
Analysis of a Novel Bacteriophage vB_AchrS_AchV4 Highlights the Diversity of Achromobacter Viruses

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phage Techniques
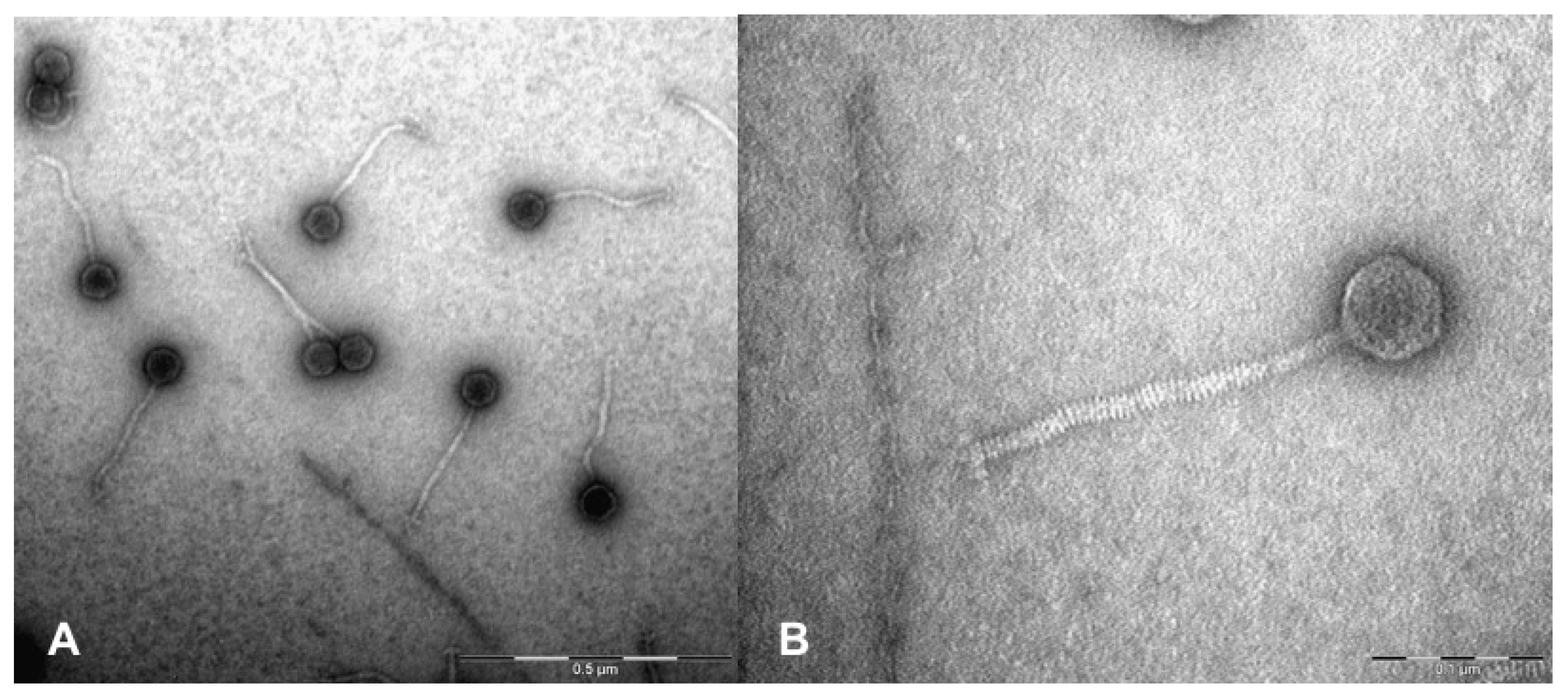
2.2. TEM Analysis
2.3. DNA Isolation
2.4. Genome Sequencing and Assembly
2.5. Genome Sequence Analysis
2.6. Phylogeny
2.7. Proteomic Analysis
3. Results
3.1. Phage Isolation and Characterization
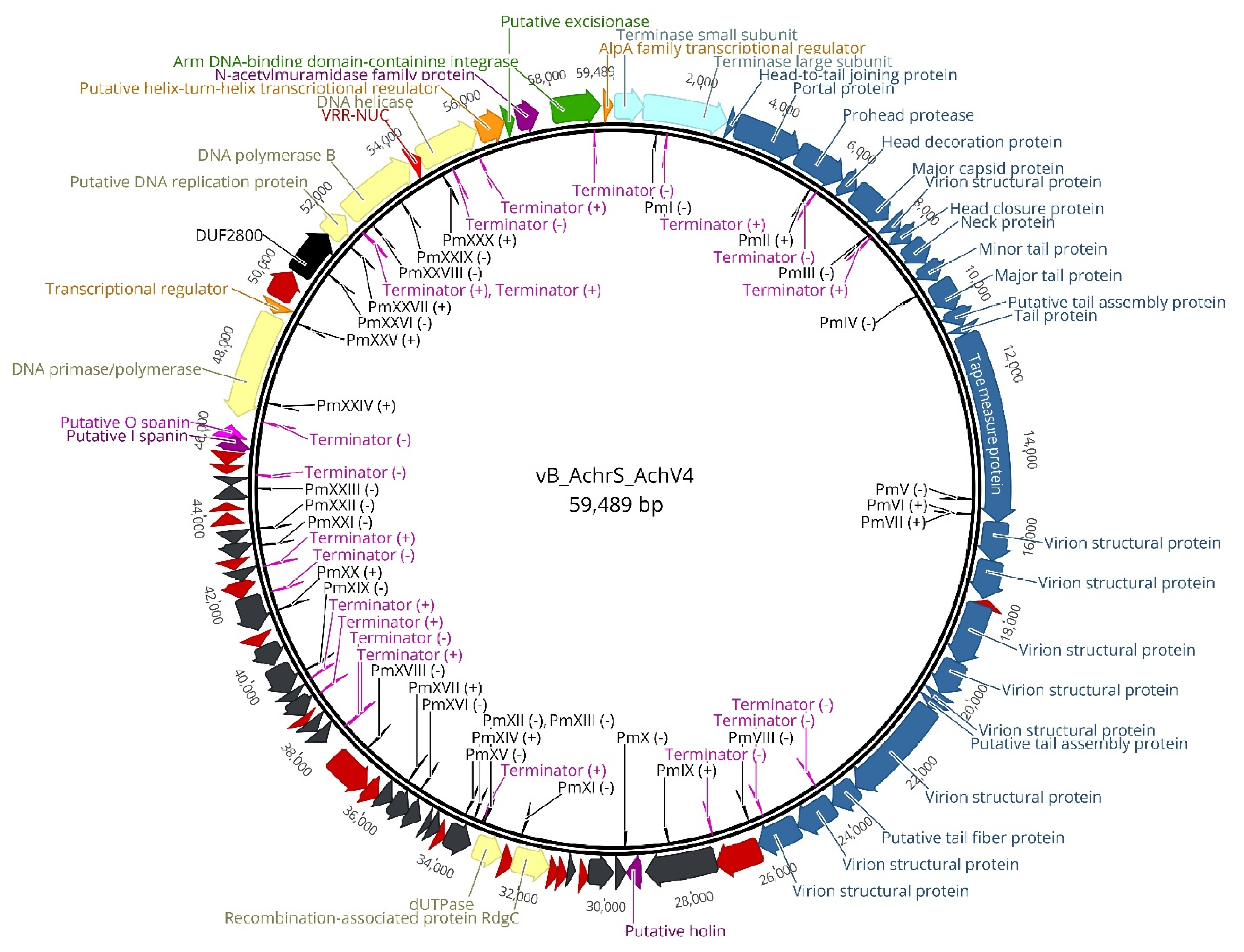
3.2. AchV4 Genome
3.2.1. Structural Module
3.2.2. DNA Packaging
3.2.3. Lysis Genes
3.2.4. DNA Metabolism
3.2.5. Lysogeny Module
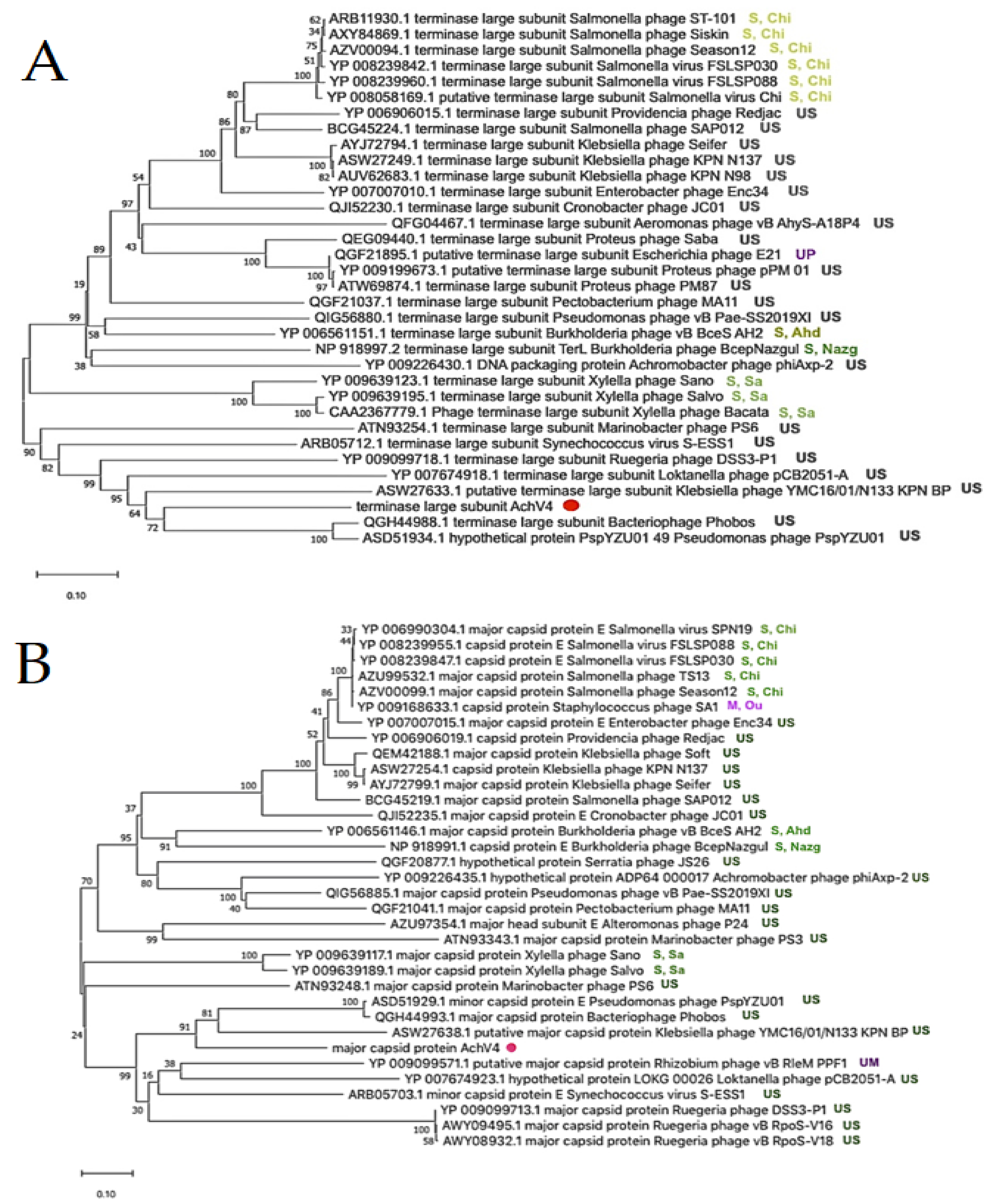
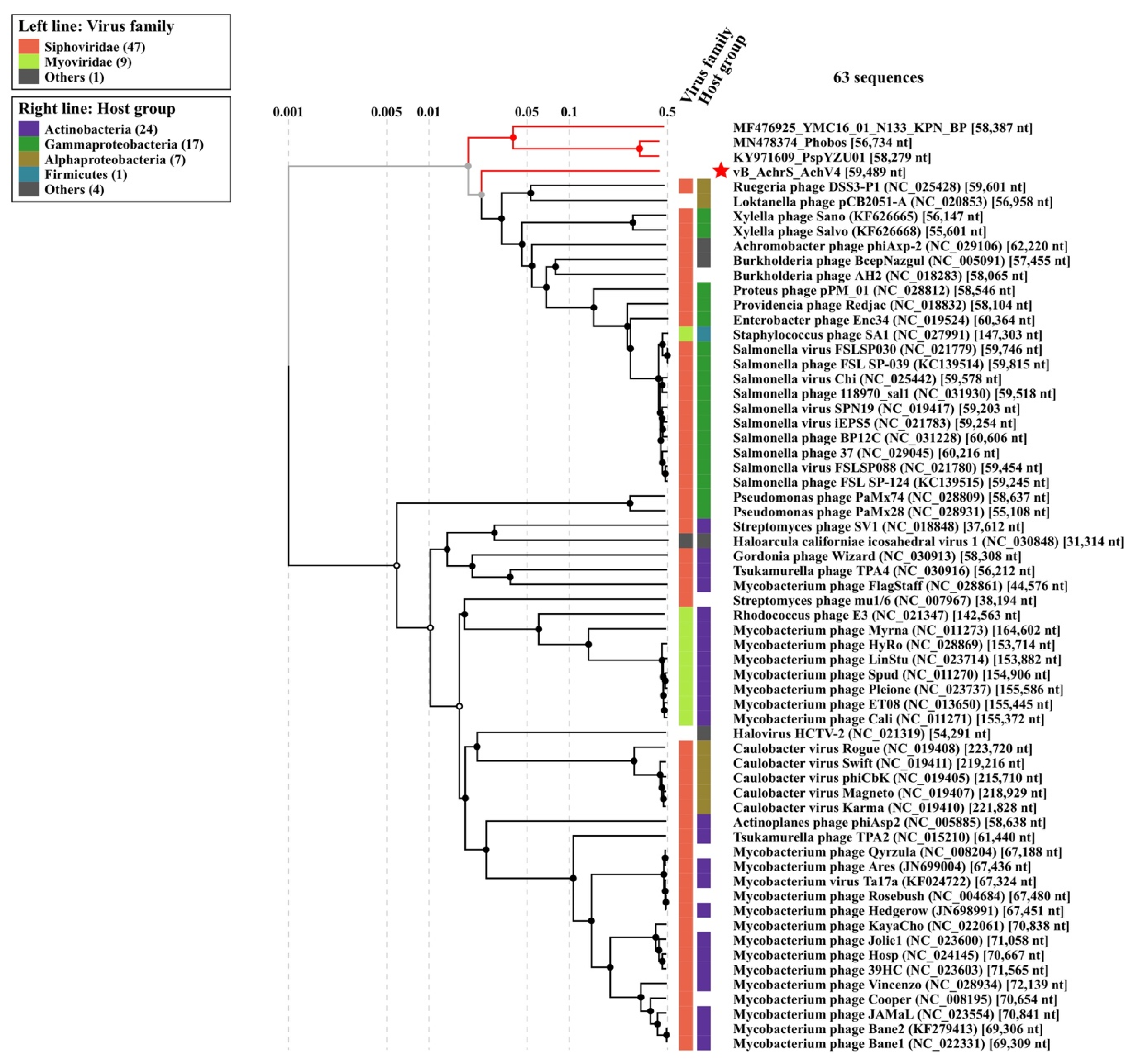
3.3. Phylogenetic Analysis
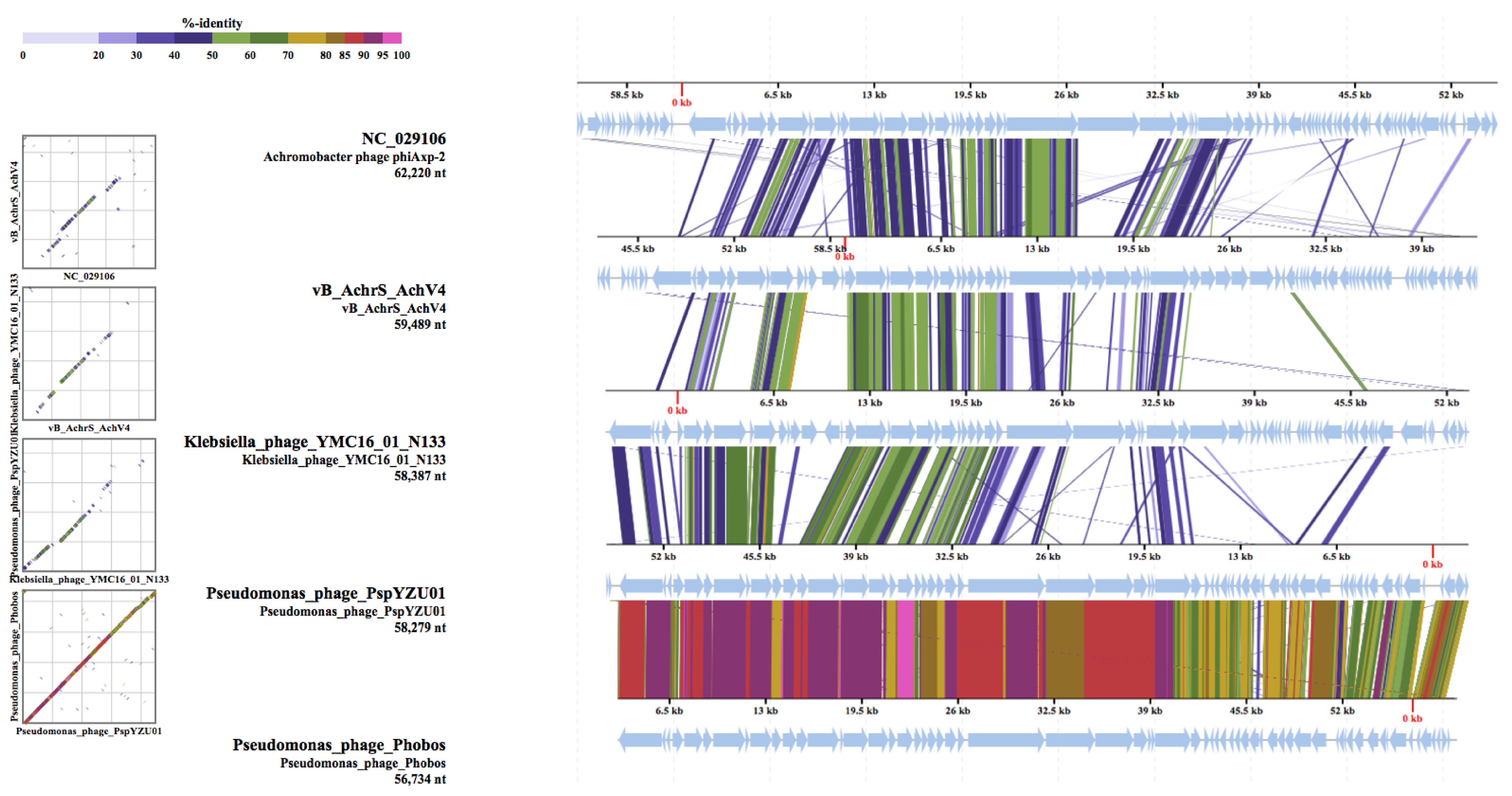
Comparison with Sequenced Achromobacter Phages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Busse, H.-J.; Auling, G. Achromobacter. In Bergey’s Manual of Systematics of Archaea and Bacteria; Trujillo, M.E., Dedysh, S., DeVos, P., Hedlund, B., Kämpfer, P., Rainey, F.A., Whitman, W.B., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar] [CrossRef]
- Swenson, C.E.; Sadikot, R.T. Achromobacter respiratory infections. Ann. Am. Thorac. Soc. 2015, 12, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Kutanovas, S.; Karvelis, L.; Vaitekūnas, J.; Stankevičiūtė, J.; Gasparavičiūtė, R.; Meškys, R. Isolation and characterization of novel pyridine dicarboxylic acid-degrading microorganisms. Chemija 2016, 27, 74–83. [Google Scholar]
- Felgate, H.; Giannopoulos, G.; Sullivan, M.J.; Gates, A.J.; Clarke, T.A.; Baggs, E.; Rowley, G.; Richardson, D.J. The impact of copper, nitrate and carbon status on the emission of nitrous oxide by two species of bacteria with biochemically distinct denitrification pathways. Environ. Microbiol. 2012, 14, 1788–1800. [Google Scholar] [CrossRef] [PubMed]
- Edwards, B.D.; Greysson-Wong, J.; Somayaji, R.; Waddell, B.; Whelan, F.J.; Storey, D.G.; Rabin, H.R.; Surette, M.G.; Parkins, M.D. Prevalence and Outcomes of Achromobacter Species Infections in Adults with Cystic Fibrosis: A North American Cohort Study. J. Clin. Microbiol. 2017, 55, 2074–2085. [Google Scholar] [CrossRef] [Green Version]
- Yabuuchi, E.; Yano, I. Achromobacter gen. nov. and Achromobacter xylosoxidans (ex Yabuuchi and Ohyama 1971) nom. rev. Int. J. Syst. Bacteriol. 1981, 31, 477–478. [Google Scholar] [CrossRef] [Green Version]
- LPSN—List of Prokaryotic Names with Standing in Nomenclature. Available online: https://lpsn.dsmz.de/genus/achromobacter (accessed on 8 January 2021).
- PubMLST—Public Databases for Molecular Typing and Microbial Genome Diversity. Available online: https://pubmlst.org/organisms/achromobacter-spp (accessed on 8 January 2021).
- Wittmann, J.; Dreiseikelmann, B.; Rohde, C.; Rohde, M.; Sikorski, J. Isolation and Characterization of Numerous Novel Phages Targeting Diverse Strains of the Ubiquitous and Opportunistic Pathogen Achromobacter xylosoxidans. PLoS ONE 2014, 9, e86935. [Google Scholar] [CrossRef]
- Li, E.; Yin, Z.; Ma, Y.; Li, H.; Lin, W.; Wei, X.; Zhao, R.; Jiang, A.; Yuan, J.; Zhao, X. Identification and molecular characterization of bacteriophage phiAxp-2 of Achromobacter xylosoxidans. Sci. Rep. 2016, 6, 34300. [Google Scholar] [CrossRef]
- Wittmann, J.; Dreiseikelmann, B.; Rohde, M.; Meier-Kolthoff, J.P.; Bunk, B.; Rohde, C. First genome sequences of Achromobacter phages reveal new members of the N4 family. Virol. J. 2014, 11, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, E.; Zhao, J.; Ma, Y.; Wei, X.; Li, H.; Lin, W.; Wang, X.; Li, C.; Shen, Z.; Zhao, R.; et al. Characterization of a novel Achromobacter xylosoxidans specific siphoviruse: PhiAxp-1. Sci. Rep. 2016, 6, 21943. [Google Scholar] [CrossRef] [Green Version]
- Essoh, C.; Vernadet, J.P.; Vergnaud, G.; Coulibaly, A.; Kakou-N’Douba, A.; N’Guetta, A.S.; Ouassa, T.; Pourcel, C. Characterization of sixteen Achromobacter xylosoxidans phages from Abidjan, Côte d’Ivoire, isolated on a single clinical strain. Arch. Virol. 2020, 165, 725–730. [Google Scholar] [CrossRef]
- Dreiseikelmann, B.; Bunk, B.; Spröer, C.; Rohde, M.; Nimtz, M.; Wittmann, J. Characterization and genome comparisons of three Achromobacter phages of the family Siphoviridae. Arch. Virol. 2017, 162, 2191–2201. [Google Scholar] [CrossRef]
- Chan, H.T.; Ku, H.; Low, Y.P.; Brown, T.; Batinovic, S.; Kabwe, M.; Petrovski, S.; Tucci, J. Characterization of Novel Lytic Bacteriophages of Achromobacter marplantensis Isolated from a Pneumonia Patient. Viruses 2020, 12, 1138. [Google Scholar] [CrossRef]
- Bartz, M.; Yao, G.; Le, T.; Liu, M.; Burrowes, B.; Gonzalez, C.; Young, R. Complete Genome Sequence of Achromobacter xylosoxidans Myophage Mano. Microbiol. Resour. Announc. 2021, 10, e01390–e01420. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, E.; Qi, Z.; Li, H.; Wei, X.; Lin, W.; Zhao, R.; Jiang, A.; Yang, H.; Yin, Z.; et al. Isolation and molecular characterisation of Achromobacter phage phiAxp-3, an N4-like bacteriophage. Sci. Rep. 2006, 6, 24776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coenye, T.; Vancanneyt, M.; Falsen, E.; Swings, J.; Vandamme, P. Achromobacter insolitus sp. nov. and Achromobacter spanius sp. nov.; from human clinical samples. Int. J. Syst. Evol. Microbiol. 2003, 53, 1819–1824. [Google Scholar] [CrossRef] [Green Version]
- Wass, T.J.; Syed-Ab-Rahman, S.F.; Carvalhais, L.C.; Ferguson, B.J.; Schenk, P.M. Complete Genome Sequence of Achromobacter spanius UQ283, a Soilborne Isolate Exhibiting Plant Growth-Promoting Properties. Microbiol. Resour. Announc. 2019, 8, e00236–e00319. [Google Scholar] [CrossRef] [Green Version]
- Carlson, K. Appendix: Working with Bacteriophages: Common Techniques and Methodological Approaches. In Bacteriophages: Biology and Applications; Kutter, E., Sulakvelidze, A., Eds.; EdiCRC Press: London, UK, 2005. [Google Scholar]
- Kaliniene, L.; Truncaitė, L.; Šimoliūnas, E.; Zajančkauskaitė, A.; Vilkaitytė, M.; Kaupinis, A.; Skapas, M.; Meškys, R. Molecular analysis of the low-temperature Escherichia coli phage vB_EcoS_NBD2. Arch. Virol. 2018, 163, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Armalytė, J.; Skerniškytė, J.; Bakienė, E.; Krasauskas, R.; Šiugždinienė, R.; Kareivienė, V.; Kerzienė, S.; Klimienė, I.; Sužiedėlienė, E.; Ružauskas, M. Microbial Diversity and Antimicrobial Resistance Profile in Microbiota from Soils of Conventional and Organic Farming Systems. Front. Microbiol. 2019, 10, 892. [Google Scholar] [CrossRef]
- Stankevičiūtė, J.; Vaitekūnas, J.; Petkevičius, V.; Gasparavičiūtė, R.; Tauraitė, D.; Meškys, R. Oxyfunctionalization of pyridine derivatives using whole cells of Burkholderia sp. MAK1. Sci. Rep. 2016, 6, 39129. [Google Scholar] [CrossRef]
- Zhao, X.; Cui, Y.; Yan, Y.; Du, Z.; Tan, Y.; Yang, H.; Bi, Y.; Zhang, P.; Zhou, L.; Zhou, D.; et al. Outer Membrane Proteins Ail and OmpF of Yersinia pestis Are Involved in the Adsorption of T7-Related Bacteriophage Yep-phi. J. Virol. 2013, 87, 12260–12269. [Google Scholar] [CrossRef] [Green Version]
- Carlson, K.; Miller, E. Experiments in T4 genetics. In Bacteriophage T4; Karam, J.D., Ed.; ASM Press: Washington, DC, USA, 1994; pp. 419–483. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2010, 27, 578–579. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.A. Sequencing, Assembling and Finishing Complete Bacteriophage Genomes. In Bacteriophages. Methods in Molecular Biology; Clokie, M.R.J., Kropinski, A.M., Lavigne, R., Eds.; Humana Press: New York, NY, USA, 2018; Volume 3, pp. 109–125. [Google Scholar] [CrossRef]
- Ramsey, J.; Rasche, H.; Maughmer, C.; Criscione, A.; Mijalis, E.; Liu, M.; Hu, J.C.; Young, R.; Gill, J.J. Galaxy and Apollo as a biologist-friendly interface for high-quality cooperative phage genome annotation. PLoS Comput. Biol. 2020, 16, e1008214. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef] [PubMed]
- McNair, K.; Bailey, B.A.; Edwards, R.A. PHACTS, a computational approach to classifying the lifestyle of phages. Bioinformatics 2012, 28, 614–618. [Google Scholar] [CrossRef] [Green Version]
- Kongari, R.; Rajaure, M.; Cahill, J.; Rasche, E.; Mijalis, E.; Berry, J.; Young, R. Phage spanins: Diversity, topological dynamics and gene convergence. BMC Bioinform. 2018, 19, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Lopes, A.; Tavares, P.; Petit, M.-A.; Guérois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genom. 2014, 15, 1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [Green Version]
- Šimoliūnas, E.; Kaliniene, L.; Stasilo, M.; Truncaitė, L.; Zajančkauskaitė, A.; Staniulis, J.; Nainys, J.; Kaupinis, A.; Valius, M.; Meškys, R. Isolation and Characterization of vB_ArS-ArV2—First Arthrobacter sp. Infecting Bacteriophage with Completely Sequenced Genome. PLoS ONE 2014, 9, e111230. [Google Scholar] [CrossRef] [Green Version]
- Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 2016, 363, fnw002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, V.B.; Feiss, M. The Bacteriophage DNA Packaging Motor. Annu. Rev. Genet. 2008, 42, 647–681. [Google Scholar] [CrossRef]
- Rao, V.B.; Feiss, M. Mechanisms of DNA Packaging by Large Double-Stranded DNA Viruses. Annu. Rev. Virol. 2015, 2, 351–378. [Google Scholar] [CrossRef] [Green Version]
- Young, R.; Wang, I.-N.; Roof, W.D. Phages will out: Strategies of host cell lysis. Trends Microbiol. 2000, 8, 120–128. [Google Scholar] [CrossRef]
- Fernandes, S.; São-José, C. Enzymes and Mechanisms Employed by Tailed Bacteriophages to Breach the Bacterial Cell Barriers. Viruses 2018, 10, 396. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Rubio, L.; Gerstmans, H.; Thorpe, S.; Mesnage, S.; Lavigne, R.; Briers, Y. DUF3380 Domain from a Salmonella Phage Endolysin Shows PotentN-Acetylmuramidase Activity. Appl. Environ. Microbiol. 2016, 82, 4975–4981. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Kumar, S.; Kumar, D.; Mishra, A.; Dewangan, R.P.; Shrivastava, P.; Ramachandran, S.; Taneja, B. The structure of Rv3717 reveals a novel amidase from Mycobacterium tuberculosis. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 2543–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klyczek, K.K.; Bonilla, J.A.; Jacobs-Sera, D.; Adair, T.L.; Afram, P.; Allen, K.G.; Archambault, M.L.; Aziz, R.M.; Bagnasco, F.G.; Ball, S.L.; et al. Tales of diversity: Genomic and morphological characteristics of forty-six Arthrobacter phages. PLoS ONE 2017, 12, e0180517. [Google Scholar] [CrossRef]
- Cahill, J.; Young, R. Phage Lysis: Multiple Genes for Multiple Barriers. Adv. Appl. Microbiol. 2019, 103, 33–70. [Google Scholar] [CrossRef]
- Drees, J.C.; Chitteni-Pattu, S.; McCaslin, D.R.; Inman, R.B.; Cox, M.M. Inhibition of RecA Protein Function by the RdgC Protein from Escherichia coli. J. Biol. Chem. 2006, 281, 4708–4717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cernooka, E.; Rumnieks, J.; Tars, K.; Kazaks, A. Structural Basis for DNA Recognition of a Single-stranded DNA-binding Protein from Enterobacter Phage Enc34. Sci. Rep. 2017, 7, 15529. [Google Scholar] [CrossRef]
- Iyer, L.M.; Koonin, E.V.; Aravind, L. Classification and evolutionary history of the single-strand annealing proteins, RecT, Redβ, ERF and RAD52. BMC Genom. 2002, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [Green Version]
- Casjens, S.R.; Hendrix, R.W. Bacteriophage lambda: Early pioneer and still relevant. Virology 2015, 479, 310–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broussard, G.W.; Oldfield, L.M.; Villanueva, V.M.; Lunt, B.L.; Shine, E.E.; Hatfull, G.F. Integration-Dependent Bacteriophage Immunity Provides Insights into the Evolution of Genetic Switches. Mol. Cell 2013, 49, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berntsson, R.P.-A.; Odegrip, R.; Sehlén, W.; Skaar, K.; Svensson, L.M.; Massad, T.; Högbom, M.; Haggård-Ljungquist, E.; Stenmark, P. Structural insight into DNA binding and oligomerization of the multifunctional Cox protein of bacteriophage P2. Nucleic Acids Res. 2013, 42, 2725–2735. [Google Scholar] [CrossRef]
- Amarillas, L.; Estrada-Acosta, M.; León-Chan, R.G.; López-Orona, C.; Lightbourn, L. Complete genome sequence of Phobos: A novel bacteriophage with unusual genomic features that infects Pseudomonas syringae. Arch. Virol. 2020, 165, 1485–1488. [Google Scholar] [CrossRef]
- Rohwer, F.; Edwards, R. The Phage Proteomic Tree: A Genome-Based Taxonomy for Phage. J. Bacteriol. 2002, 184, 4529–4535. [Google Scholar] [CrossRef] [Green Version]
- Adriaenssens, E.M.; Edwards, R.P.; Nash, J.H.; Mahadevan, P.; Seto, D.; Ackermann, H.-W.; Lavigne, R.; Kropinski, A.M. Integration of genomic and proteomic analyses in the classification of the Siphoviridae family. Virology 2015, 477, 144–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, L.C.; Bonis, B.; Phan, C.Q.; Kent, L.A.; Lee, A.K.; Hunter, R.C. A putative enoyl-CoA hydratase contributes to biofilm formation and the antibiotic tolerance of Achromobacter xylosoxidans. NPJ Biofilms Microbiomes 2019, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Busse, H.-J.; Stolz, A. Achromobacter, Alcaligenes and Related Genera. In The Prokaryotes; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 675–700. [Google Scholar] [CrossRef]
- Abdel-Rahman, H.M.; Salem, A.A.; Moustafa, M.M.A.; El-Garhy, H.A.S. A novice Achromobacter sp. EMCC1936 strain acts as a plant-growth-promoting agent. Acta Physiol. Plant. 2017, 39, 61. [Google Scholar] [CrossRef]
- Hendrix, R.W.; Smith, M.C.M.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary relationships among diverse bacteriophages and prophages: All the world’s a phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef] [Green Version]
- Mushegian, A.R. Are There 1031 Virus Particles on Earth, or More, or Fewer? J. Bacteriol. 2020, 202, e00052. [Google Scholar] [CrossRef]
- Bailly-Bechet, M.; Vergassola, M.; Rocha, E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007, 17, 1486–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfull, G.F. Dark Matter of the Biosphere: The Amazing World of Bacteriophage Diversity. J. Virol. 2015, 89, 8107–8110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cresawn, S.G.; Pope, W.H.; Jacobs-Sera, D.; Bowman, C.A.; Russell, D.A.; Dedrick, R.M.; Adair, T.; Anders, K.R.; Ball, S.; Bollivar, D.; et al. Comparative Genomics of Cluster O Mycobacteriophages. PLoS ONE 2015, 10, e0118725. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Putative Function | MW (KDa) | Peptide Count | Sequence Coverage (%) |
|---|---|---|---|---|
| AchV4_0004 | Portal protein | 61.537 | 35 | 64.69 |
| AchV4_0005 | Prohead protease | 45.031 | 23 | 47.42 |
| AchV4_0006 | Head decoration protein | 14.105 | 8 | 91.79 |
| AchV4_0007 | Major capsid protein | 38.315 | 65 | 93.95 |
| AchV4_0008 | Virion structural protein | 12.299 | 24 | 82.46 |
| AchV4_0009 | Head closure protein | 14.922 | 12 | 81.06 |
| AchV4_0010 | Neck protein | 22.423 | 17 | 74.63 |
| AchV4_0012 | Major tail protein | 28.341 | 31 | 75.77 |
| AchV4_0015 | Tape measure protein | 167.818 | 232 | 83.54 |
| AchV4_0016 | Virion structural protein | 36.358 | 12 | 61.79 |
| AchV4_0017 | Virion structural protein | 33.351 | 20 | 61.86 |
| AchV4_0019 | Virion structural protein | 57.374 | 24 | 50.58 |
| AchV4_0020 | Virion structural protein | 35.315 | 27 | 61.76 |
| AchV4_0021 | Virion structural protein | 6.989 | 2 | 53.13 |
| AchV4_0023 | Virion structural protein | 97.678 | 53 | 69.37 |
| AchV4_0024 | Putative tail fiber protein | 26.079 | 13 | 90.83 |
| AchV4_0025 | Virion structural protein | 35.935 | 22 | 75.08 |
| AchV4_0026 | Virion structural protein | 37.575 | 14 | 35.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaliniene, L.; Noreika, A.; Kaupinis, A.; Valius, M.; Jurgelaitis, E.; Lazutka, J.; Meškienė, R.; Meškys, R. Analysis of a Novel Bacteriophage vB_AchrS_AchV4 Highlights the Diversity of Achromobacter Viruses. Viruses 2021, 13, 374. https://doi.org/10.3390/v13030374
Kaliniene L, Noreika A, Kaupinis A, Valius M, Jurgelaitis E, Lazutka J, Meškienė R, Meškys R. Analysis of a Novel Bacteriophage vB_AchrS_AchV4 Highlights the Diversity of Achromobacter Viruses. Viruses. 2021; 13(3):374. https://doi.org/10.3390/v13030374
Chicago/Turabian StyleKaliniene, Laura, Algirdas Noreika, Algirdas Kaupinis, Mindaugas Valius, Edvinas Jurgelaitis, Justas Lazutka, Rita Meškienė, and Rolandas Meškys. 2021. "Analysis of a Novel Bacteriophage vB_AchrS_AchV4 Highlights the Diversity of Achromobacter Viruses" Viruses 13, no. 3: 374. https://doi.org/10.3390/v13030374