
Phylodynamics and Codon Usage Pattern Analysis of Broad Bean Wilt Virus 2


Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Isolates
2.2. Recombination Analysis
2.3. Phylogenetic and Evolution Dynamic Analysis
2.4. Nucleotide Composition Analysis
2.5. Effective Number of Codon (ENC) Analysis
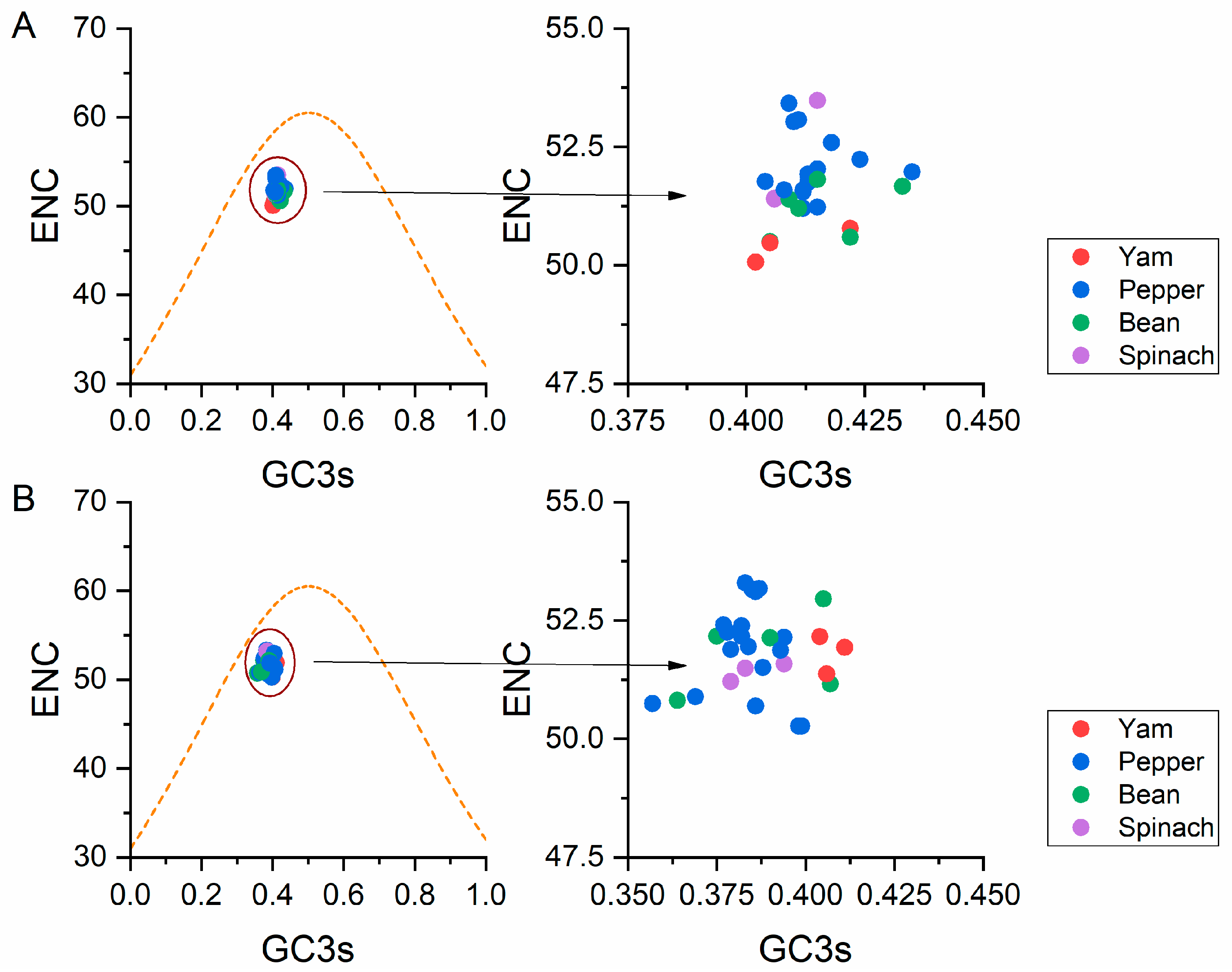
2.6. ENC-Plot Analysis
2.7. Relative Synonymous Codon Usage (RSCU) Analysis
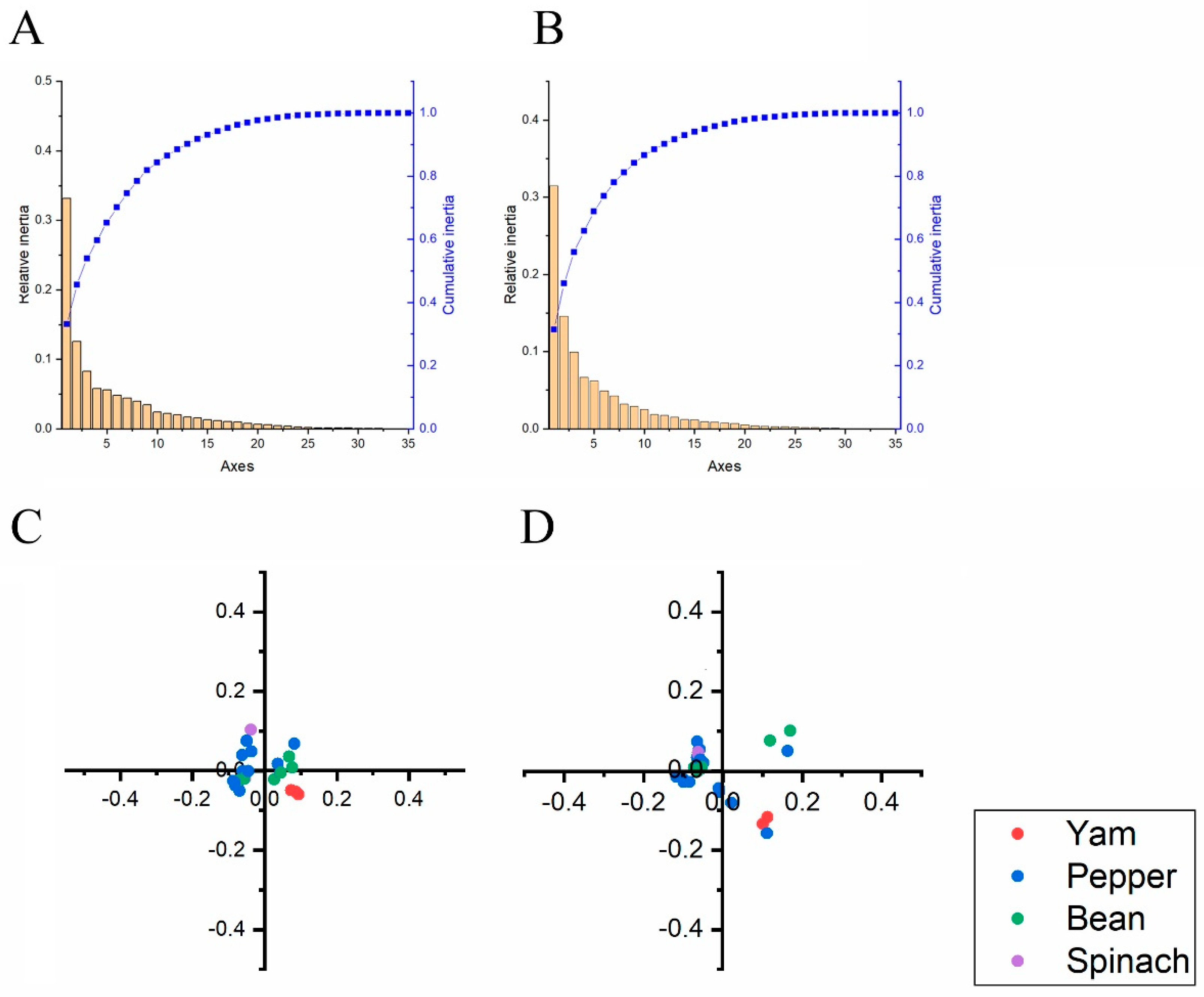
2.8. Principal Component (PCA) Analysis
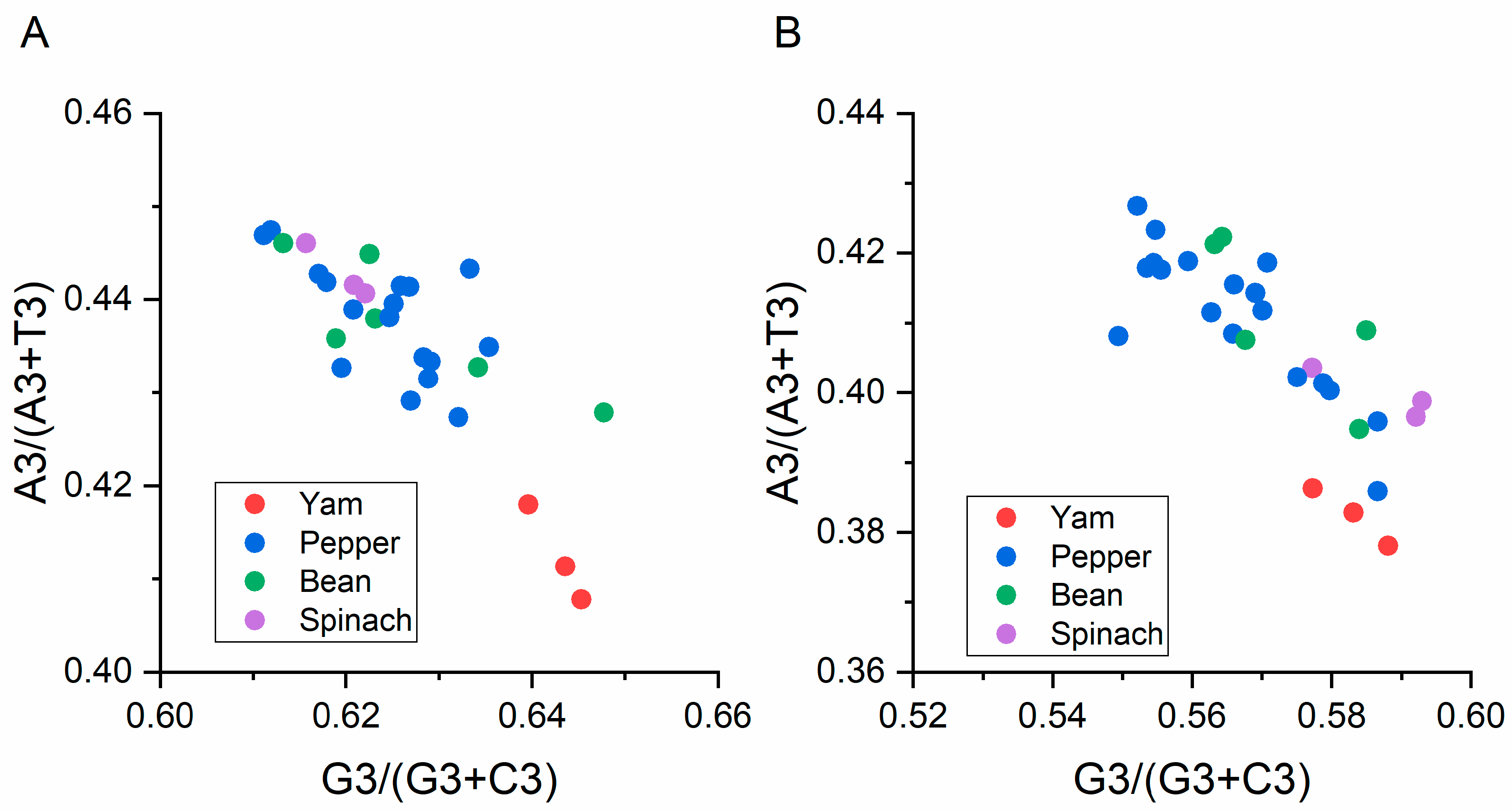
2.9. Parity Rule 2 Analysis (PR2)
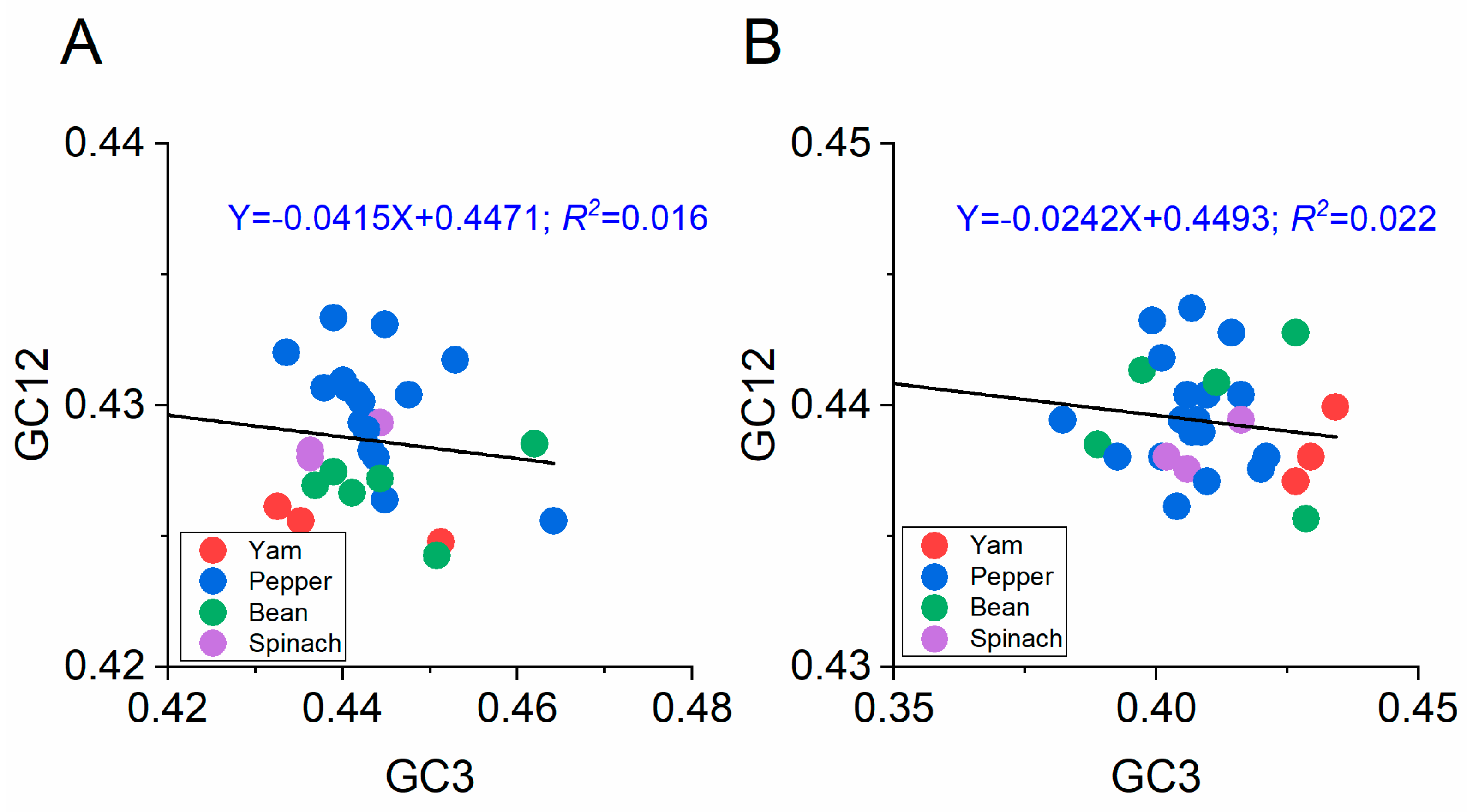
2.10. Neutrality Analysis
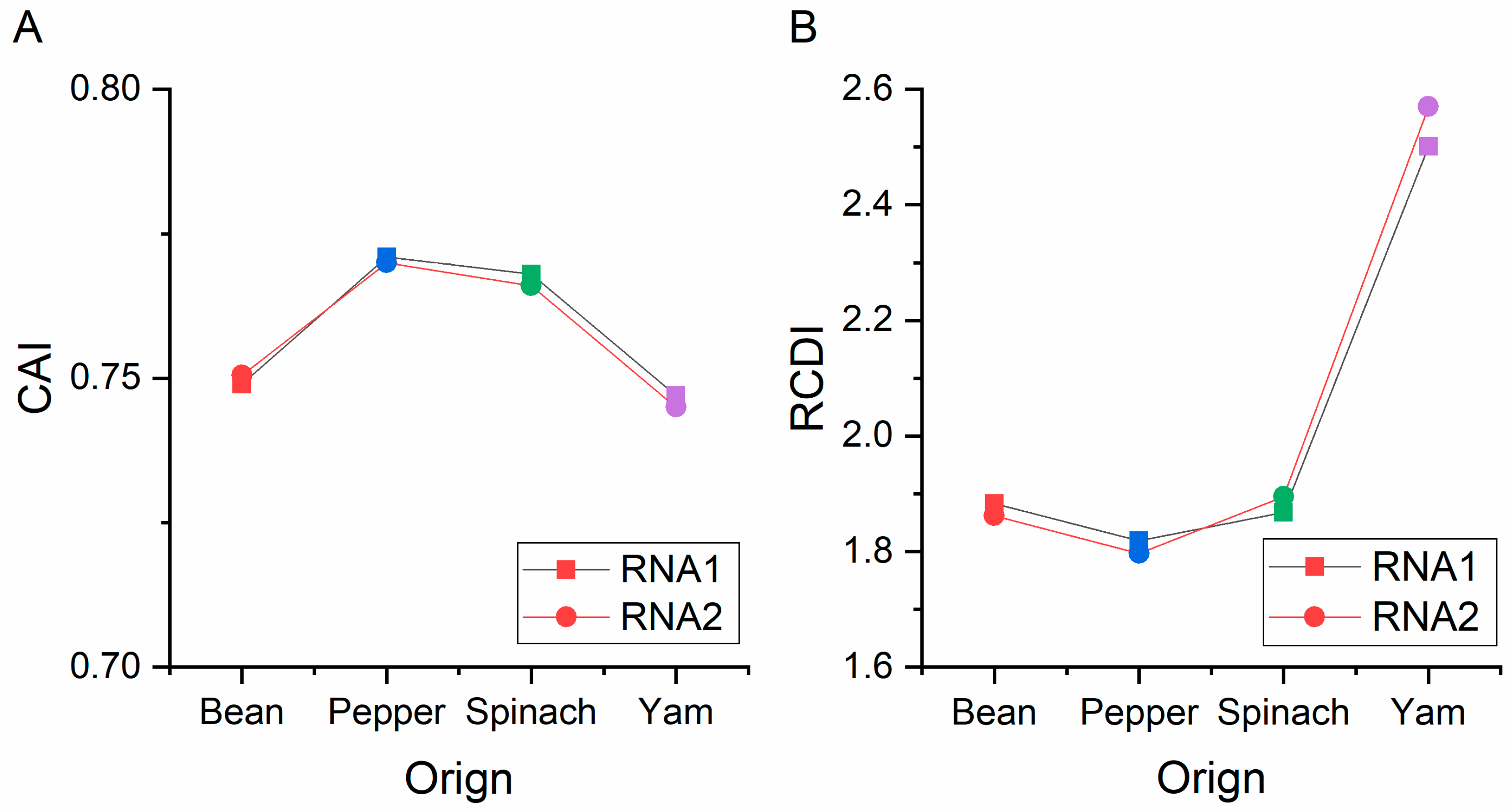
2.11. Codon Adaptation Index (CAI) Analysis
2.12. Relative Codon Deoptimization Index (RCDI) Analysis
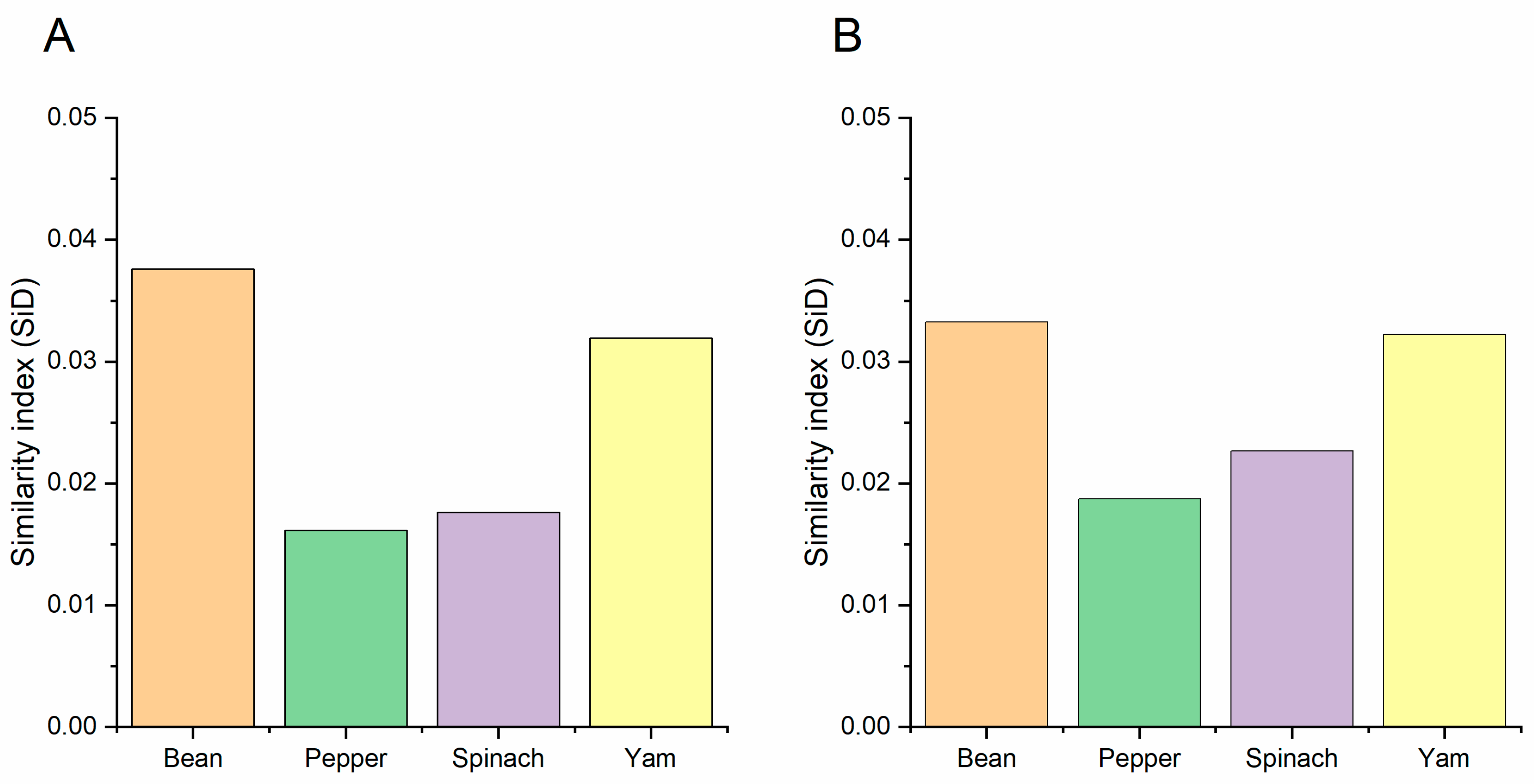
2.13. Similarity Index (SiD) Analysis
2.14. Gravy and Aroma Statistics
2.15. Statistical Analysis
3. Results
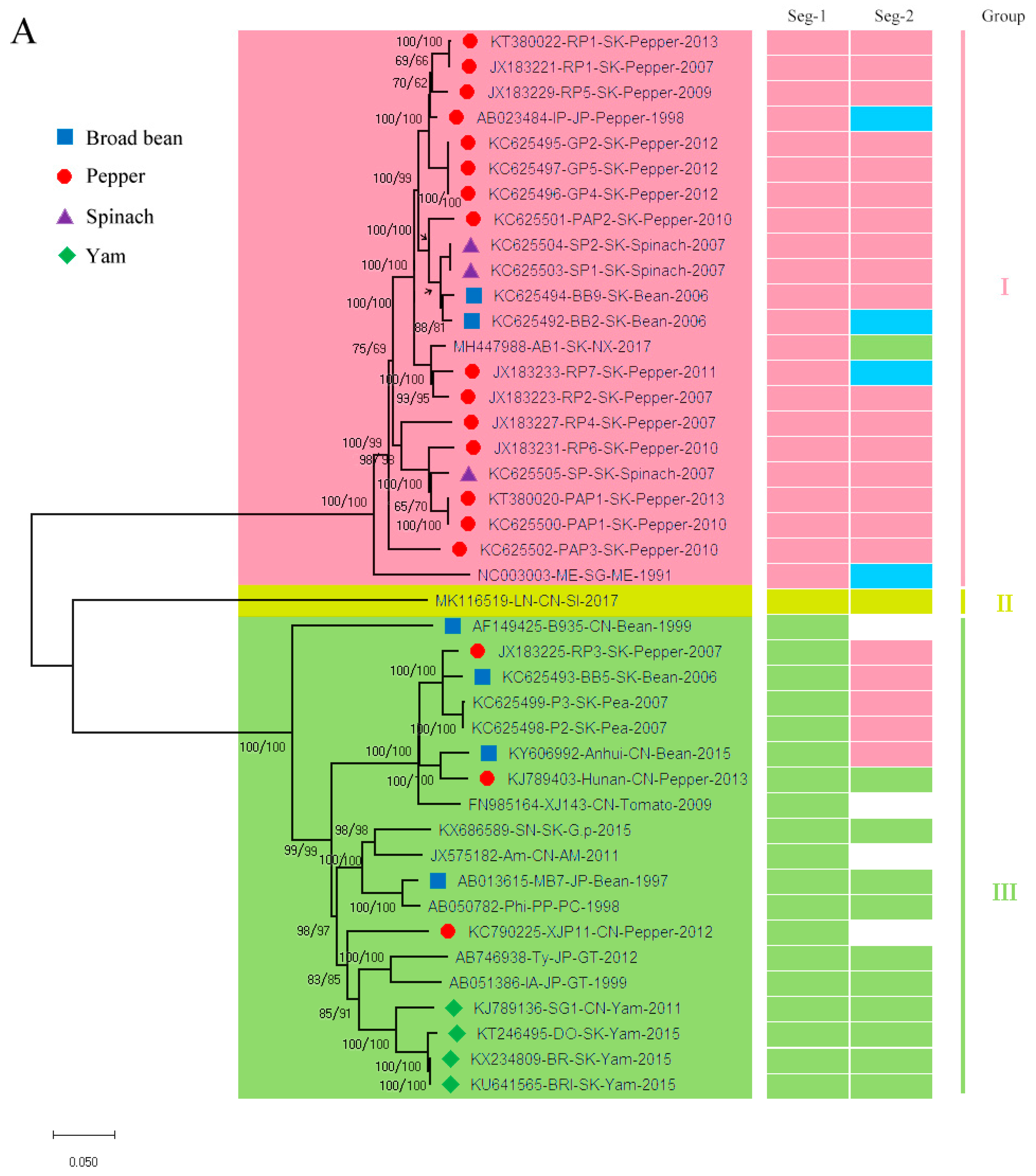
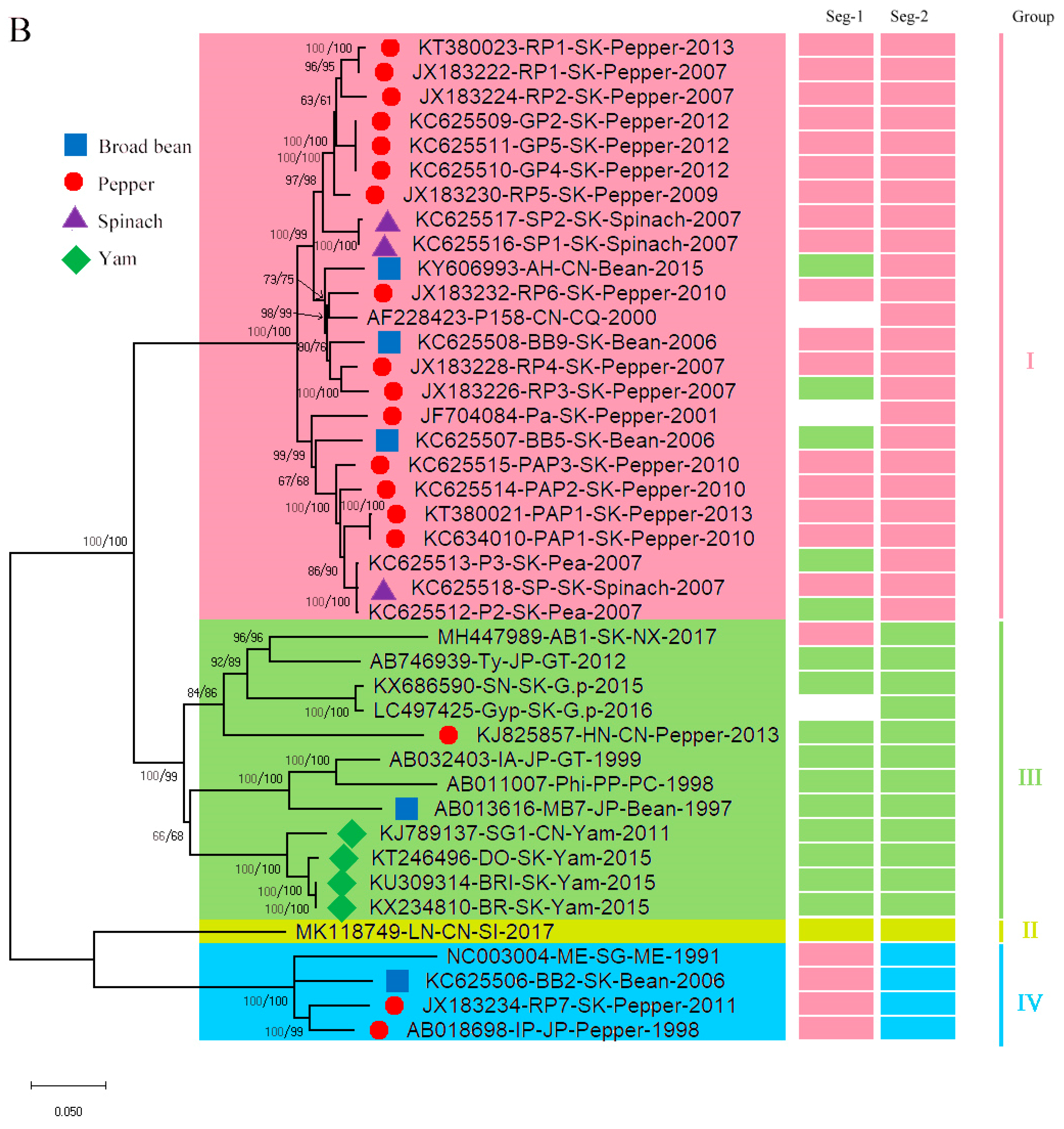
3.1. Recombination and Phylogenetic Analysis
3.2. Reassortment Analysis
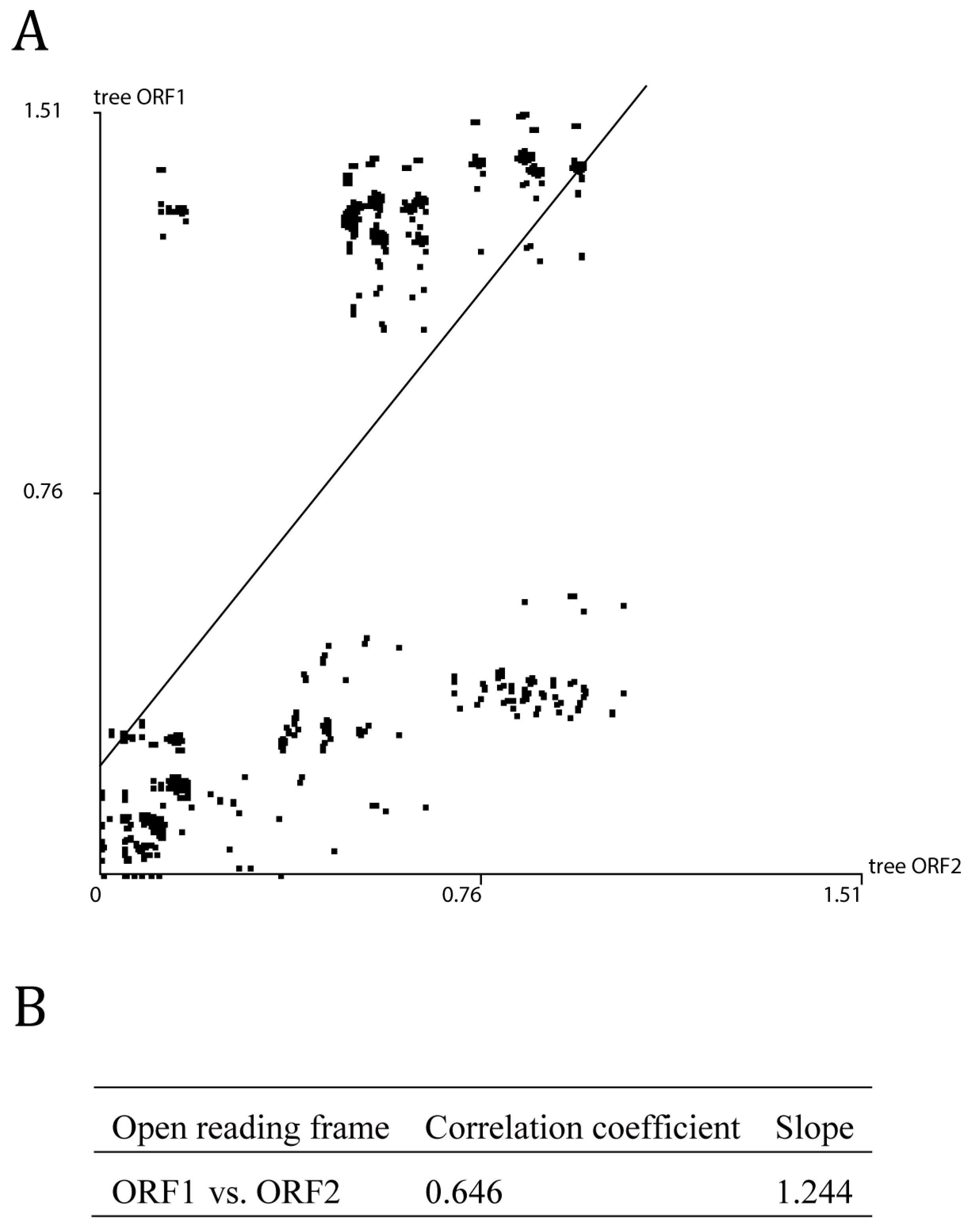
3.3. Evolutionary Dynamic Analysis
3.4. Nucleotide Bias Analysis
3.5. Codon Usage Bias of BBWV-2
3.5.1. Trends in Codon Usage Variations
3.5.2. ENC-Plot Analysis
3.5.3. Neutrality Plot
3.5.4. Parity Analysis
3.6. Codon Usage Adaptation in BBWV-2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castrovilli, S.; Savino, V.; Castellano, M.A.; Engelbrecht, D.J. Characterization of a grapevine isolate of Broad bean wilt virus. Phytopathol. Mediterr. 1985, 24, 35–40. [Google Scholar]
- Ferrer, R.M.; Ferriol, I.; Moreno, P.; Guerri, J.; Rubio, L. Genetic variation and evolutionary analysis of Broad bean wilt virus 2. Arch. Virol. 2011, 156, 1445–1450. [Google Scholar] [CrossRef]
- Fortass, M.; Bos, L. Survey of faba bean (Vicia faba L.) for viruses in Morocco. Neth. J. Plant. Pathol. 1991, 97, 369–380. [Google Scholar] [CrossRef]
- Kondo, T.; Fuji, S.; Yamashita, K.; Kang, D.K.; Chang, M.U. Broad bean wilt virus 2 in yams. J. Gen. Plant. Pathol. 2005, 71, 441–443. [Google Scholar] [CrossRef]
- Sui, C.; Wei, J.H.; Zhan, Q.Q.; Zhang, J. First report of Broad bean wilt virus 2 infecting Bupleurum chinense in China. Plant Dis. 2009, 93, 844. [Google Scholar] [CrossRef]
- Fuji, S.; Mochizuki, N.; Fujinaga, M.; Ikeda, M.; Shinoda, K.; Uematsu, S.; Furuya, H.; Naito, H.; Fukumoto, F. Incidence of viruses in Alstroemeria plants cultivated in Japan and characterization of Broad bean wilt virus 2, Cucumber mosaic virus and Youcai mosaic virus. J. Gen. Plant Pathol. 2007, 73, 216–221. [Google Scholar] [CrossRef]
- Hasegawa, M.; Yasunaga, T.; Miyata, T. Secondary structure of MS2 phage RNA and bias in code word usage. Nucleic Acids Res. 1979, 7, 2073–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Tuohy, T.M.F.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [PubMed]
- Comeron, J.M.; Aguadé, M. An evaluation of measures of synonymous codon usage bias. J. Mol. Evol. 1998, 47, 268–274. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Cowe, E. Synonymous codon usage in Saccharomyces cerevisiae. Yeast 1991, 7, 657–678. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.R.; Papamichail, D.; Skiena, S.; Futcher, B.; Wimmer, E.; Mueller, S. Virus attenuation by genome-scale changes in codon pair bias. Science 2008, 320, 1784–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duret, L.; Mouchiroud, D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc. Natl. Acad. Sci. USA 1999, 96, 4482–4487. [Google Scholar] [CrossRef] [Green Version]
- Fuglsang, A. Accounting for background nucleotide composition when measuring codon usage bias: Brilliant Idea, difficult in practice. Mol. Biol. Evol. 2006, 23, 1345–1347. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Sueoka, N. Translation-coupled violation of Parity Rule 2 in human genes is not the cause of heterogeneity of the DNA G+C content of third codon position. Gene 1999, 238, 53–58. [Google Scholar] [CrossRef]
- Xu, X.; Liu, Q.; Fan, L.; Cui, X.; Zhou, X. Analysis of synonymous codon usage and evolution of begomoviruses. J. Zhejiang Univ. Sci. B. 2008, 9, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Biswas, K.; Palchoudhury, S.; Chakraborty, P.; Bhattacharyya, U.; Ghosh, D.; Debnath, P.; Ramadugu, C.; Keremane, M.; Khetarpal, R.; Lee, R. Codon usage bias analysis of Citrus tristeza virus: Higher codon adaptation to Citrus reticulata host. Viruses 2019, 11, 331. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Dong, Z.; Gan, H. Comprehensive codon usage analysis of rice black-streaked dwarf virus based on P8 and P10 protein coding sequences. Infect. Genet. Evol. 2020, 86, 104601. [Google Scholar] [CrossRef]
- He, M.; Guan, S.Y.; He, C.Q. Evolution of rice stripe virus. Mol. Phylogenet. Evol. 2017, 109, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Das, S.; Saha, B.; Sarkar, P.; Karmakar, A.; Saha, A.; Saha, D.; Saha, A. Phylogeny and synonymous codon usage pattern of Papaya ringspot virus coat protein gene in the sub-Himalayan region of north-east India. Can. J. Microbiol. 2015, 61, 555–564. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Gan, H.; Liang, X. Analysis of synonymous codon usage bias in Potato virus M and its adaption to hosts. Viruses 2019, 11, 752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Dong, Z.; Gan, H. Genetic changes and host adaptability in sugarcane mosaic virus based on complete genome sequences. Mol. Phylogenet. Evol. 2020, 149, 106848. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.O.; Kobayashi, A.; Nakano, M.; Hagiwara, K.; Honda, Y.; Omura, T. Analysis of genetic relations between Broad bean wilt virus 1 and Broad bean wilt virus 2. J. Gen. Plant. Pathol. 2003, 69, 320–326. [Google Scholar] [CrossRef]
- Koh, L.H.; Cooper, J.I.; Wong, S.M. Complete sequences and phylogenetic analyses of a Singapore isolate of broad bean wilt fabavirus. Arch. Virol. 2001, 146, 135–147. [Google Scholar] [CrossRef]
- Kwak, H.A.; Lee, Y.J.; Kim, J.; Kim, M.K.; Kim, J.S.; Choi, H.S.; Seo, J.K. A determinant of disease symptom severity is located in RNA2 of Broad bean wilt virus 2. Virus Res. 2016, 211, 25–28. [Google Scholar] [CrossRef]
- Nakamura, S.; Iwai, T.; Honkura, R. Complete nucleotide sequence and genome organization of Broad bean wilt virus 2. Jpn. J. Phytopathol. 1998, 64, 565–568. [Google Scholar] [CrossRef]
- Qi, Y.; Zhou, X.; Li, D. Complete nucleotide sequence and infectious cDNA clone of the RNA1 of a Chinese isolate of Broad bean wilt virus 2. Virus Genes 2000, 20, 201–207. [Google Scholar] [CrossRef]
- Qi, Y.; Zhou, X.; Xue, C.; Li, D. Nucleotide sequence of RNA2 and polyprotein processing sites of a Chinese isolate of Broad bean wilt virus. Prog. Nat. Sci. 2000, 10, 684–686. [Google Scholar]
- Xie, L.; Shang, W.; Liu, C.; Zhang, Q.; Zhou, X.P. Mutual association of Broad bean wilt virus 2 VP37-derived tubules and plasmodesmata obtained from cytological observation. Sci. Rep. 2016, 6, 21552. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.W.; Wilm, A.; Lopez, R. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Rybicki, E.P. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Salminen, M.O.; Carr, J.K.; Burke, D.S.; McCutchan, F.E. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res. Hum. Retrovir. 1995, 11, 1423–1425. [Google Scholar] [CrossRef]
- Sawyer, S.A. GENECONV: A Computer Package for the Statistical Detection of Gene Conversion; Department of Mathematics, Washington University in St. Louis: St. Louis, MO, USA, 1999; Available online: http://www.math.wustl.edu/sawyer (accessed on 19 January 2021).
- Smith, J. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-Scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Page, R.D.M. Tree View: An application to display phylogenetic trees on personal computers. Bioinformatics 1996, 12, 357–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Ramsden, C.; Holmes, E.C.; Charleston, M.A. Hantavirus evolution in relation to its rodent and insectivore hosts: No evidence for codivergence. Mol. Biol. Evol. 2008, 26, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Duchêne, S.; Duchêne, D.; Holmes, E.C.; Ho, S.Y.W. The performance of the date-randomization test in phylogenetic analyses of time-structured virus data. Mol. Biol. Evol. 2015, 32, 1895–1906. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M.; Nasrullah, I.; Qamar, R.; Tong, Y. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerg. Microbes Infect. 2016, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gerton, J.L.; DeRisi, J.; Shroff, R.; Lichten, M.; Brown, P.O.; Petes, T.D. Global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2000, 97, 11383–11390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steel, M. The phylogenetic handbook: A practical approach to phylogenetic analysis and hypothesis testing edited by Lemey, P.; Salemi, M. and Vandamme, A.M. Biometrics 2010, 66, 324–325. [Google Scholar] [CrossRef]
- Jacquot, M.; Rao, P.P.; Yadav, S.; Nomikou, K.; Maan, S.; Jyothi, Y.K.; Reddy, N.; Putty, K.; Hemadri, D.; Singh, K.P. Contrasting selective patterns across the segmented genome of bluetongue virus in a global reassortment hotspot. Virus Evol. 2019, 5, 1–14. [Google Scholar] [CrossRef]
- McDonald, S.M.; Nelson, M.I.; Turner, P.E.; Patton, J.T. Reassortment in segmented RNA viruses: Mechanisms and outcomes. Nat. Rev. Microbiol. 2016, 14, 448–460. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Liu, X.; Du, Z.; Hou, H.; Wang, X.; Wang, F.; Yang, J. Bayesian phylodynamic analysis reveals the dispersal patterns of Tobacco mosaic virus in China. Virology 2019, 528, 110–117. [Google Scholar] [CrossRef]
- He, Z.; Chen, W.; Yasaka, R.; Chen, C.; Chen, X. Temporal analysis and adaptive evolution of the global population of potato virus M. Infect. Genet. Evol. 2019, 73, 167–174. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Tomitaka, Y.; Ho, S.Y.W.; Duchêne, S.; Vetten, H.J.; Lesemann, D.; Walsh, J.A.; Gibbs, A.J.; Ohshima, K. Turnip mosaic potyvirus probably first spread to Eurasian brassica crops from wild orchids about 1000 years ago. PLoS ONE 2013, 8, e55336. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Dong, T.; Wu, W.; Chen, W.; Liu, X.; Li, L. Evolutionary rates and phylogeographical analysis of Odontoglossum ringspot virus based on the 166 coat protein gene sequences. Plant. Pathol. J. 2019, 35, 498–507. [Google Scholar] [PubMed]
- Mao, Y.; Sun, X.; Shen, J.; Gao, F.; Qiu, G.; Wang, T.; Nie, X.; Zhang, W.; Gao, Y.; Bai, Y. Molecular evolutionary analysis of Potato virus Y infecting potato based on the VPg gene. Front. Microbiol. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Zhao, J.; Xing, G.; Li, G.; Wang, R.; Wang, Z. Genetic analysis and evolutionary changes of Porcine circovirus 2. Mol. Phylogenet. Evol. 2019, 139, 106520. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, H.; Wang, S.; Xing, G.; Zhang, C.; Zhang, W.; Liu, J.; Zhang, J.; Su, S.; Zhou, J. Insights into the genetic and host adaptability of emerging porcine circovirus 3. Virulence 2018, 9, 1301–1313. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Wang, R.; Zhang, L.; Shen, B.; Wang, N.; Xu, Q.; He, W.; He, W.; Li, G.; Su, S. Evolutionary changes of the novel Influenza D virus hemagglutinin-esterase fusion gene revealed by the codon usage pattern. Virulence 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, L.; He, W.; Zhang, X.; Wen, B.; Wang, C.; Xu, Q.; Li, G.; Zhou, J.; Veit, M. Genetic evolution and molecular selection of the HE gene of Influenza C virus. Viruses 2019, 11, 167. [Google Scholar] [CrossRef] [Green Version]
- Torres-Pérez, F.; Palma, R.E.; Hjelle, B.; Holmes, E.C.; Cook, J.A. Spatial but not temporal co-divergence of a virus and its mammalian host. Mol. Ecol. 2011, 20, 4109–4122. [Google Scholar] [CrossRef] [Green Version]
- Rodelo-Urrego, M.; Pagán, I.; González-Jara, P.; Betancourt, M.; Moreno-Letelier, A.; Ayllón, M.A.; Fraile, A.; Piñero, D.; García-Arenal, F. Landscape heterogeneity shapes host-parasite interactions and results in apparent plant-virus codivergence. Mol. Ecol. 2013, 22, 2325–2340. [Google Scholar] [CrossRef]
- Irwin, N.R.; Bayerlová, M.; Missa, O.; Martínková, N. Complex patterns of host switching in new world arenaviruses. Mol. Ecol. 2012, 21, 4137–4150. [Google Scholar] [CrossRef]
- Puigbò, P.; Aragonès, L.; Garcia-Vallvé, S. RCDI/eRCDI: A web-server to estimate codon usage deoptimization. BMC Res. Notes 2010, 3, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.K.; Shin, O.J.; Kwak, H.R.; Kim, M.K.; Choi, H.S.; Lee, S.H.; Kim, J.S. First Report of Broad bean wilt virus 2 in Leonurus sibiricus in Korea. Plant Dis. 2014, 98, 1748. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, G.; Tomita, R.; Kobayashi, K.; Sekine, K.T. Establishment of an agroinoculation system for Broad bean wilt virus 2. Arch. Virol. 2013, 158, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, T.; Okumura, A.; Takeda, I.; Miura, Y.; Suzuki, K. Nucleotide sequence and synthesis of infectious RNA from cloned cDNA of broad bean wilt virus 2 RNA 2. Arch. Virol. 2000, 145, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.O.; Nakano, M.; Kashiwazaki, S.; Naito, T.; Mikoshiba, Y.; Shiota, A.; Kameya-Iwaki, M.; Honda, Y. Sequence analysis of RNA-2 of different isolates of Broad bean wilt virus confirms the existence of two distinct species. Arch. Virol. 1999, 144, 1429–1438. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Segment | Isolate | Sequence Used to Infer Major Parent | Sequence Used to Infer Minor Parent | Recombination Site a | Recombination Detecting Program (p-Value b) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RDP | GENECONV | BOOTSCAN | MAXCHI | CHIMAERA | SISCAN | 3SEQ | |||||
| RNA1 | KF498696 | UN c | FN985164 | 1311–2836 | 2.399 × 10−7 | 7.723 × 10−1 | 4.485 × 10−8 | 5.120 × 10−5 | 3.670 × 10−2 | 2.467 × 10−11 | 1.869 × 10−6 |
| KM076648 | KC625492 | AB023484 | 1644–5592 | 2.711 × 10−141 | 9.401 × 10−134 | 5.393 × 10−133 | 7.798 × 10−44 | 1.023 × 10−30 | 1.281 × 10−55 | 8.626 × 10−13 | |
| RNA2 | JQ855708 | KJ825857 | KC625506 | 236–1144 | 2.057 × 10−26 | 1.165 × 10−19 | 9.826 × 10−25 | 1.522 × 10−16 | 5.628 × 10−14 | 1.524 × 10−33 | 1.096 × 10−12 |
| KM076649 | JX183234 | KJ825857 | 148–3166 | 3.811 × 10−23 | 1.442 × 10−15 | 6.082 × 10−20 | 1.152 × 10−7 | 1.394 × 10−6 | 1.938 × 10−9 | 2.193 × 10−12 | |
| HQ283389 | KJ825857 | KC625506 | 236–1132 | 1.132 × 10−30 | 3.734 × 10−22 | 8.940 × 10−29 | 7.192 × 10−22 | 5.962 × 10−16 | 2.644 × 10−26 | 1.096 × 10−12 | |
| KF498697 | KC625506 | LC497425 | 2561–3166 | 6.698 × 10−8 | - | 3.281 × 10−8 | 5.828 × 10−6 | 1.533 × 10−2 | 5.191 × 10−12 | 8.114 × 10−4 | |
| GQ202215 | KF498697 | KX686590 | 1484–2576 | 5.190 × 10−13 | 1.640 × 10−8 | 3.624 × 10−15 | 6.479 × 10−13 | 4.696 × 10−6 | 3.525 × 10−22 | 1.096 × 10−12 | |
| HQ283390 | AB018698 | LC497425 | 1278–3166 | 6.174 × 10−22 | 9.652 × 10−18 | 1.257 × 10−12 | 8.796 × 10−23 | 5.680 × 10−12 | 1.397 × 10−16 | 4.858 × 10−40 | |
| Codon | aa | ORF1 | ORF2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Broad Bean (n = 6) | Pepper (n = 18) | Spinach (n = 3) | Yam (n = 4) | All (n = 42) | Broad Bean (n = 5) | Pepper (n = 18) | Spinach (n = 3) | Yam (n = 4) | All (n = 41) | ||
| TTT | F | 1.38 * | 1.37 | 1.38 | 1.35 | 1.35 | 1.25 | 1.35 | 1.28 | 1.25 | 1.32 |
| TTC | F | 0.62 | 0.63 | 0.62 | 0.65 | 0.65 | 0.75 | 0.65 | 0.73 | 0.76 | 0.68 |
| TTA | L | 0.76 | 0.80 | 0.76 | 0.71 | 0.79 | 0.73 | 0.72 | 0.58 | 0.66 | 0.72 |
| TTG | L | 2.97 | 2.72 | 2.86 | 3.02 | 2.82 | 2.66 | 2.50 | 2.77 | 2.81 | 2.66 |
| CTT | L | 0.95 | 0.94 | 0.85 | 1.13 | 0.96 | 1.20 | 1.30 | 1.35 | 1.20 | 1.21 |
| CTC | L | 0.42 | 0.45 | 0.48 | 0.44 | 0.44 | 0.31 | 0.27 | 0.20 | 0.25 | 0.31 |
| CTA | L | 0.29 | 0.32 | 0.31 | 0.22 | 0.31 | 0.31 | 0.33 | 0.29 | 0.32 | 0.33 |
| CTG | L | 0.61 | 0.77 | 0.74 | 0.47 | 0.68 | 0.80 | 0.79 | 0.82 | 0.75 | 0.77 |
| ATT | I | 1.50 | 1.53 | 1.53 | 1.60 | 1.54 | 1.40 | 1.35 | 1.27 | 1.43 | 1.36 |
| ATC | I | 0.64 | 0.57 | 0.61 | 0.54 | 0.59 | 0.62 | 0.69 | 0.72 | 0.72 | 0.68 |
| ATA | I | 0.86 | 0.89 | 0.86 | 0.86 | 0.87 | 0.98 | 0.96 | 1.01 | 0.84 | 0.96 |
| GTT | V | 1.33 | 1.38 | 1.37 | 1.22 | 1.37 | 1.32 | 1.19 | 1.29 | 1.28 | 1.29 |
| GTC | V | 0.58 | 0.57 | 0.58 | 0.70 | 0.57 | 0.56 | 0.67 | 0.54 | 0.61 | 0.63 |
| GTA | V | 0.38 | 0.34 | 0.45 | 0.40 | 0.36 | 0.54 | 0.55 | 0.58 | 0.50 | 0.51 |
| GTG | V | 1.71 | 1.71 | 1.61 | 1.69 | 1.7 | 1.58 | 1.59 | 1.59 | 1.62 | 1.57 |
| TCT | S | 1.07 | 1.15 | 1.15 | 1.15 | 1.12 | 1.26 | 1.27 | 1.51 | 1.2 | 1.28 |
| TCC | S | 0.54 | 0.59 | 0.58 | 0.41 | 0.54 | 0.58 | 0.62 | 0.47 | 0.65 | 0.6 |
| TCA | S | 1.70 | 1.46 | 1.60 | 1.66 | 1.57 | 1.23 | 1.23 | 1.19 | 1.03 | 1.19 |
| TCG | S | 0.55 | 0.64 | 0.52 | 0.63 | 0.61 | 0.35 | 0.30 | 0.32 | 0.46 | 0.35 |
| AGT | S | 1.18 | 1.05 | 1.00 | 1.36 | 1.14 | 1.50 | 1.53 | 1.46 | 1.58 | 1.53 |
| AGC | S | 0.96 | 1.11 | 1.16 | 0.79 | 1.02 | 1.09 | 1.05 | 1.06 | 1.08 | 1.05 |
| CCT | P | 1.38 | 1.14 | 1.29 | 1.57 | 1.28 | 1.69 | 1.71 | 1.76 | 1.30 | 1.66 |
| CCC | P | 0.77 | 0.85 | 0.74 | 0.66 | 0.78 | 0.70 | 0.67 | 0.51 | 1.03 | 0.72 |
| CCA | P | 1.43 | 1.62 | 1.44 | 1.51 | 1.54 | 1.17 | 1.23 | 1.22 | 0.98 | 1.18 |
| CCG | P | 0.42 | 0.39 | 0.53 | 0.26 | 0.39 | 0.43 | 0.38 | 0.51 | 0.69 | 0.44 |
| ACT | T | 1.27 | 1.25 | 1.23 | 1.33 | 1.26 | 1.41 | 1.43 | 1.42 | 1.64 | 1.44 |
| ACC | T | 0.53 | 0.48 | 0.47 | 0.51 | 0.49 | 0.52 | 0.49 | 0.49 | 0.30 | 0.47 |
| ACA | T | 1.57 | 1.63 | 1.71 | 1.49 | 1.59 | 1.50 | 1.42 | 1.38 | 1.50 | 1.43 |
| ACG | T | 0.64 | 0.64 | 0.59 | 0.66 | 0.65 | 0.58 | 0.66 | 0.72 | 0.56 | 0.65 |
| GCT | A | 1.32 | 1.31 | 1.28 | 1.59 | 1.37 | 1.26 | 1.32 | 1.36 | 1.37 | 1.32 |
| GCC | A | 0.70 | 0.72 | 0.77 | 0.57 | 0.69 | 0.62 | 0.62 | 0.59 | 0.79 | 0.64 |
| GCA | A | 1.47 | 1.40 | 1.39 | 1.34 | 1.4 | 1.5 | 1.45 | 1.34 | 1.26 | 1.45 |
| GCG | A | 0.51 | 0.57 | 0.56 | 0.50 | 0.54 | 0.62 | 0.61 | 0.71 | 0.58 | 0.58 |
| TAT | Y | 1.12 | 1.12 | 1.18 | 1.14 | 1.14 | 1.09 | 1.06 | 1.11 | 0.99 | 1.05 |
| TAC | Y | 0.88 | 0.88 | 0.82 | 0.86 | 0.86 | 0.91 | 0.94 | 0.89 | 1.01 | 0.95 |
| CAT | H | 1.42 | 1.40 | 1.33 | 1.51 | 1.46 | 1.46 | 1.45 | 1.52 | 1.35 | 1.39 |
| CAC | H | 0.58 | 0.60 | 0.67 | 0.49 | 0.54 | 0.54 | 0.55 | 0.49 | 0.65 | 0.61 |
| CAA | Q | 1.10 | 1.10 | 1.04 | 1.11 | 1.10 | 1.26 | 1.20 | 1.17 | 1.37 | 1.23 |
| CAG | Q | 0.90 | 0.90 | 0.96 | 0.89 | 0.92 | 0.74 | 0.80 | 0.83 | 0.63 | 0.77 |
| AAT | N | 1.36 | 1.37 | 1.40 | 1.30 | 1.35 | 1.37 | 1.38 | 1.45 | 1.48 | 1.39 |
| AAC | N | 0.65 | 0.63 | 0.60 | 0.70 | 0.65 | 0.63 | 0.62 | 0.55 | 0.52 | 0.61 |
| AAA | K | 0.96 | 0.97 | 0.98 | 0.86 | 0.95 | 1.09 | 1.09 | 1.03 | 1.01 | 1.06 |
| AAG | K | 1.04 | 1.03 | 1.02 | 1.14 | 1.05 | 0.91 | 0.91 | 0.97 | 0.99 | 0.94 |
| GAT | D | 1.48 | 1.54 | 1.53 | 1.48 | 1.51 | 1.53 | 1.53 | 1.54 | 1.54 | 1.54 |
| GAC | D | 0.52 | 0.46 | 0.47 | 0.52 | 0.49 | 0.47 | 0.47 | 0.46 | 0.46 | 0.46 |
| GAA | E | 0.88 | 0.94 | 1.02 | 0.93 | 0.93 | 1.00 | 1.05 | 1.03 | 0.73 | 1.01 |
| GAG | E | 1.12 | 1.06 | 0.99 | 1.07 | 1.07 | 1.01 | 0.95 | 0.97 | 1.27 | 0.99 |
| TGT | C | 1.06 | 1.00 | 1.04 | 1.12 | 1.04 | 1.38 | 1.31 | 1.08 | 1.69 | 1.29 |
| TGC | C | 0.94 | 1.01 | 0.96 | 0.88 | 0.96 | 0.62 | 0.69 | 0.92 | 0.31 | 0.71 |
| CGT | R | 0.54 | 0.52 | 0.56 | 0.72 | 0.57 | 1.30 | 1.19 | 1.09 | 1.13 | 1.15 |
| CGC | R | 0.67 | 0.79 | 0.82 | 0.52 | 0.69 | 1.14 | 1.29 | 1.30 | 0.99 | 1.2 |
| CGA | R | 0.59 | 0.58 | 0.70 | 0.39 | 0.59 | 0.29 | 0.31 | 0.25 | 0.35 | 0.36 |
| CGG | R | 0.47 | 0.55 | 0.42 | 0.55 | 0.49 | 0.21 | 0.14 | 0.21 | 0.12 | 0.16 |
| AGA | R | 2.03 | 2.04 | 2.03 | 1.88 | 1.97 | 2.16 | 2.45 | 2.64 | 1.91 | 2.3 |
| AGG | R | 1.69 | 1.52 | 1.47 | 1.95 | 1.69 | 0.90 | 0.62 | 0.50 | 1.51 | 0.82 |
| GGT | G | 0.89 | 1.10 | 1.11 | 1.13 | 1.04 | 1.08 | 1.12 | 1.16 | 1.52 | 1.18 |
| GGC | G | 0.84 | 0.73 | 0.79 | 0.65 | 0.75 | 0.78 | 0.73 | 0.73 | 0.64 | 0.74 |
| GGA | G | 1.56 | 1.46 | 1.45 | 1.51 | 1.49 | 1.29 | 1.32 | 1.29 | 1.20 | 1.32 |
| GGG | G | 0.71 | 0.72 | 0.65 | 0.72 | 0.72 | 0.84 | 0.83 | 0.812 | 0.65 | 0.77 |
| Gene | ENC | GC3s | GC | Axis1 | Axis2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| r | p | r | p | r | p | r | p | r | p | ||
| ORF1 | Gravy | 0.13091 ns | 0.40274 | 0.01371 ns | 0.93045 | 0.05954 ns | 0.7045 | −0.32221 * | 0.0351 | −0.25199 ns | 0.10307 |
| Aromo | 0.47618 ** | 0.00125 | −0.21277 ns | 0.17073 | 0.26115 ns | 0.09074 | −0.85596 ** | 2.58 × 10−13 | 0.03121 ns | 0.8425 | |
| ORF2 | Gravy | 0.02381 ns | 0.88105 | 0.25202 ns | 0.10738 | 0.20002 ns | 0.20408 | −0.43133 ** | 0.00435 | −0.37632 * | 0.01404 |
| Aromo | −0.38519 * | 0.01177 | −0.10812 ns | 0.49553 | −0.22767 ns | 0.14705 | −0.27126 ns | 0.08227 | −0.27379 ns | 0.07934 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Z.; Dong, Z.; Qin, L.; Gan, H. Phylodynamics and Codon Usage Pattern Analysis of Broad Bean Wilt Virus 2. Viruses 2021, 13, 198. https://doi.org/10.3390/v13020198
He Z, Dong Z, Qin L, Gan H. Phylodynamics and Codon Usage Pattern Analysis of Broad Bean Wilt Virus 2. Viruses. 2021; 13(2):198. https://doi.org/10.3390/v13020198
Chicago/Turabian StyleHe, Zhen, Zhuozhuo Dong, Lang Qin, and Haifeng Gan. 2021. "Phylodynamics and Codon Usage Pattern Analysis of Broad Bean Wilt Virus 2" Viruses 13, no. 2: 198. https://doi.org/10.3390/v13020198