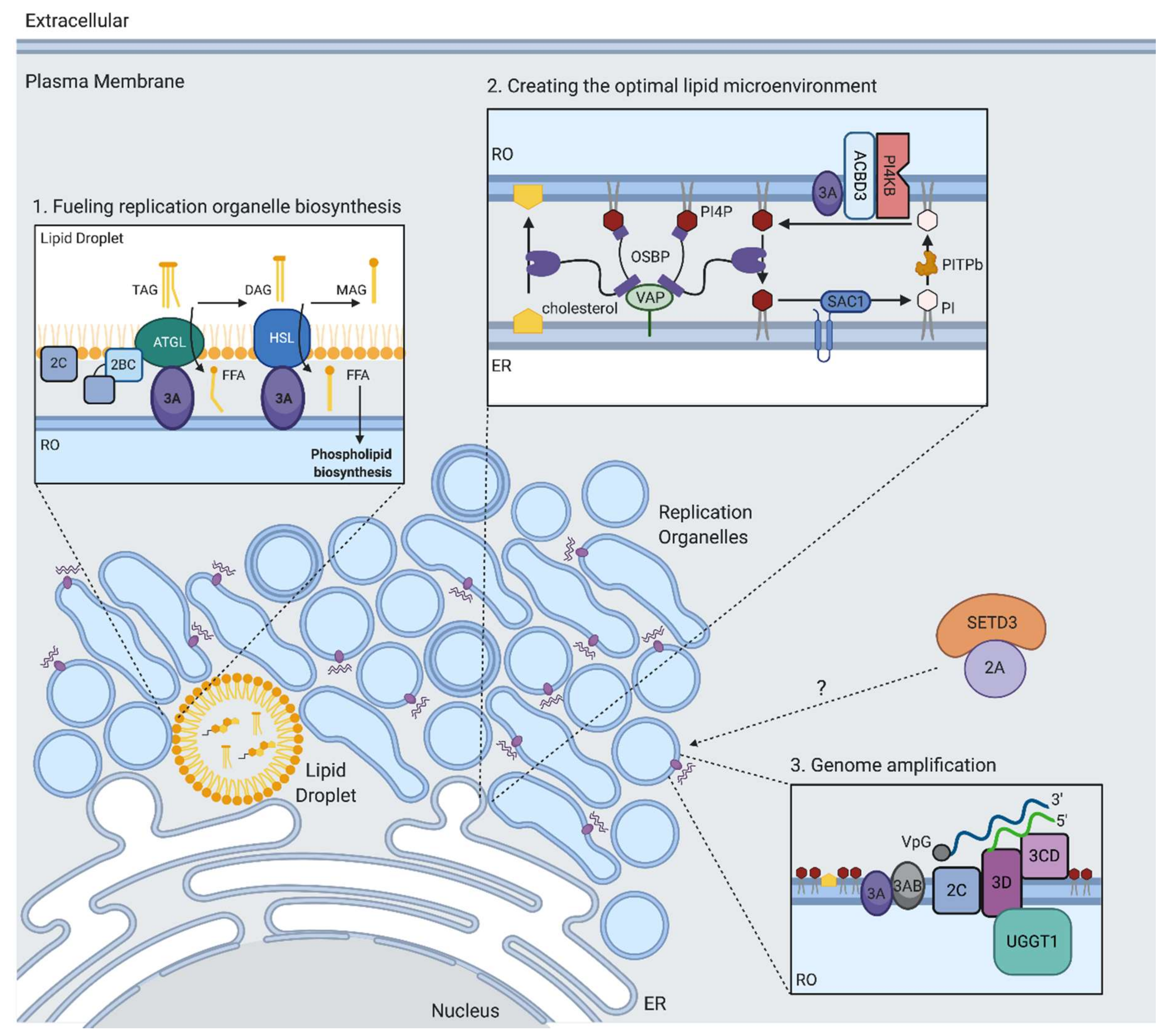
11. Replication Organelle Biogenesis and Phospholipid Biosynthesis
12. Creating the Optimal Lipid Microenvironment for RNA Replication
13. Autophagy in the EV Life Cycle
14. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cassidy, H.; Poelman, R.; Knoester, M.; Van Leer-Buter, C.C.; Niesters, H.G.M. Enterovirus D68—The new polio? Front. Microbiol. 2018, 9, 2677. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Forgie, S.; Robinson, J.L. Non-polio Enterovirus detection with acute flaccid paralysis: A systematic review. J. Med. Virol. 2018, 90, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-S.; Lee, H.-C.; Lee, K.-M.; Gong, Y.-N.; Shih, S.-R. Enterovirus and encephalitis. Front. Microbiol. 2020, 11, 261. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, H.; Schroten, H.; Tenenbaum, T. Enterovirus infections of the central nervous system in children: An update. Pediatr. Infect. Dis. J. 2016, 35, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Owino, C.O.; Chu, J.J.H. Recent advances on the role of host factors during non-poliovirus enteroviral infections. J. Biomed. Sci. 2019, 26, 47. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 2018, 17, 35–56. [Google Scholar] [CrossRef]
- Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Carette, J.E. A CRISPR toolbox to study virus-host interactions. Nat. Rev. Microbiol. 2017, 15, 351–364. [Google Scholar] [CrossRef]
- Pillay, S.; Carette, J.E. Hunting viral receptors using haploid cells. Annu. Rev. Virol. 2015, 2, 219–239. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.S.; Wojcechowskyj, J.A.; Eckhardt, M.; Krogan, N.J. Comparative mapping of host-pathogen protein-protein interactions. Curr. Opin. Microbiol. 2015, 27, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, B.; Lee, L.Y.; Lakadamyali, M.; Rust, M.J.; Zhuang, X.; Hogle, J.M. Imaging poliovirus entry in live cells. PLoS Biol. 2007, 5, e183. [Google Scholar] [CrossRef]
- Racaniello, V.R. Early events in poliovirus infection: Virus-receptor interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 11378–11381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uprety, P.; Graf, E.H. Enterovirus infection and acute flaccid myelitis. Curr. Opin. Virol. 2020, 40, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Schubert, R.D.; Hawes, I.A.; Ramachandran, P.S.; Ramesh, A.; Crawford, E.D.; Pak, J.E.; Wu, W.; Cheung, C.K.; O’Donovan, B.D.; Tato, C.M.; et al. Pan-viral serology implicates enteroviruses in acute flaccid myelitis. Nat. Med. 2019, 25, 1748–1752. [Google Scholar] [CrossRef] [PubMed]
- Baggen, J.; Thibaut, H.J.; Staring, J.; Jae, L.T.; Liu, Y.; Guo, H.; Slager, J.J.; de Bruin, J.W.; van Vliet, A.L.; Blomen, V.A.; et al. Enterovirus D68 receptor requirements unveiled by haploid genetics. Proc. Natl. Acad. Sci. USA 2016, 113, 1399–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, A.B.; Warren, A.L.; Racaniello, V.R. Neurotropism of enterovirus D68 isolates is i-dependent of sialic acid and is not a recently acquired phenotype. mBio 2019, 10, e02370-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Sheng, J.; Baggen, J.; Meng, G.; Xiao, C.; Thibaut, H.J.; Van Kuppeveld, F.J.M.; Rossmann, M.G. Sialic acid-dependent cell entry of human enterovirus D68. Nat. Commun. 2015, 6, 8865. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Lee, K.; Bae, S.; Park, J.; Lee, C.K.; Kim, M.; Kim, E.; Kim, M.; Kim, S.; Kim, C.; et al. CRISPR/Cas9-mediated gene knockout screens and target identification via whole-genome sequencing uncover host genes required for picornavirus infection. J. Biol. Chem. 2017, 292, 10664–10671. [Google Scholar] [CrossRef] [Green Version]
- Dermody, T.S.; Kirchner, E.; Guglielmi, K.M.; Stehle, T. Immunoglobulin superfamily virus receptors and the evolution of adaptive immunity. PLoS Pathog. 2009, 5, e1000481. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Guo, H.; Chang, J.; Yu, Y.; Liu, G.; Zhang, N.; Willard, S.H.; Zheng, S.; Yu, X.F. ICAM-5/telencephalin is a functional entry receptor for enterovirus D68. Cell Host Microbe 2016, 20, 631–641. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Zhu, R.; Xu, L.; He, M.; Yan, X.; Liu, D.; Yin, Z.; Wu, Y.; Li, Y.; Yang, L.; et al. Atomic structures of enterovirus D68 in complex with two monoclonal antibodies define distinct mechanisms of viral neutralization. Nat. Microbiol. 2019, 4, 124–133. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Yamashita, Y.; Li, J.; Hanagata, N.; Minowa, T.; Takemura, T.; Koike, S. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat. Med. 2009, 15, 798–801. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Ohka, S.; Fujii, K.; Koike, S. Functional comparison of SCARB2 and PSGL1 as receptors for enterovirus 71. J. Virol. 2013, 87, 3335–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Koike, S. Cellular receptors for enterovirus A71. J. Biomed. Sci. 2020, 27, 23. [Google Scholar] [CrossRef] [PubMed]
- Qing, J.; Wang, Y.; Sun, Y.; Huang, J.; Yan, W.; Wang, J.; Su, D.; Ni, C.; Li, J.; Rao, Z.; et al. Cyclophilin A associates with enterovirus-71 virus capsid and plays an essential role in viral infection as an uncoating regulator. PLoS Pathog. 2014, 10, e1004422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, M.L.; Jia, L.; Yip, C.C.Y.; Chan, J.F.W.; Teng, J.L.L.; Chan, K.H.; Cai, J.P.; Zhang, C.; Zhang, A.J.; Wong, W.M.; et al. Human tryptophanyl-tRNA synthetase is an IFN-γ-inducible entry factor for enterovirus. J. Clin. Investig. 2018, 128, 5163–5177. [Google Scholar] [CrossRef]
- Too, I.H.K.; Bonne, I.; Tan, E.L.; Chu, J.J.H.; Alonso, S. Prohibitin plays a critical role in Enterovirus 71 neuropathogenesis. PLoS Pathog. 2018, 14, e1006778. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.W.; Poh, C.L.; Sam, I.C.; Chan, Y.F. Enterovirus 71 uses cell surface heparan sulfate glycosaminoglycan as an attachment receptor. J. Virol. 2013, 87, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Du, N.; Cong, H.; Tian, H.; Zhang, H.; Zhang, W.; Song, L.; Tien, P. Cell surface vimentin is an attachment receptor for enterovirus 71. J. Virol. 2014, 88, 5816–5833. [Google Scholar] [CrossRef] [Green Version]
- Staring, J.; Hengel, L.G.V.D.; Raaben, M.; Blomen, V.A.; Carette, J.E.; Brummelkamp, T.R. KREMEN1 is a host entry receptor for a major group of enteroviruses. Cell Host Microbe 2018, 23, 636–643.e5. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Peng, R.; Song, H.; Tong, Z.; Qu, X.; Liu, S.; Zhao, X.; Chai, Y.; Wang, P.; Gao, G.F.; et al. Molecular basis of Coxsackievirus A10 entry using the two-in-one attachment and uncoating receptor KRM1. Proc. Natl. Acad. Sci. USA 2020, 117, 18711–18718. [Google Scholar] [CrossRef]
- Bergelson, J.M.; Chan, M.; Solomon, K.R.; John, N.F.S.; Lin, H.; Finberg, R.W. Decay-accelerating factor (CD55), a glycosylphosphatidylinositol-anchored complement regulatory protein, is a receptor for several echoviruses. Proc. Natl. Acad. Sci USA 1994, 91, 6245–6248. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Lin, F.; Chipman, P.R.; Bator, C.M.; Baker, T.S.; Shoham, M.; Kuhn, R.J.; Medof, M.E.; Rossmann, M.G. Structure of decay-accelerating factor bound to echovirus 7: A virus-receptor complex. Proc. Natl. Acad. Sci. USA 2002, 99, 10325–10329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevaliez, S.; Balanant, J.; Maillard, P.; Lone, Y.-C.; Lemonnier, F.A.; Delpeyroux, F. Role of class I human leukocyte antigen molecules in early steps of echovirus infection of rhabdomyosarcoma cells. Virology 2008, 381, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Zhang, G.; Liu, S.; Chen, X.; Peng, R.; Dai, L.; Qu, X.; Li, S.; Song, H.; Gao, Z.; et al. Human neonatal Fc receptor is the cellular uncoating receptor for enterovirus B. Cell 2019, 177, 1553–1565.e16. [Google Scholar] [CrossRef]
- Morosky, S.; Wells, A.I.; Lemon, K.; Evans, A.S.; Schamus, S.; Bakkenist, C.J.; Coyne, C. The neonatal Fc receptor is a pan-echovirus receptor. Proc. Natl. Acad. Sci. USA 2019, 116, 3758–3763. [Google Scholar] [CrossRef] [Green Version]
- Racaniello, V. One hundred years of poliovirus pathogenesis. Virology 2006, 344, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hixon, A.M.; Yu, G.; Leser, J.S.; Yagi, S.; Clarke, P.; Chiu, C.Y.; Tyler, K.L. A mouse model of paralytic myelitis caused by enterovirus D68. PLoS Pathog. 2017, 13, e1006199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hixon, A.M.; Clarke, P.; Tyler, K.L. Contemporary circulating enterovirus D68 Strains infect and undergo retrograde axonal transport in spinal motor neurons independent of sialic acid. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.M.; Hixon, A.M.; Oldfield, L.M.; Zhang, Y.; Novotny, M.; Wang, W.; Das, S.R.; Shabman, R.S.; Tyler, K.L.; Scheuermann, R.H. Contemporary circulating enterovirus D68 strains have acquired the capacity for viral entry and replication in human neuronal cells. mBio 2018, 9, e01954-18. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.W.; Yu, S.L.; Shao, H.Y.; Lin, H.-Y.; Liu, C.-C.; Hsiao, K.-N.; Chitra, E.; Tsou, Y.-L.; Chang, H.-W.; Sia, C.; et al. Human SCARB2 transgenic mice as an infectious animal model for enterovirus 71. PLoS ONE 2013, 8, e57591. [Google Scholar] [CrossRef] [Green Version]
- Fujii, K.; Nagata, N.; Sato, Y.; Ong, K.C.; Wong, K.T.; Yamayoshi, S.; Shimanuki, M.; Shitara, H.; Taya, C.; Koike, S. Transgenic mouse model for the study of enterovirus 71 neuropathogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 14753–14758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Liu, Q.; Wu, X.; Chen, P.; Wu, X.; Guo, Y.; Liu, S.; Liang, Z.-L.; Fan, C.; Wang, Y. A safe and sensitive enterovirus A71 infection model based on human SCARB2 knock-in mice. Vaccine 2016, 34, 2729–2736. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dong, W.; Quan, X.; Ma, C.; Qin, C.; Zhang, L. Transgenic expression of human P-selectin glycoprotein ligand-1 is not sufficient for enterovirus 71 infection in mice. Arch. Virol. 2012, 157, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Tijsma, A.; Mirabelli, C.; Baggen, J.; Wahedi, M.; Franco, D.; De Palma, A.; Leyssen, P.; Verbeken, E.; van Kuppeveld, F.J.M.; et al. Intra-host emergence of an enterovirus A71 variant with enhanced PSGL1 usage and neurovirulence. Emerg. Microbes. Infect. 2019, 8, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Jiao, X.Y.; Guo, L.; Huang, D.Y.; Chang, X.L.; Qiu, Q.C. Distribution of EV71 receptors SCARB2 and PSGL-1 in human tissues. Virus Res. 2014, 190, 40–52. [Google Scholar] [CrossRef]
- Wang, W.; Sun, J.; Wang, N.; Sun, Z.; Ma, Q.; Li, J.; Zhang, M.; Xu, J. Enterovirus A71 capsid protein VP1 increases blood–brain barrier permeability and virus receptor vimentin on the brain endothelial cells. J. Neurovirol. 2020, 26, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Cao, Y.; Su, W.; Huang, S.; Lu, W.; Zhou, Y.; Gao, J.; Zhao, W.; Zhang, B.; Wu, X. Enterovirus A71 VP1 variation A289T decreases the central nervous system infectivity via attenuation of interactions between VP1 and vimentin in vitro and in vivo. Viruses 2019, 11, 467. [Google Scholar] [CrossRef] [Green Version]
- Tseligka, E.D.; Sobo, K.; Stoppini, L.; Cagno, V.; Abdul, F.; Piuz, I.; Meylan, P.; Huang, S.; Constant, S.; Tapparel, C. A VP1 mutation acquired during an enterovirus 71 disseminated infection confers heparan sulfate binding ability and modulates ex vivo tropism. PLoS Pathog. 2018, 14, e1007190. [Google Scholar] [CrossRef]
- Earley, D.F.; Bailly, B.; Maggioni, A.; Kundur, A.R.; Thomson, R.J.; Chang, C.-W.; Von Itzstein, M. Efficient blocking of enterovirus 71 infection by heparan sulfate analogues acting as decoy receptors. ACS Infect. Dis. 2019, 5, 1708–1717. [Google Scholar] [CrossRef]
- Kobayashi, K.; Mizuta, K.; Koike, S. Heparan sulfate attachment receptor is a major selection factor for attenuated enterovirus 71 mutants during cell culture adaptation. PLoS Pathog. 2020, 16, e1008428. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, C.; Wu, S.; Chen, X.; Shi, Y.; Zhou, B.; Zhang, L.; Zhang, F.; Wang, Z.; Zhang, Y.; et al. A novel finding for enterovirus virulence from the capsid protein VP1 of EV71 circulating in mainland China. Virus Genes 2014, 48, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Sudaka, Y.; Takashino, A.; Imura, A.; Fujii, K.; Koike, S. Amino acid variation at VP1-145 of enterovirus 71 determines attachment receptor usage and neurovirulence in human scavenger receptor B2 transgenic mice. J. Virol. 2018, 92, e00681-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, C.; Suzuki, T.; Kotani, O.; Iwata-Yoshikawa, N.; Nagata, N.; Ami, Y.; Wakita, T.; Nishimura, Y.; Shimizu, H. The role of VP1 amino acid residue 145 of enterovirus 71 in viral fitness and pathogenesis in a cynomolgus monkey model. PLoS Pathog. 2015, 11, e1005033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tee, H.K.; Tan, C.W.; Yogarajah, T.; Lee, M.H.P.; Chai, H.J.; Hanapi, N.A.; Yusof, S.R.; Ong, K.C.; Lee, V.S.; Sam, I.C.; et al. Electrostatic interactions at the five-fold axis alter heparin-binding phenotype and drive enterovirus A71 virulence in mice. PLoS Pathog. 2019, 15, e1007863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuss, S.K.; Best, G.T.; Etheredge, C.A.; Pruijssers, A.J.; Frierson, J.M.; Hooper, L.V.; Dermody, T.S.; Pfeiffer, J.K. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 2011, 334, 249–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, C.M.; Acevedo, M.A.W.; McCune, B.T.; Pfeiffer, J.K. Related enteric viruses have different requirements for host microbiota in mice. J. Virol. 2019, 93, e01339-19. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Jesudhasan, P.R.; Pfeiffer, J.K. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe 2014, 15, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Erickson, A.K.; Jesudhasan, P.R.; Mayer, M.J.; Narbad, A.; Winter, S.E.; Pfeiffer, J.K. Bacteria facilitate enteric virus co-infection of mammalian cells and promote genetic recombination. Cell Host Microbe 2018, 23, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, E.R.; Nguyen, Y.; Sasaki, J.; Pfeiffer, J.K. Bacterial stabilization of a panel of picornaviruses. mSphere 2019, 4, e00183-19. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, M.A.W.; Pfeiffer, J.K. Microbiota-independent antiviral effects of antibiotics on poliovirus and coxsackievirus. Virology 2020, 546, 20–24. [Google Scholar] [CrossRef]
- Duan, G.; Yang, H.; Shi, L.; Sun, W.; Sui, M.; Zhang, R.; Wang, X.; Wang, F.; Zhang, W.; Xi, Y.; et al. Serum inflammatory cytokine levels correlate with hand-foot-mouth disease severity: A nested serial case-control study. PLoS ONE 2014, 9, e112676. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Liao, Y.; Gao, Y.; Jiang, G.; Wang, L.; Zhang, Y.; Fan, S.; Xu, X.; Li, Q.-H. Mechanism for the lethal effect of enterovirus A71 intracerebral injection in neonatal mice. Lab. Investig. 2019, 100, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Su, R.; Wang, W.; Liang, Y.; Zeng, X.; Shereen, M.A.; Bashir, N.; Zhang, Q.; Zhao, L.; Wu, K.; et al. EV71 infection induces neurodegeneration via activating TLR7 signaling and IL-6 production. PLoS Pathog. 2019, 15, e1008142. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.-D.; Mai, J.-N.; He, L.-Y.; Li, P.-Q.; Chen, W.-X.; Yan, J.-J.; Zhu, W.-D.; Deng, L.; Wei, D.; Liu, D.-H.; et al. Glucocorticoids prevent enterovirus 71 capsid protein VP1 induced calreticulin surface exposure by alleviating neuronal ER stress. Neurotox. Res. 2017, 31, 204–217. [Google Scholar] [CrossRef]
- Staring, J.; Von Castelmur, E.; Blomen, V.A.; Hengel, L.G.V.D.; Brockmann, M.; Baggen, J.; Thibaut, H.J.; Nieuwenhuis, J.; Janssen, H.; Van Kuppeveld, F.J.M.; et al. PLA2G16 represents a switch between entry and clearance of Picornaviridae. Nat. Cell Biol. 2017, 541, 412–416. [Google Scholar] [CrossRef]
- Thurston, T.L.M.; Wandel, M.P.; Von Muhlinen, N.; Foeglein, Á.; Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nat. Cell Biol. 2012, 482, 414–418. [Google Scholar] [CrossRef]
- Jiang, H.; Leung, C.; Tahan, S.; Wang, D. Entry by multiple picornaviruses is dependent on a pathway that includes TNK2, WASL, and NCK1. eLife 2019, 8, e50276. [Google Scholar] [CrossRef]
- Martinez-Salas, E.; Francisco-Velilla, R.; Fernandez-Chamorro, J.; Lozano, G.; Diaz-Toledano, R. Picornavirus IRES elements: RNA structure and host protein interactions. Virus Res. 2015, 206, 62–73. [Google Scholar] [CrossRef]
- Lai, M.C.; Chen, H.H.; Xu, P.; Wang, R.Y.L. Translation control of Enterovirus A71 gene expression. J. Biomed. Sci. 2020, 27, 22. [Google Scholar] [CrossRef]
- Su, Y.S.; Tsai, A.H.; Ho, Y.F.; Huang, S.Y.; Liu, Y.C.; Hwang, L.H. Stimulation of the internal ribosome entry site (IRES)-dependent translation of enterovirus 71 by DDX3X RNA helicase and viral 2A and 3C proteases. Front. Microbiol. 2018, 9, 1324. [Google Scholar] [CrossRef]
- Chen, Y.M.; Ou, B.T.; Chen, C.Y.; Chan, H.H.; Chen, C.J.; Wang, R.Y. Staufen1 protein participates positively in the viral RNA replication of enterovirus 71. Viruses 2019, 11, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diep, J.; Ooi, Y.S.; Wilkinson, A.W.; Peters, C.E.; Foy, E.; Johnson, J.R.; Zengel, J.; Ding, S.; Weng, K.F.; Laufman, O.; et al. Enterovirus pathogenesis requires the host methyltransferase SETD3. Nat. Microbiol. 2019, 4, 2523–2537. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.W.; Diep, J.; Dai, S.; Liu, S.; Ooi, Y.S.; Song, D.; Li, T.M.; Horton, J.R.; Zhang, X.; Liu, C.; et al. SETD3 is an actin histidine methyltransferase that prevents primary dystocia. Nature 2019, 565, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, S.; Seliga, A.K.; Vertommen, D.; Terreri, M.; Ishikawa, T.; Grabowska, I.; Tiebe, M.; Teleman, A.A.; Jagielski, A.K.; Veiga-da-Cunha, M.; et al. SETD3 protein is the actin-specific histidine N-methyltransferase. eLife 2018, 7, e37921. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.-N.; Jheng, J.-R.; Arnold, J.J.; Wang, J.-R.; Cameron, C.E.; Shih, S.-R. UGGT1 enhances enterovirus 71 pathogenicity by promoting viral RNA synthesis and viral replication. PLoS Pathog. 2017, 13, e1006375. [Google Scholar] [CrossRef] [PubMed]
- Limpens, R.W.A.L.; Van Der Schaar, H.M.; Kumar, D.; Koster, A.J.; Snijder, E.J.; Van Kuppeveld, F.J.M.; Bárcena, M. The transformation of enterovirus replication structures: A three-dimensional study of single- and double-membrane compartments. mBio 2011, 2, e00166-11. [Google Scholar] [CrossRef] [Green Version]
- Belov, G.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Melia, C.E.; Peddie, C.J.; De Jong, A.W.M.; Snijder, E.J.; Collinson, L.M.; Koster, A.J.; Van Der Schaar, H.M.; Van Kuppeveld, F.J.M.; Bárcena, M. Origins of enterovirus replication organelles established by whole-cell electron microscopy. mBio 2019, 10, e00951-19. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.; Bartenschlager, R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef]
- Viktorova, E.G.; Nchoutmboube, J.A.; Ford-Siltz, L.A.; Iverson, E.; Belov, G.A. Phospholipid synthesis fueled by lipid droplets drives the structural development of poliovirus replication organelles. PLoS Pathog. 2018, 14, e1007280. [Google Scholar] [CrossRef] [Green Version]
- Laufman, O.; Perrino, J.; Andino, R. Viral generated inter-organelle contacts redirect lipid flux for genome replication. Cell 2019, 178, 275–289.e16. [Google Scholar] [CrossRef] [PubMed]
- Nchoutmboube, J.A.; Viktorova, E.G.; Scott, A.J.; Ford, L.A.; Pei, Z.; Watkins, P.A.; Ernst, R.K.; Belov, G.A. Increased long chain acyl-coa synthetase activity and fatty acid import is linked to membrane synthesis for development of picornavirus replication organelles. PLoS Pathog. 2013, 9, e1003401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roulin, P.S.; Lötzerich, M.; Torta, F.; Tanner, L.B.; Van Kuppeveld, F.J.; Wenk, M.R.; Greber, U.F. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-golgi interface. Cell Host Microbe 2014, 16, 677–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Schaar, H.M.; Dorobantu, C.M.; Albulescu, L.; Strating, J.R.; Van Kuppeveld, F.J. Fat(al) attraction: Picornaviruses usurp lipid transfer at membrane contact sites to create replication organelles. Trends Microbiol. 2016, 24, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, N.-Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.-H.; Takvorian, P.M.; Pau, C.; Van Der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godi, A.; Pertile, P.; Meyers, R.; Marra, P.; Di Tullio, G.; Iurisci, C.; Luini, A.; Corda, D.; De Matteis, M.A. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat. Cell Biol. 1999, 1, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Altan-Bonnet, N.; Kovtunovych, G.; Jackson, C.L.; Lippincott-Schwartz, J.; Ehrenfeld, E. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J. Virol. 2006, 81, 558–567. [Google Scholar] [CrossRef] [Green Version]
- Lanke, K.H.; van der Schaar, H.M.; Belov, G.A.; Feng, Q.; Duijsings, D.; Jackson, C.L.; Ehrenfeld, E.; van Kuppeveld, F.J. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J. Virol. 2009, 83, 11940–11949. [Google Scholar] [CrossRef] [Green Version]
- Greninger, A.L.; Knudsen, G.M.; Betegon, M.; Burlingame, A.L.; Derisi, J.L. The 3A protein from multiple picornaviruses utilizes the golgi adaptor protein ACBD3 to recruit PI4KIIIβ. J. Virol. 2012, 86, 3605–3616. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, J.; Ishikawa, K.; Arita, M.; Taniguchi, K. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 2012, 31, 754–766. [Google Scholar] [CrossRef]
- Lyoo, H.; Van Der Schaar, H.M.; Dorobantu, C.M.; Rabouw, H.H.; Strating, J.R.P.M.; Van Kuppeveld, F.J. ACBD3 is an essential pan-enterovirus host factor that mediates the interaction between viral 3A protein and cellular protein PI4KB. mBio 2019, 10, e02742-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greninger, A.L.; Knudsen, G.M.; Betegon, M.; Burlingame, A.L.; DeRisi, J.L. ACBD3 Interaction with TBC1 Domain 22 Protein Is Differentially Affected by Enteroviral and Kobuviral 3A protein binding. mBio 2013, 4, e00098-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomen, V.A.; Májek, P.; Jae, L.T.; Bigenzahn, J.W.; Nieuwenhuis, J.; Staring, J.; Sacco, R.; Van Diemen, F.R.; Olk, N.; Stukalov, A.; et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350, 1092–1096. [Google Scholar] [CrossRef]
- McPhail, J.A.; Lyoo, H.; Pemberton, J.G.; Hoffmann, R.M.; van Elst, W.; Strating, J.; Jenkins, M.L.; Stariha, J.T.B.; Powell, C.J.; Boulanger, M.J.; et al. Characterization of the c10orf76-PI4KB complex and its necessity for Golgi PI4P levels and enterovirus replication. EMBO Rep. 2020, 21, e48441. [Google Scholar] [CrossRef] [PubMed]
- Arita, M. Phosphatidylinositol-4 kinase III beta and oxysterol-binding protein accumulate unester-ified cholesterol on poliovirus-induced membrane structure. Microbiol. Immunol. 2014, 58, 239–256. [Google Scholar] [CrossRef]
- Banerjee, S.; Aponte-Diaz, D.; Yeager, C.; Sharma, S.D.; Ning, G.; Oh, H.S.; Han, Q.; Umeda, M.; Hara, Y.; Wang, R.Y.L.; et al. Hijacking of multiple phospholipid biosynthetic pathways and induction of membrane biogenesis by a picornaviral 3CD protein. PLoS Pathog. 2018, 14, e1007086. [Google Scholar] [CrossRef] [Green Version]
- Shengjuler, D.; Chan, Y.M.; Sun, S.; Moustafa, I.M.; Li, Z.-L.; Gohara, D.W.; Buck, M.; Cremer, P.S.; Boehr, D.D.; Cameron, C.E. The RNA-binding site of poliovirus 3C protein doubles as a phosphoinositide-binding domain. Structure 2017, 25, 1875–1886.e7. [Google Scholar] [CrossRef] [Green Version]
- Melia, C.E.; van der Schaar, H.M.; Lyoo, H.; Limpens, R.; Feng, Q.; Wahedi, M.; Overheul, G.J.; van Rij, R.P.; Snijder, E.J.; Koster, A.J.; et al. Escaping host factor PI4KB inhibition: Enterovirus genomic RNA replication in the absence of replication organelles. Cell Rep. 2017, 21, 587–599. [Google Scholar] [CrossRef] [Green Version]
- Albulescu, L.; Wubbolts, R.; Van Kuppeveld, F.J.; Strating, J.R.P.M. Cholesterol shuttling is important for RNA replication of coxsackievirus B3 and encephalomyocarditis virus. Cell. Microbiol. 2015, 17, 1144–1156. [Google Scholar] [CrossRef]
- Ilnytska, O.; Santiana, M.; Hsu, N.Y.; Du, W.L.; Chen, Y.H.; Viktorova, E.G.; Belov, G.; Brinker, A.; Storch, J.; Moore, C.; et al. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe 2013, 14, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Strating, J.R.; van der Linden, L.; Albulescu, L.; Bigay, J.; Arita, M.; Delang, L.; Leyssen, P.; van der Schaar, H.M.; Lanke, K.H.; Thibaut, H.J.; et al. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep. 2015, 10, 600–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa-Sasaki, K.; Nagashima, S.; Taniguchi, K.; Sasaki, J. Model of OSBP-mediated cholesterol supply to Aichi virus RNA replication sites involving protein-protein interactions among viral proteins, ACBD3, OSBP, VAP-A/B, and SAC1. J. Virol. 2018, 92, e01952-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesmin, B.; Bigay, J.; Von Filseck, J.M.; Lacas-Gervais, S.; Drin, G.; Antonny, B. A Four-Step Cycle Driven by PI(4)P Hydrolysis Directs Sterol/PI(4)P Exchange by the ER-Golgi Tether OSBP. Cell 2013, 155, 830–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siltz, L.A.F.; Viktorova, E.G.; Zhang, B.; Kouiavskaia, D.; Dragunsky, E.; Chumakov, K.; Isaacs, L.; Belov, G. New small-molecule inhibitors effectively blocking picornavirus replication. J. Virol. 2014, 88, 11091–11107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Schaar, H.M.; Leyssen, P.; Thibaut, H.J.; De Palma, A.; Van Der Linden, L.; Lanke, K.H.W.; Lacroix, C.; Verbeken, E.; Conrath, K.; MacLeod, A.M.; et al. A novel, broad-spectrum inhibitor of enterovirus replication that targets host cell factor phosphatidylinositol 4-kinase IIIβ. Antimicrob. Agents Chemother. 2013, 57, 4971–4981. [Google Scholar] [CrossRef] [Green Version]
- Arita, M.; Kojima, H.; Nagano, T.; Okabe, T.; Wakita, T.; Shimizu, H. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J. Virol. 2011, 85, 2364–2372. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, A.M.; Mitchell, D.R.; Palmer, N.J.; Van De Poël, H.; Conrath, K.; Andrews, M.; Leyssen, P.; Neyts, J. Identification of a series of compounds with potent antiviral activity for the treatment of enterovirus infections. ACS Med. Chem. Lett. 2013, 4, 585–589. [Google Scholar] [CrossRef] [Green Version]
- Arita, M.; Kojima, H.; Nagano, T.; Okabe, T.; Wakita, T.; Shimizu, H. Oxysterol-binding protein family I is the target of minor enviroxime-like compounds. J. Virol. 2013, 87, 4252–4260. [Google Scholar] [CrossRef] [Green Version]
- Albulescu, L.; Bigay, J.; Biswas, B.; Weber-Boyvat, M.; Dorobantu, C.M.; Delang, L.; van der Schaar, H.M.; Jung, Y.S.; Neyts, J.; Olkkonen, V.M.; et al. Uncovering oxysterol-binding protein (OSBP) as a target of the anti-enteroviral compound TTP-8307. Antiviral. Res. 2017, 140, 37–44. [Google Scholar] [CrossRef]
- Roberts, B.L.; Severance, Z.C.; Bensen, R.C.; Le, A.T.; Kothapalli, N.R.; Nuñez, J.I.; Ma, H.; Wu, S.; Standke, S.J.; Yang, Z.; et al. Transient compound treatment induces a multigenerational reduction of oxysterol-binding protein (OSBP) levels and prophylactic antiviral activity. ACS Chem. Biol. 2019, 14, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Roberts, B.L.; Severance, Z.C.; Bensen, R.C.; Le-McClain, A.T.; Malinky, C.A.; Mettenbrink, E.M.; Nuñez, J.I.; Reddig, W.J.; Blewett, E.L.; Burgett, A.W.G. Differing activities of oxysterol-binding protein (OSBP) targeting anti-viral compounds. Antiviral. Res. 2019, 170, 104548. [Google Scholar] [CrossRef] [PubMed]
- Albulescu, L.; Strating, J.R.P.M.; Thibaut, H.J.; Van Der Linden, L.; Shair, M.D.; Neyts, J.; Van Kuppeveld, F.J. Broad-range inhibition of enterovirus replication by OSW-1, a natural compound targeting OSBP. Antivir. Res. 2015, 117, 110–114. [Google Scholar] [CrossRef] [PubMed]
- van der Schaar, H.M.; van der Linden, L.; Lanke, K.H.; Strating, J.R.; Pürstinger, G.; de Vries, E.; de Haan, C.A.; Neyts, J.; van Kuppeveld, F.J. Coxsackievirus mutants that can bypass host factor PI4KIIIβ and the need for high levels of PI4P lipids for replication. Cell Res. 2012, 22, 1576–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arita, M.; Bigay, J. Poliovirus evolution toward independence from the phosphatidylinositol-4 kinase III β/oxysterol-binding protein family I pathway. ACS Infect. Dis. 2019, 5, 962–973. [Google Scholar] [CrossRef]
- Lamarche, M.J.; Borawski, J.; Bose, A.; Capacci-Daniel, C.; Colvin, R.; Dennehy, M.; Ding, J.; Dobler, M.; Drumm, J.; Gaither, L.A.; et al. Anti-hepatitis C virus activity and toxicity of type III phosphatidylinositol-4-kinase beta inhibitors. Antimicrob. Agents Chemother. 2012, 56, 5149–5156. [Google Scholar] [CrossRef] [Green Version]
- Spickler, C.; Lippens, J.; Laberge, M.K.; Desmeules, S.; Bellavance, É.; Garneau, M.; Guo, T.; Hucke, O.; Leyssen, P.; Neyts, J.; et al. Phosphatidylinositol 4-kinase III beta is essential for replication of human rhinovirus and its inhibition causes a lethal phenotype in vivo. Antimicrob. Agents Chemother. 2013, 57, 3358–3368. [Google Scholar] [CrossRef] [Green Version]
- Shim, A.; Song, J.H.; Kwon, B.E.; Lee, J.J.; Ahn, J.H.; Kim, Y.J.; Rhee, K.J.; Chang, S.Y.; Cha, Y.; Lee, Y.S.; et al. Therapeutic and prophylactic activity of itraconazole against human rhinovirus infection in a murine model. Sci. Rep. 2016, 6, 23110. [Google Scholar] [CrossRef] [Green Version]
- Mohamud, Y.; Shi, J.; Poon, T.; Qu, J.; Xue, Y.C.; Deng, H.; Zhang, J.; Luo, H. Enteroviral infection inhibits autophagic flux via disruption of the SNARE complex to enhance viral replication. SSRN Electron. J. 2018, 22, 3292–3303. [Google Scholar] [CrossRef]
- Too, I.H.K.; Yeo, H.; Sessions, O.M.; Yan, B.; Libau, E.A.; Howe, J.L.C.; Lim, Z.Q.; Suku-Maran, S.; Ong, W.-Y.; Chua, K.B.; et al. Enterovirus 71 infection of motor neuron-like NSC-34 cells undergoes a non-lytic exit pathway. Sci. Rep. 2016, 6, 36983. [Google Scholar] [CrossRef]
- Abernathy, E.; Mateo, R.; Majzoub, K.; Van Buuren, N.; Bird, S.W.; Carette, J.E.; Kirkegaard, K. Differential and convergent utilization of autophagy components by positive-strand RNA viruses. PLoS Biol. 2019, 17, e2006926. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.-C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.E.; Rossignol, E.D.; Chang, D.; Zaia, J.; Forrester, I.; Raja, K.; Winbigler, H.; Nicastro, D.; Jackson, W.T.; Bullitt, E. Complexity and ultrastructure of infectious extracellular vesicles from cells infected by non-enveloped virus. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.K.; Saulsbery, H.M.; Velazquez, A.F.C.; Jackson, W.T. Enteroviruses remodel autophagic trafficking through regulation of host SNARE proteins to promote virus replication and cell exit. Cell Rep. 2018, 22, 3304–3314. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.I.; Lin, J.Y.; Chiang, H.C.; Huang, P.-N.; Lin, Q.D.; Shih, S.R. Exosomes facilitate transmission of enterovirus A71 from human intestinal epithelial cells. J. Infect. Dis. 2020, 222, 456–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Wu, J.; Fang, D.; Qiu, Y.; Zou, X.; Jia, X.; Yin, Y.; Shen, L.; Mao, L. Exosomes cloak the virion to transmit Enterovirus 71 nonlytic ally. Virulence 2020, 11, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-Y.; Huang, H.I. Autophagy is induced and supports virus replication in Enterovirus A71-infected human primary neuronal cells. Sci. Rep. 2020, 10, 15234. [Google Scholar] [CrossRef]
- Lee, Y.; Wang, P.-S.; Wang, J.R.; Liu, H.-S. Enterovirus 71-induced autophagy increases viral replication and pathogenesis in a suckling mouse model. J. Biomed. Sci. 2014, 21, 80. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, M.A.W.; Pfeiffer, J.K. Microbiota-immune system interactions and enteric virus infection. Curr. Opin. Virol. 2020, 46, 15–19. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peters, C.E.; Carette, J.E. Return of the Neurotropic Enteroviruses: Co-Opting Cellular Pathways for Infection. Viruses 2021, 13, 166. https://doi.org/10.3390/v13020166
Peters CE, Carette JE. Return of the Neurotropic Enteroviruses: Co-Opting Cellular Pathways for Infection. Viruses. 2021; 13(2):166. https://doi.org/10.3390/v13020166
Chicago/Turabian StylePeters, Christine E., and Jan E. Carette. 2021. "Return of the Neurotropic Enteroviruses: Co-Opting Cellular Pathways for Infection" Viruses 13, no. 2: 166. https://doi.org/10.3390/v13020166
APA StylePeters, C. E., & Carette, J. E. (2021). Return of the Neurotropic Enteroviruses: Co-Opting Cellular Pathways for Infection. Viruses, 13(2), 166. https://doi.org/10.3390/v13020166