
Hepatitis B Virus DNA Integration: In Vitro Models for Investigating Viral Pathogenesis and Persistence



Abstract
1. Introduction
1.1. Natural History of Chronic Hepatitis B
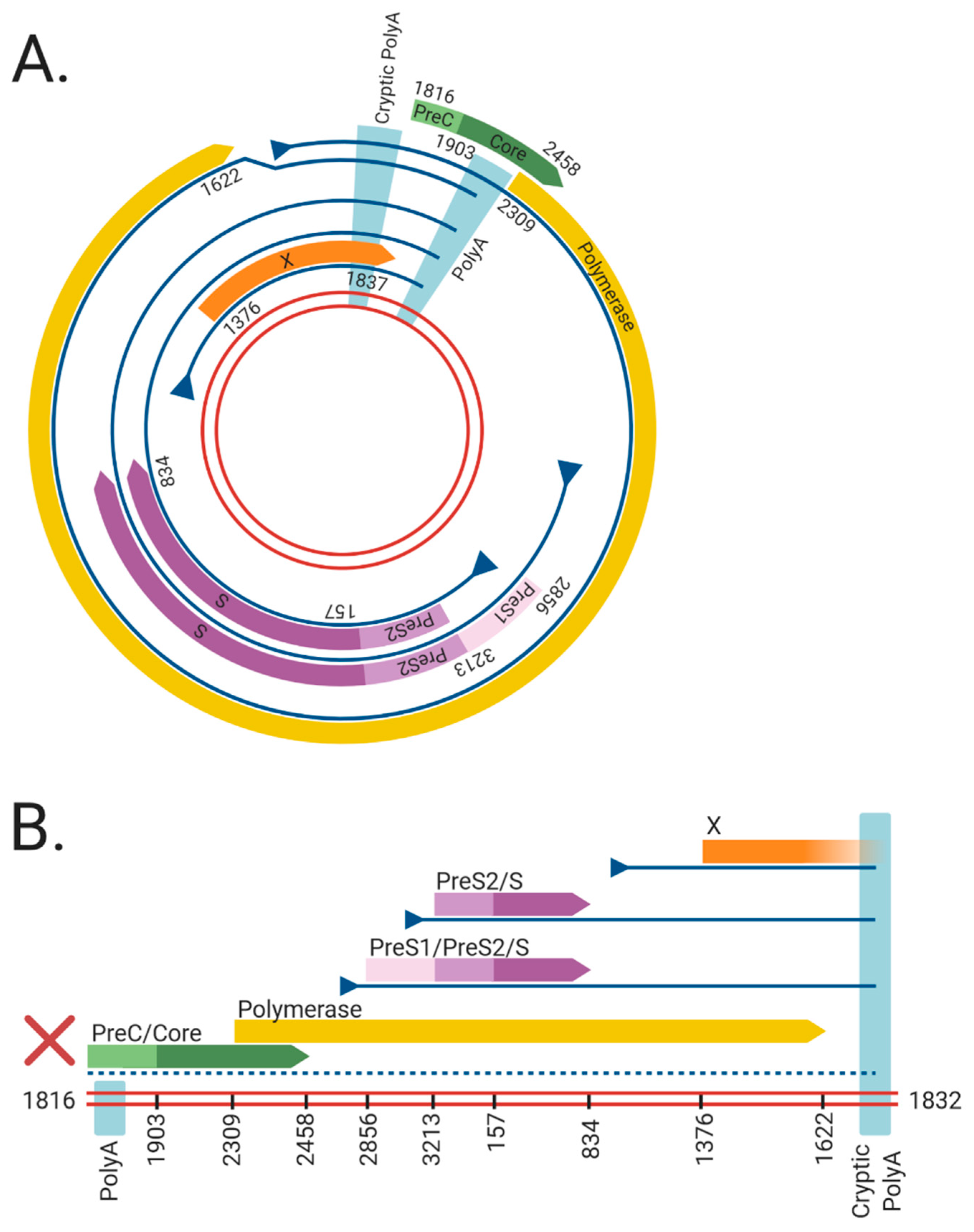
1.2. HBV Structure
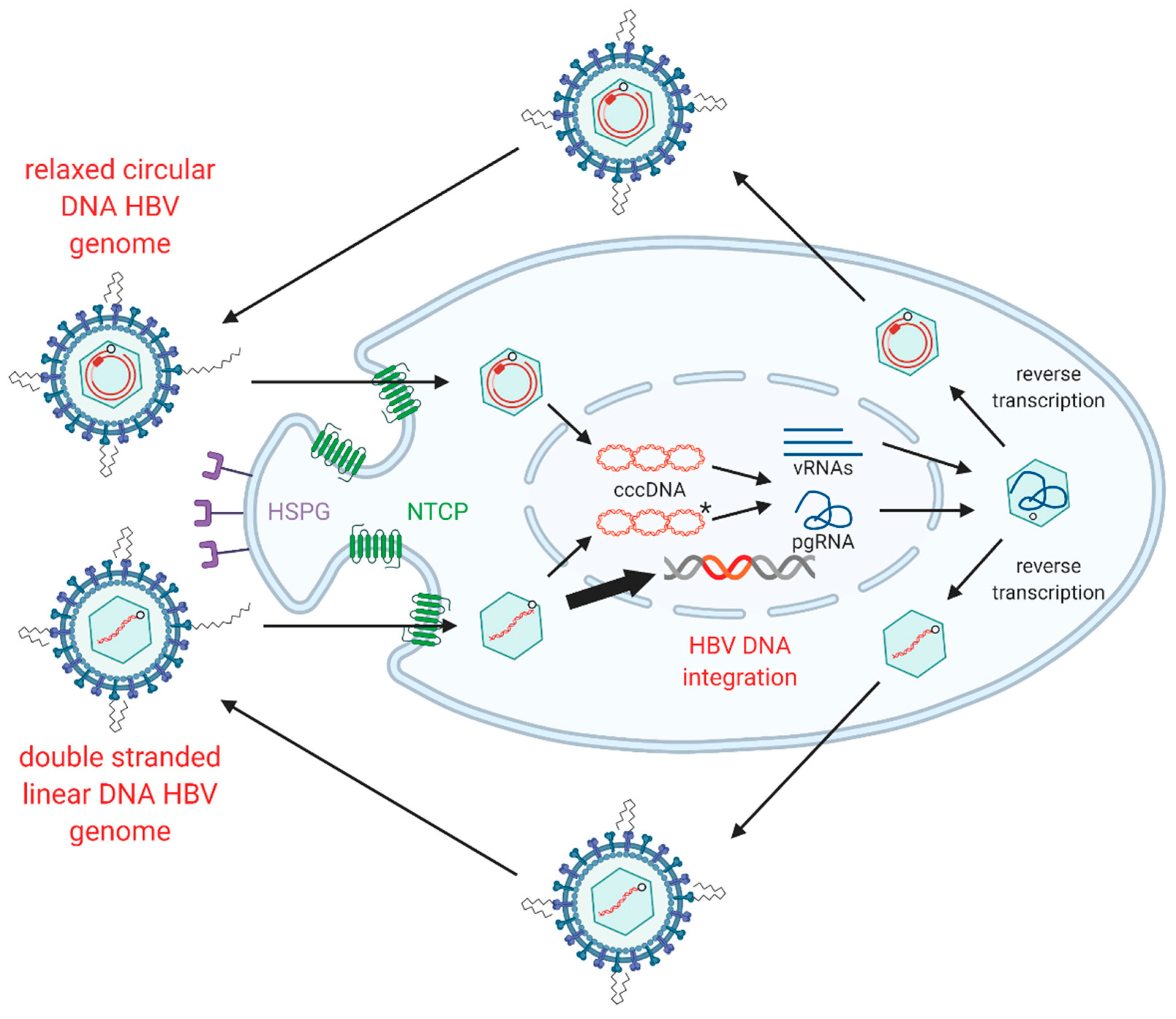
1.3. HBV Replication
1.4. HBV dslDNA and HBV DNA Integration
1.5. In Vitro Models of HBV DNA Integration

{kind=link}
{kind=link}
{kind=link}
| Model System | Type | Infectious HBV Produced? | Forms New HBV DNA Integrations? | Refs |
|---|---|---|---|---|
| PLC/PRF/5 | Tumour-derived cell line 1 | No | No | [44,45] |
| Hep3B | Tumour-derived cell line 1 | No | No | [46,47] |
| HepG2.2.15 | Engineered HBV-producer cell line 2 | Yes | Yes | [52,53] |
| HepAD38 | Engineered HBV-producer cell line 2 | Yes | Unknown | [54] |
| Transfection of HBV over-length constructs | HBV transfection 3 | Yes | Unknown | [55,56,57,58,59] |
| Transfection of HBV monomeric DNA | HBV transfection 3 | Yes | Unknown | [3,60,61] |
| Transfection of HBV virion DNA | HBV transfection 3 | Yes | Yes | [62] |
| Transfection of in vitro transcribed HBV pgRNA | HBV transfection 3 | Yes | Unknown | [63] |
| Huh7-NTCP | HBV infection 4 | Yes | Yes | [48,51] |
| HepG2-NTCP | HBV infection 4 | Yes | Yes | [48,51] |
| HepaRG | HBV infection 4 | Yes | Yes | [48,51] |
| HepaRG-NTCP | HBV infection 4 | Yes | Yes | [48,51] |
| Primary human hepatocytes | HBV infection 4 | Yes | Yes | [48,51] |
2. Molecular Mechanisms of HBV DNA Integration
2.1. HBV DNA Integration Occurs upon de Novo Infection
2.2. Molecular Pathways Involved in Integration
2.3. Sites of Integration in the Host Genome
3. The Role of Integration in HBV-Associated HCC
3.1. Cis-Mediated Mechanisms
3.2. Trans-Mediated Mechanisms
4. Integrated HBV DNA as a Source of HBV Surface Antigen
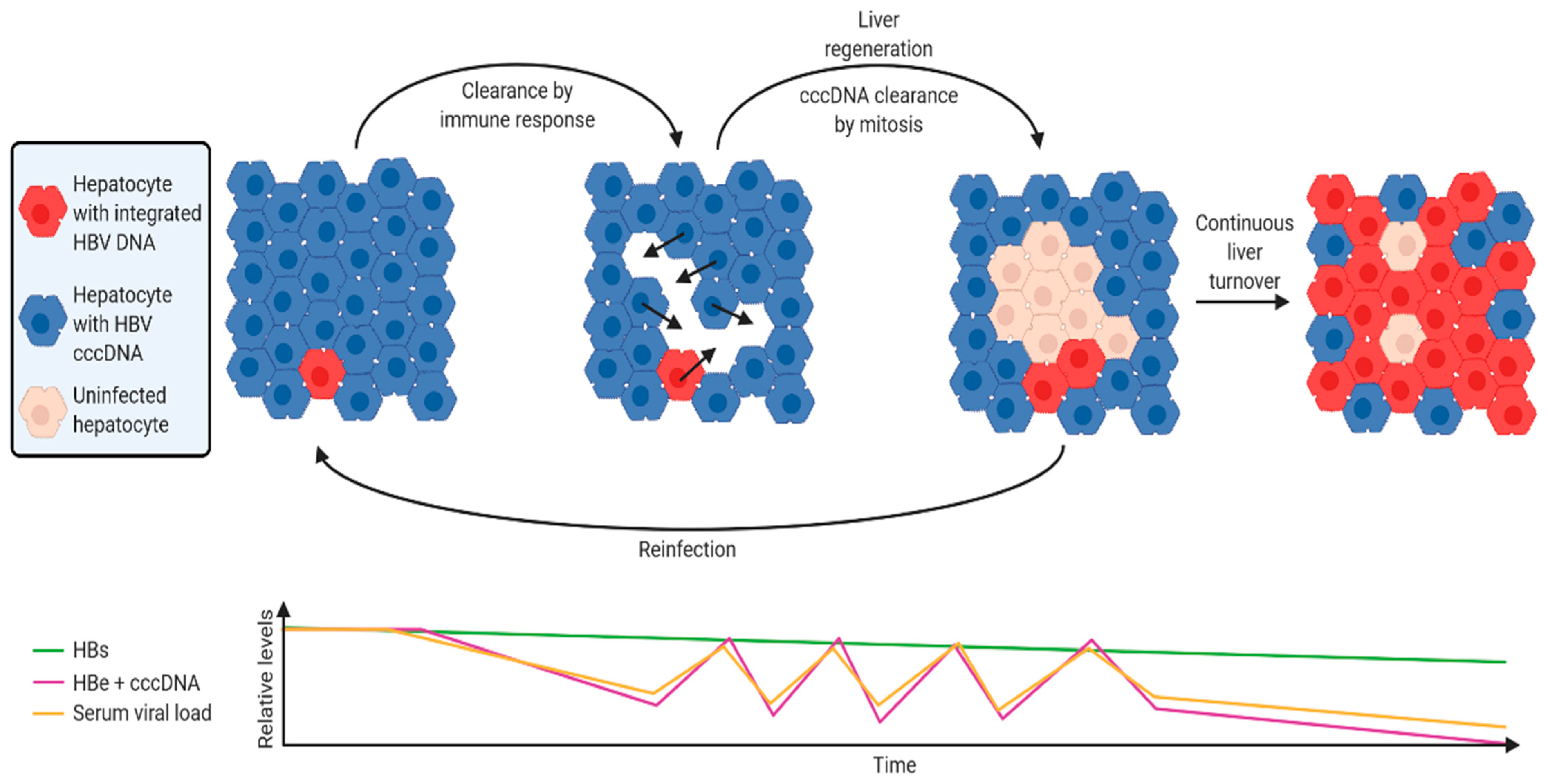
4.1. A Hypothetical Model of HBV DNA Integration Dynamics during Chronic Infection
- The virus infects the whole liver following the initial infection. In this initial phase, the majority of HBsAg is derived from cccDNA, as integrated HBV DNA is rare (1 integration per ~10,000 cells, as shown in in vitro [48,51] and in vivo models). Ongoing nuclear import and integration in chronically-infected cells is rare [48,49,50,51], so integration rate remains relatively stable. New infections (which contribute to new HBV integrations) are suppressed by super-infection exclusion effected through LHBs expression by infected cells (discovered in in vitro models of duck HBV infection [121]).
- Upon activation of the anti-HBV immune response, HBV e antigen and HBV polymerase (which are coded by cccDNA, but not integrated HBV DNA) are the main antigens that are targeted [122].
- In parallel, replicative space increases due to loss of cells expressing HBsAg from cccDNA. cccDNA is lost with mitosis [48,50,123] and daughter cells are susceptible to reinfection (and therefore new integration events). Cells infected by rcDNA-containing virions that form cccDNA are selected against, while cells infected by dslDNA-containing viruses and integrate are not subject to this selection pressure.
- The ratio of cccDNA to integrated HBV DNA frequency therefore decreases. Correspondingly, the ratio of both HBV e antigen and serum HBV DNA to HBsAg decreases (as observed in patient sera [124]).
- A new equilibrium emerges where the level of immune suppression by HBsAg [115] counteracts the immune stimulation by productively-infected hepatocytes, reducing the amount of HBeAg secreted and intrahepatic cccDNA.
- Long-term exposure to the stably high levels of HBsAg can deplete HBV-targeting T-cells [116]. In this way, HBV integration maintains a persistent infection.
4.2. Implications on Therapy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Global Hepatitis Report 2017; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- The Polaris Observatory Collaborators. Global Prevalence, Treatment, and Prevention of Hepatitis B Virus Infection in 2016: A Modelling Study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar] [CrossRef]
- Mutz, P.; Metz, P.; Lempp, F.A.; Bender, S.; Qu, B.; Schöneweis, K.; Seitz, S.; Tu, T.; Restuccia, A.; Frankish, J.; et al. HBV Bypasses the Innate Immune Response and Does Not Protect HCV From Antiviral Activity of Interferon. Gastroenterology 2018, 154, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Block, J.M.; Wang, S.; Cohen, C.; Douglas, M.W. The Lived Experience of Chronic Hepatitis B: A Broader View of Its Impacts and Why We Need a Cure. Viruses 2020, 12, 515. [Google Scholar] [CrossRef] [PubMed]
- Allard, N.; MacLachlan, J.H.; Cowie, B.C. The cascade of care for Australians living with chronic hepatitis B: Measuring access to diagnosis, management and treatment. Aust. N. Z. J. Public Health 2015, 39, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.M.; Osinubi, A.; Nelson, N.P.; Thompson, W.W. The hepatitis B care cascade using administrative claims data, 2016. Am. J. Manag. Care 2020, 26, 331–338. [Google Scholar]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Jilbert, A.R. Conceptual models for the initiation of hepatitis B virus-associated hepatocellular carcinoma. Liver Int. 2015, 35, 1786–1800. [Google Scholar] [CrossRef]
- Tu, T.; Bühler, S.; Bartenschlager, R. Chronic viral hepatitis and its association with liver cancer. Biol. Chem. 2017, 398, 817–837. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Zhao, X.-L.; Yang, J.-R.; Lin, S.-Z.; Ma, H.; Guo, F.; Yang, R.-F.; Zhang, H.-H.; Han, J.-C.; Wei, L.; Pan, X.-B. Serum viral duplex-linear DNA proportion increases with the progression of liver disease in patients infected with HBV. Gut 2015, 65, 502–511. [Google Scholar] [CrossRef]
- Chen, M.S.; Billaud, J.-N.; Sällberg, M.; Guidotti, L.G.; Chisari, F.V.; Jones, J.; Hughes, J.; Milich, D.R. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc. Natl. Acad. Sci. USA 2004, 101, 14913–14918. [Google Scholar] [CrossRef]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Leistner, C.M.; Gruen-Bernhard, S.; Glebe, D. Role of glycosaminoglycans for binding and infection of hepatitis B virus. Cell. Microbiol. 2007, 10, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Sureau, C.; Salisse, J. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus A-determinant. Hepatology 2012, 57, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Fälth, M.; Stindt, J.; Königer, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Herrscher, C.; Pastor, F.; Burlaud-Gaillard, J.; Dumans, A.; Seigneuret, F.; Moreau, A.; Patient, R.; Eymieux, S.; De Rocquigny, H.; Hourioux, C.; et al. Hepatitis B virus entry into HepG2-NTCP cells requires clathrin-mediated endocytosis. Cell. Microbiol. 2020, 22, e13205. [Google Scholar] [CrossRef]
- Umetsu, T.; Inoue, J.; Kogure, T.; Kakazu, E.; Ninomiya, M.; Iwata, T.; Takai, S.; Nakamura, T.; Sano, A.; Shimosegawa, T. Inhibitory effect of silibinin on hepatitis B virus entry. Biochem. Biophys. Rep. 2018, 14, 20–25. [Google Scholar] [CrossRef]
- Rabe, B.; Delaleau, M.; Bischof, A.; Foss, M.; Sominskaya, I.; Pumpens, P.; Cazenave, C.; Castroviejo, M.; Kann, M. Nuclear Entry of Hepatitis B Virus Capsids Involves Disintegration to Protein Dimers followed by Nuclear Reassociation to Capsids. PLoS Pathog. 2009, 5, e1000563. [Google Scholar] [CrossRef]
- Yang, W.; Summers, J. Illegitimate replication of linear hepadnavirus DNA through nonhomologous recombination. J. Virol. 1995, 69, 4029–4036. [Google Scholar] [CrossRef]
- Yang, W.; Mason, W.S.; Summers, J. Covalently closed circular viral DNA formed from two types of linear DNA in woodchuck hepatitis virus-infected liver. J. Virol. 1996, 70, 4567–4575. [Google Scholar] [CrossRef]
- Yang, W.; Summers, J. Infection of ducklings with virus particles containing linear double-stranded duck hepatitis B virus DNA: Illegitimate replication and reversion. J. Virol. 1998, 72, 8710–8717. [Google Scholar] [CrossRef] [PubMed]
- Budzinska, M.A.; Shackel, N.A.; Urban, S.; Tu, T. Sequence analysis of integrated hepatitis B virus DNA during HBeAg-seroconversion. Emerg. Microbes Infect. 2018, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Ploss, A. Core components of DNA lagging strand synthesis machinery are essential for hepatitis B virus cccDNA for-mation. Nat. Microbiol. 2020, 5, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Que, L.; Shimadu, M.; Koura, M.; Ishihara, Y.; Wakae, K.; Nakamura, T.; Watashi, K.; Wakita, T.; Muramatsu, M. Flap endonuclease 1 is involved in cccDNA formation in the hepatitis B virus. PLoS Pathog. 2018, 14, e1007124. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Sheraz, M.; McGrane, M.; Chang, J.; Guo, J.-T. DNA Polymerase alpha is essential for intracellular amplification of hepatitis B virus covalently closed circular DNA. PLoS Pathog. 2019, 15, e1007742. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA Polymerase kappa Is a Key Cellular Factor for the Formation of Covalently Closed Circular DNA of Hepatitis B Virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef]
- Long, Q.; Yan, R.; Hu, J.; Cai, D.; Mitra, B.; Kim, E.S.; Marchetti, A.; Zhang, H.; Wang, S.; Liu, Y.; et al. The role of host DNA ligases in hepadnavirus covalently closed circular DNA formation. PLoS Pathog. 2017, 13, e1006784. [Google Scholar] [CrossRef]
- Sheraz, M.; Cheng, J.; Tang, L.; Chang, J.; Guo, J.-T. Cellular DNA Topoisomerases Are Required for the Synthesis of Hepatitis B Virus Covalently Closed Circular DNA. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Königer, C.; Wingert, I.; Marsmann, M.; Rösler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef]
- Cui, X.; McAllister, R.; Boregowda, R.; Sohn, J.A.; Ledesma, F.C.; Caldecott, K.W.; Seeger, C.; Hu, J. Does Tyrosyl DNA Phosphodiesterase-2 Play a Role in Hepatitis B Virus Genome Repair? PLoS ONE 2015, 10, e0128401. [Google Scholar] [CrossRef]
- Haines, K.M.; Loeb, D.D. The Sequence of the RNA Primer and the DNA Template Influence the Initiation of Plus-strand DNA Synthesis in Hepatitis B Virus. J. Mol. Biol. 2007, 370, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Lewellyn, E.B.; Loeb, D.D. Base Pairing between cis-Acting Sequences Contributes to Template Switching during Plus-Strand DNA Synthesis in Human Hepatitis B Virus. J. Virol. 2007, 81, 6207–6215. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Summers, J. Integration of hepadnavirus DNA in infected liver: Evidence for a linear precursor. J. Virol. 1999, 73, 9710–9717. [Google Scholar] [CrossRef] [PubMed]
- Schütz, T.; Kairat, A.; Schröder, C.H. DNA Sequence Requirements for the Activation of a CATAAA Polyadenylation Signal within the Hepatitis B Virus X Reading Frame: Rapid Detection of Truncated Transcripts. Virology 1996, 223, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Wooddell, C.I.; Lik-Yuen, C.H.; Chan, H.L.-Y.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017, 9, eaan0241. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Ji, L.; Maguire, M.L.; Loeb, D.D. cis-Acting Sequences That Contribute to the Synthesis of Relaxed-Circular DNA of Human Hepatitis B Virus. J. Virol. 2004, 78, 716–723. [Google Scholar] [CrossRef]
- Wang, G.H.; Seeger, C. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 1993, 67, 6507–6512. [Google Scholar] [CrossRef]
- Oropeza, C.E.; McLachlan, A. Complementarity between epsilon and phi sequences in pregenomic RNA influences hepatitis B virus replication efficiency. Virology 2007, 359, 371–381. [Google Scholar] [CrossRef]
- Nassal, M.; Rieger, A. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for dis-continuous first-strand DNA synthesis. J. Virol. 1996, 70, 2764–2773. [Google Scholar] [CrossRef]
- Bill, C.A.; Summers, J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc. Natl. Acad. Sci. USA 2004, 101, 11135–11140. [Google Scholar] [CrossRef]
- Summers, J.; Mason, W.S. Residual integrated viral DNA after hepadnavirus clearance by nucleoside analog therapy. Proc. Natl. Acad. Sci. USA 2003, 101, 638–640. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Liu, C.; Aldrich, C.E.; Litwin, S.; Yeh, M.M. Clonal Expansion of Normal-Appearing Human Hepatocytes during Chronic Hepatitis B Virus Infection. J. Virol. 2010, 84, 8308–8315. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Bey, E.; Whitcutt, J.M.; Gear, J.H. Adaptation of cells derived from human malignant tumours to growth In Vitro. S. Afr. J. Med Sci. 1976, 41, 89–98. [Google Scholar] [PubMed]
- Dejean, A.; Bréchot, C.; Tiollais, P.; Wain-Hobson, S. Characterization of integrated hepatitis B viral DNA cloned from a human hepatoma and the hepatoma-derived cell line PLC/PRF/5. Proc. Natl. Acad. Sci. USA 1983, 80, 2505–2509. [Google Scholar] [CrossRef] [PubMed]
- Aden, D.P.; Fogel, A.; Plotkin, S.; Damjanov, I.; Knowles, B.B. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nat. Cell Biol. 1979, 282, 615–616. [Google Scholar] [CrossRef]
- Knowles, B.B.; Howe, C.C.; Aden, D.P. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science 1980, 209, 497–499. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B virus DNA integration occurs early in the viral life cycle in an In Vitro infection model via NTCP-dependent uptake of enveloped virus particles. J. Virol. 2018. [Google Scholar] [CrossRef]
- Volz, T.; Allweiss, L.; Ben Ḿbarek, M.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; Lütgehetmann, M.; et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef]
- Allweiss, L.; Volz, T.; Giersch, K.; Kah, J.; Raffa, G.; Petersen, J.; Lohse, A.W.; Beninati, C.; Pollicino, T.; Urban, S.; et al. Proliferation of primary human hepatocytes and prevention of hepatitis B virus reinfection efficiently deplete nuclear cccDNA In Vivo. Gut 2018, 67, 542–552. [Google Scholar] [CrossRef]
- Tu, T.; Zehnder, B.; Qu, B.; Urban, S. De novo synthesis of Hepatitis B virus nucleocapsids is dispensable for the maintenance and transcriptional regulation of cccDNA. JHEP Rep. 2020. [Google Scholar] [CrossRef]
- Dandri, M.; Burda, M.R.; Bürkle, A.; Zuckerman, D.M.; Will, H.; Rogler, C.E.; Greten, H.; Petersen, J. Increase in de novo HBV DNA integrations in response to oxidative DNA damage or inhibition of poly (ADP-ribosyl) ation. Hepatology 2002, 35, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Ruan, P.; Dai, X.; Sun, J.; He, C.; Huang, C.; Zhou, R.; Chemin, I. Integration of hepatitis B virus DNA into p21-activated kinase 3 (PAK3) gene in HepG2.2.15 cells. Virus Genes 2020, 56, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Ladner, S.K.; Otto, M.J.; Barker, C.S.; Zaifert, K.; Wang, G.H.; Guo, J.T.; Seeger, C.; King, R.W. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 1997, 41, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Sureau, C.; Romet-Lemonne, J.-L.; Mullins, J.I.; Essex, M. Production of hepatitis B virus by a differentiated human hepatoma cell line after transfection with cloned circular HBV DNA. Cell 1986, 47, 37–47. [Google Scholar] [CrossRef]
- Sureau, C.; Eichberg, J.W.; Hubbard, G.B.; Romet-Lemonne, J.L.; Essex, M. A molecularly cloned hepatitis B virus produced In Vitro is infectious in a chimpanzee. J. Virol. 1988, 62, 3064–3067. [Google Scholar] [CrossRef]
- Sells, M.A.; Zelent, A.Z.; Shvartsman, M.; Acs, G. Replicative intermediates of hepatitis B virus in HepG2 cells that produce infectious virions. J. Virol. 1988, 62, 2836–2844. [Google Scholar] [CrossRef]
- Acs, G.; Sells, M.A.; Purcell, R.H.; Price, P.; Engle, R.; Shapiro, M.; Popper, H. Hepatitis B virus produced by transfected Hep G2 cells causes hepatitis in chimpanzees. Proc. Natl. Acad. Sci. USA 1987, 84, 4641–4644. [Google Scholar] [CrossRef]
- Will, H.; Cattaneo, R.; Koch, H.-G.; Darai, G.; Schaller, H.; Schellekens, H.; Van Eerd, P.M.C.A.; Deinhardt, F. Cloned HBV DNA causes hepatitis in chimpanzees. Nat. Cell Biol. 1982, 299, 740–742. [Google Scholar] [CrossRef]
- Guo, X.; Chen, P.; Hou, X.; Xu, W.; Wang, D.; Wang, T.-Y.; Zhang, L.; Zheng, G.; Gao, Z.-L.; He, C.-Y.; et al. The recombined cccDNA produced using minicircle technology mimicked HBV genome in structure and function closely. Sci. Rep. 2016, 6, 25552. [Google Scholar] [CrossRef]
- Gunther, S.; Sommer, G.; Von Breunig, F.; Iwanska, A.; Kalinina, T.; Sterneck, M.; Will, H. Amplification of full-length hepatitis B virus genomes from samples from patients with low levels of viremia: Fre-quency and functional consequences of PCR-introduced mutations. J. Clin. Microbiol. 1998, 36, 531–538. [Google Scholar] [CrossRef]
- Tu, T.; Zehnder, B.; Levy, M.; Micali, G.; Tran, L.; Dabere, O.; Main, N.; Shackel, N.; Urban, S. Hepatitis B virus (HBV) DNA integration is not driven by viral proteins. Jahrestag. Dtsch. Arb. Stud. Leb. 2019, 57. [Google Scholar] [CrossRef]
- Yu, Y.; Schneider, W.M.; Michailidis, E.; Acevedo, A.; Ni, Y.; Ambrose, C.; Zou, P.; Kabbani, M.; Quirk, C.; Jahan, C.; et al. An RNA-based system to study hepatitis B virus replication and select drug-resistance mutations. bioRxiv 2019. [Google Scholar] [CrossRef]
- Marion, P.L.; Salazar, F.H.; Alexander, J.J.; Robinson, W.S. State of hepatitis B viral DNA in a human hepatoma cell line. J Virol. 1980, 33, 795–806. [Google Scholar] [CrossRef]
- Shafritz, D.A.; Shouval, D.; Sherman, H.I.; Hadziyannis, S.J.; Kew, M.C. Integration of Hepatitis B Virus DNA into the Genome of Liver Cells in Chronic Liver Disease and Hepatocellular Carcinoma. N. Engl. J. Med. 1981, 305, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Hino, O.; Kitagawa, T.; Koike, K.; Kobayashi, M.; Hara, M.; Mori, W.; Nakashima, T.; Hattori, N.; Sugano, H. Detection of hepatitis B virus DNA in hepatocellular carcinomas in Japan. Hepatology 1984, 4, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Fowler, M.J.; Greenfield, C.; Chu, C.M.; Karayiannis, P.; Dunk, A.; Lok, A.S.; Lai, C.L.; Yeoh, E.K.; Monjardino, J.P.; Wankya, B.M. Integration of HBV-DNA may not be a prerequisite for the maintenance of the state of malignant transformation. An analysis of 110 liver biopsies. J. Hepatol. 1986, 2, 218–229. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Harrison, T.J.; Lee, C.-S.; Chen, D.-S.; Zuckerman, A.J. Detection of hepatitis B virus dna in hepatocellular carcinoma: Analysis by hybridization with subgenomic dna fragments. Hepatology 1988, 8, 518–523. [Google Scholar] [CrossRef]
- Esumi, M.; Tanaka, Y.; Tozuka, S.; Shikata, T. Clonal state of human hepatocellular carcinoma and non-tumorous hepatocytes. Cancer Chemother. Pharmacol. 1989, 23, S1–S3. [Google Scholar] [CrossRef]
- Bréchot, C. Pathogenesis of hepatitis B virus—related hepatocellular carcinoma: Old and new paradigms. Gastroenterology 2004, 127, S56–S61. [Google Scholar] [CrossRef]
- Paterlini-Bréchot, P.; Saigo, K.; Murakami, Y.; Chami, M.; Gozuacik, D.; Mugnier, C.; Lagorce, D.; Bréchot, C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 2003, 22, 3911–3916. [Google Scholar] [CrossRef]
- Twist, E.M.; Clark, H.F.; Aden, D.P.; Knowles, B.B.; Plotkin, S.A. Integration pattern of hepatitis B virus DNA sequences in human hepatoma cell lines. J Virol. 1981, 37, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Koshy, R.; Maupas, P.; Muller, R.; Hofschneider, P.H. Detection of Hepatitis B Virus-specific DNA in the Genomes of Human Hepatocellular Carcinoma and Liver Cirrhosis Tissues. J. Gen. Virol. 1981, 57, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Edman, J.C.; Gray, P.; Valenzuela, P.; Rall, L.B.; Rutter, W.J. Integration of hepatitis B virus sequences and their expression in a human hepatoma cell. Nat. Cell Biol. 1980, 286, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Brechot, C.; Pourcel, C.; Louise, A.; Rain, B.; Tiollais, P. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nat. Cell Biol. 1980, 286, 533–535. [Google Scholar] [CrossRef]
- Budzinska, M.A.; Shackel, N.A.; Urban, S.; Tu, T. Cellular Genomic Sites of Hepatitis B Virus DNA Integration. Genes 2018, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Mason, W.S.; Clouston, A.D.; Shackel, N.A.; McCaughan, G.W.; Yeh, M.M.; Schiff, E.R.; Ruszkiewicz, A.R.; Chen, J.W.; Harley, H.A.J.; et al. Clonal expansion of hepatocytes with a selective advantage occurs during all stages of chronic hepatitis B virus infection. J. Viral Hepat. 2015, 22, 737–753. [Google Scholar] [CrossRef]
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.L.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA Integration and Clonal Hepatocyte Expansion in Chronic Hepatitis B Patients Considered Immune Tolerant. Gastroenterology 2016, 151, 986–998.e4. [Google Scholar] [CrossRef]
- Kimbi, G.C.; Kramvis, A.; Kew, M.C. Integration of hepatitis B virus DNA into chromosomal DNA during acute hepatitis B. World J. Gastroenterol. 2005, 11, 6416–6421. [Google Scholar] [CrossRef]
- Scotto, J.; Hadchouel, M.; Hery, C.; Alvarez, F.; Yvart, J.; Tiollais, P.; Bernard, O.; Brechot, C. Hepatitis B virus DNA in children’s liver diseases: Detection by blot hybridisation in liver and serum. Gut 1983, 24, 618–624. [Google Scholar] [CrossRef]
- Yaginuma, K.; Kobayashi, H.; Kobayashi, M.; Morishima, T.; Matsuyama, K.; Koike, K. Multiple integration site of hepatitis B virus DNA in hepatocellular carcinoma and chronic active hepatitis tissues from children. J. Virol. 1987, 61, 1808–1813. [Google Scholar] [CrossRef]
- Summers, J.; Jilbert, A.R.; Yang, W.; Aldrich, C.E.; Saputelli, J.; Litwin, S.; Toll, E.; Mason, W.S. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc. Natl. Acad. Sci. USA 2003, 100, 11652–11659. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, R.; Churchill, N.D.; Mulrooney-Cousins, P.M.; Michalak, T.I. Initial sites of hepadnavirus integration into host genome in human hepatocytes and in the woodchuck model of hepatitis B-associated hepatocellular carcinoma. Oncogenesis 2017, 6, e317. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-H.; Liu, X.; Yan, H.-X.; Li, W.-Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.-P.; Zhuang, X.-H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.-C.; Sun, T.; Ching, A.K.; He, M.; Li, J.-W.; Wong, A.M.; Na Co, N.; Chan, A.W.; Li, P.-S.; Lung, R.W.; et al. Viral-Human Chimeric Transcript Predisposes Risk to Liver Cancer Development and Progression. Cancer Cell 2014, 25, 335–349. [Google Scholar] [CrossRef]
- Yoo, S.; Wang, W.; Wang, Q.; Fiel, M.I.; Lee, E.; Hiotis, S.P.; Zhu, J. A pilot systematic genomic comparison of recurrence risks of hepatitis B virus-associated hepatocellular carcinoma with low- and high-degree liver fibrosis. BMC Med. 2017, 15, 214. [Google Scholar] [CrossRef] [PubMed]
- Bréchot, C.; Gozuacik, D.; Murakami, Y.; Paterlini-Bréchot, P. Molecular bases for the development of hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC). Semin. Cancer Biol. 2000, 10, 211–231. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef]
- Sung, W.-K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mu-tations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- Yang, X.; Wu, L.; Lin, J.; Wang, A.; Wan, X.; Wu, Y.; Robson, S.C.; Sang, X.; Zhao, H. Distinct hepatitis B virus integration patterns in hepatocellular carcinoma and adjacent normal liver tissue. Int. J. Cancer 2017, 140, 1324–1330. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Mai, G.; Villiger, P.; Oberholzer, J.; Salmon, P.; Morel, P.; Buhler, L.; Trono, D. Treatment of acetaminophen-induced acute liver failure in the mouse with conditionally immortalized human hepatocytes. J. Hepatol. 2005, 43, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Totsugawa, T.; Yong, C.; Rivas-Carrillo, J.D.; Soto-Gutierrez, A.; Navarro-Alvarez, N.; Noguchi, H.; Okitsu, T.; Westerman, K.A.; Kohara, M.; Reth, M.; et al. Survival of liver failure pigs by transplantation of reversibly immortalized human hepatocytes with Tamoxi-fen-mediated self-recombination. J. Hepatol. 2007, 47, 74–82. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsuruga, Y.; Kiyono, T.; Matsushita, M.; Takahashi, T.; Kasai, H.; Todo, S.; Matsumoto, S. Establishment of immortalized human hepatocytes by introduction of HPV16 E6/E7 and hTERT as cell sources for liver cell-based therapy. Cell Transplant. 2008, 17, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Miyazaki, J.-I.; Hino, O.; Tomita, N.; Chisaka, O.; Matsubara, K.; Yamamura, K. Expression and replication of hepatitis B virus genome in transgenic mice. Proc. Natl. Acad. Sci. USA 1989, 86, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Matzke, B.; Schaller, H.; Chisari, F.V. High-level hepatitis B virus replication in transgenic mice. J. Virol. 1995, 69, 6158–6169. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.A.; Lee, H.W.; Kim, I.H.; Park, S.Y.; Sinn, D.H.; Yu, J.H.; Seo, Y.S.; Um, S.H.; Lee, J.I.; Lee, K.S.; et al. Extremely low risk of hepatocellular carcinoma development in patients with chronic hepatitis B in immune-tolerant phase. Aliment. Pharmacol. Ther. 2020, 52, 196–204. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, W.-L.; Louie, S.G.; Yen, T.S.B.; Ou, J.-H.J. Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice. Hepatol. 2006, 45, 16–21. [Google Scholar] [CrossRef]
- Slagle, B.L.; Andrisani, O.M.; Bouchard, M.J.; Lee, C.G.L.; Ou, J.-H.J.; Siddiqui, A. Technical standards for hepatitis B virus X protein (HBx) research. Hepatology 2015, 61, 1416–1424. [Google Scholar] [CrossRef]
- Chisari, F.V.; Klopchin, K.; Moriyama, T.; Pasquinelli, C.; Dunsford, H.A.; Sell, S.; Pinkert, C.A.; Brinster, R.L.; Palmiter, R.D. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell 1989, 59, 1145–1156. [Google Scholar] [CrossRef]
- Chisari, F.V.; Pinkert, C.A.; Milich, D.R.; Filippi, P.; McLachlan, A.; Palmiter, R.D.; Brinster, R.L. A transgenic mouse model of the chronic hepatitis B surface antigen carrier state. Science 1985, 230, 1157–1160. [Google Scholar] [CrossRef]
- Babinet, C.; Farza, H.; Morello, D.; Hadchouel, M.; Pourcel, C.; Mortlock, R.A.; Froelich, P.N. Specific expression of hepatitis B surface antigen (HBsAg) in transgenic mice. Science 1985, 230, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Decorsiere, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Riviere, L.; Quioc-Salomon, B.; Fallot, G.; Halgand, B.; Féray, C.; Buendia, M.-A.; Neuveut, C. Hepatitis B virus replicating in hepatocellular carcinoma encodes HBx variants with preserved ability to antagonize restriction by Smc5/6. Antiviral Res. 2019, 172, 104618. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Freytag von Loringhoven, A.; Kahmann, R.; Hofschneider, P.H.; Koshy, R. The genetic organization of integrated hepatitis B virus DNA in the human hepatoma cell line PLC/PRF/5. Nucleic Acids Res. 1984, 12, 6871–6886. [Google Scholar] [CrossRef]
- Liu, W.-C.; Wu, I.-C.; Lee, Y.-C.; Lin, C.-P.; Cheng, J.-H.; Lin, Y.-J.; Yen, C.-J.; Cheng, P.-N.; Li, P.-F.; Cheng, Y.-T.; et al. Hepatocellular carcinoma-associated single-nucleotide variants and deletions identified by the use of genome-wide high-throughput analysis of hepatitis B virus. J. Pathol. 2017, 243, 176–192. [Google Scholar] [CrossRef]
- Yen, T.T.-C.; Yang, A.; Chiu, W.-T.; Li, T.-N.; Wang, L.-H.; Wu, Y.-H.; Wang, H.-C.; Chen, L.; Wang, W.-C.; Huang, W.; et al. Hepatitis B virus PreS2-mutant large surface antigen activates store-operated calcium entry and promotes chromosome instability. Oncotarget 2016, 7, 23346–23360. [Google Scholar] [CrossRef]
- Hsieh, Y.-H.; Chang, Y.-Y.; Su, I.-J.; Yen, C.-J.; Liu, Y.-R.; Liu, R.-J.; Hsieh, W.-C.; Tsai, H.-W.; Wang, L.H.-C.; Huang, W. Hepatitis B virus pre-S2 mutant large surface protein inhibits DNA double-strand break repair and leads to genome instability in hepatocarcinogenesis. J. Pathol. 2015, 236, 337–347. [Google Scholar] [CrossRef]
- Lai, M.-W.; Liang, K.-H.; Lin, W.-R.; Huang, Y.-H.; Huang, S.-F.; Chen, T.-C.; Yeh, C.-T. Hepatocarcinogenesis in transgenic mice carrying hepatitis B virus pre-S/S gene with the sW172* mutation. Oncogenesis 2016, 5, e273. [Google Scholar] [CrossRef][Green Version]
- Yen, C.-S.; Ai, Y.-L.; Tsai, H.-W.; Chan, S.-H.; Cheng, K.-H.; Lee, Y.-P.; Kao, C.-W.; Wang, Y.-C.; Chen, Y.-L.; Lin, C.-H.; et al. Hepatitis B virus surface gene pre-S2mutant as a high-risk serum marker for hepatoma recurrence after curative hepatic resection. Hepatology 2018, 68, 815–826. [Google Scholar] [CrossRef]
- Tsai, H.-W.; Lin, Y.-J.; Wu, H.-C.; Chang, T.-T.; Wu, I.-C.; Cheng, P.-N.; Yen, C.-J.; Chan, S.-H.; Huang, W.; Su, I.-J. Resistance of ground glass hepatocytes to oral antivirals in chronic hepatitis B patients and implication for the development of hepatocellular carcinoma. Oncotarget 2016, 7, 27724–27734. [Google Scholar] [CrossRef]
- Su, I.-J.; Wang, H.-C.; Wu, H.-C.; Huang, W.-Y. Ground glass hepatocytes contain pre-S mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J. Gastroenterol. Hepatol. 2008, 23, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Lu, C.; Chen, W.; Yao, W.; Wang, H.; Chang, T.; Lei, H.; Shiau, A.; Su, I. Prevalence and significance of hepatitis B virus (HBV) pre-S mutants in serum and liver at different replicative stages of chronic HBV infection. Hepatology 2001, 33, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Ghosh, S.; Shimakawa, Y.; Ramou, N.; Garcia, P.S.; Dubois, A.; Guillot, C.; Kakwata-Nkor Deluce, N.; Tilloy, V.; Durand, G.; et al. Hepatitis B virus preS2Delta38-55 variants: A newly identified risk factor for hepatocellular carcinoma. JHEP Rep. 2020, 2, 100144. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhai, N.; Wang, Z.; Song, H.; Yang, Y.; Cui, A.; Li, T.; Wang, G.; Niu, J.; Crispe, I.N.; et al. Regulatory NK cells mediated between immunosuppressive monocytes and dysfunctional T cells in chronic HBV infection. Gut 2018, 67, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Le Bert, N.; Gill, U.S.; Hong, M.; Kunasegaran, K.; Tan, D.Z.; Ahmad, R.; Cheng, Y.; Dutertre, C.-A.; Heinecke, A.; Rivino, L.; et al. Effects of Hepatitis B Surface Antigen on Virus-Specific and Global T Cells in Patients with Chronic Hepatitis B Virus infection. Gastroenterology 2020, 159, 652–664. [Google Scholar] [CrossRef]
- Michler, T.; Kosinska, A.D.; Festag, J.; Bunse, T.; Su, J.; Ringelhan, M.; Imhof, H.; Grimm, D.; Steiger, K.; Mogler, C.; et al. Knockdown of Virus Antigen Expression Increases Therapeutic Vaccine Efficacy in High-Titer Hepatitis B Virus Carrier Mice. Gastroenterology 2020, 158, 1762–1775.e9. [Google Scholar] [CrossRef]
- Freitas, N.; Cunha, C.; Menne, S.; Gudima, S.O. Envelope Proteins Derived from Naturally Integrated Hepatitis B Virus DNA Support Assembly and Release of Infectious Hepatitis Delta Virus Particles. J. Virol. 2014, 88, 5742–5754. [Google Scholar] [CrossRef]
- Freitas, N.; Lukash, T.; Gunewardena, S.; Chappell, B.; Slagle, B.L.; Gudima, S.O. Relative Abundance of Integrant-Derived Viral RNAs in Infected Tissues Harvested from Chronic Hepatitis B Virus Carriers. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Goyal, A.; Liao, L.E.; Perelson, A.S. Within-host mathematical models of hepatitis B virus infection past, present, and future. Curr. Opin. Syst. Biol. 2019, 18, 27–35. [Google Scholar] [CrossRef]
- Walters, K.-A.; Joyce, M.A.; Addison, W.R.; Fischer, K.P.; Tyrrell, D.L.J. Superinfection Exclusion in Duck Hepatitis B Virus Infection Is Mediated by the Large Surface Antigen. J. Virol. 2004, 78, 7925–7937. [Google Scholar] [CrossRef]
- Chua, C.G.; Mehrotra, A.; Mazzulli, T.; Wong, D.K.; Feld, J.J.; Janssen, H.L.A.; Gehring, A.J. Optimized ex vivo stimulation identifies multi-functional HBV-specific T cells in a majority of chronic hepatitis B patients. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lutgehetmann, M.; Volz, T.; Köpke, A.; Broja, T.; Tigges, E.; Lohse, A.W.; Fuchs, E.; Murray, J.M.; Petersen, J.; Dandri, M. In Vivo proliferation of hepadnavirus-infected hepatocytes induces loss of covalently closed circular DNA in mice. Hepatology 2010, 52, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.B.; Malmström, S.; Hannoun, C.; Norkrans, G.; Lindh, M. Mechanisms downstream of reverse transcription reduce serum levels of HBV DNA but not of HBsAg in chronic hepatitis B virus infection. Virol. J. 2015, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, T.; Zhang, H.; Urban, S. Hepatitis B Virus DNA Integration: In Vitro Models for Investigating Viral Pathogenesis and Persistence. Viruses 2021, 13, 180. https://doi.org/10.3390/v13020180
Tu T, Zhang H, Urban S. Hepatitis B Virus DNA Integration: In Vitro Models for Investigating Viral Pathogenesis and Persistence. Viruses. 2021; 13(2):180. https://doi.org/10.3390/v13020180
Chicago/Turabian StyleTu, Thomas, Henrik Zhang, and Stephan Urban. 2021. "Hepatitis B Virus DNA Integration: In Vitro Models for Investigating Viral Pathogenesis and Persistence" Viruses 13, no. 2: 180. https://doi.org/10.3390/v13020180
APA StyleTu, T., Zhang, H., & Urban, S. (2021). Hepatitis B Virus DNA Integration: In Vitro Models for Investigating Viral Pathogenesis and Persistence. Viruses, 13(2), 180. https://doi.org/10.3390/v13020180