
Design, Synthesis and Characterization of HIV-1 CA-Targeting Small Molecules: Conformational Restriction of PF74

 , , ,
, , , 
Abstract
:
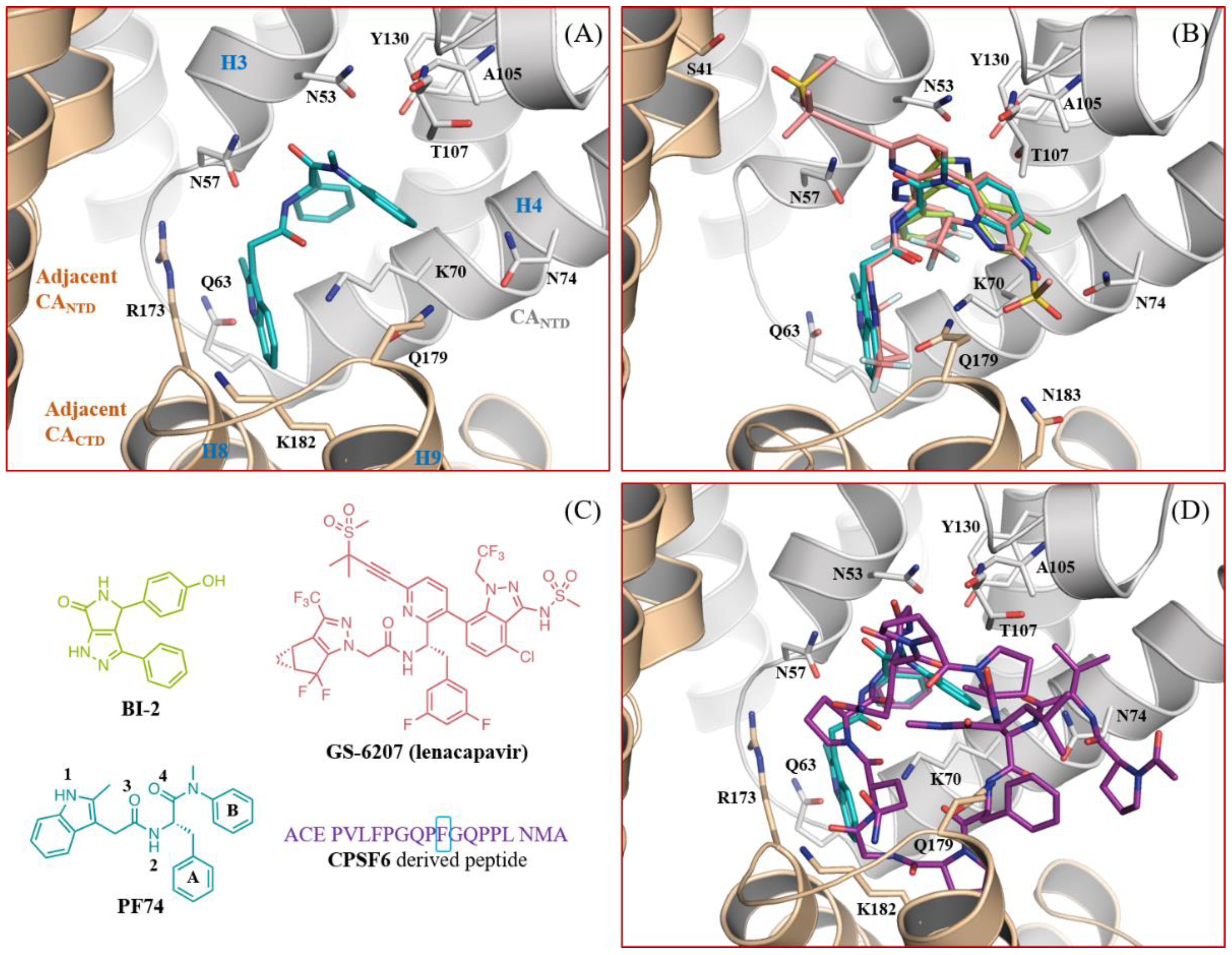
1. Introduction
2. Materials and Methods
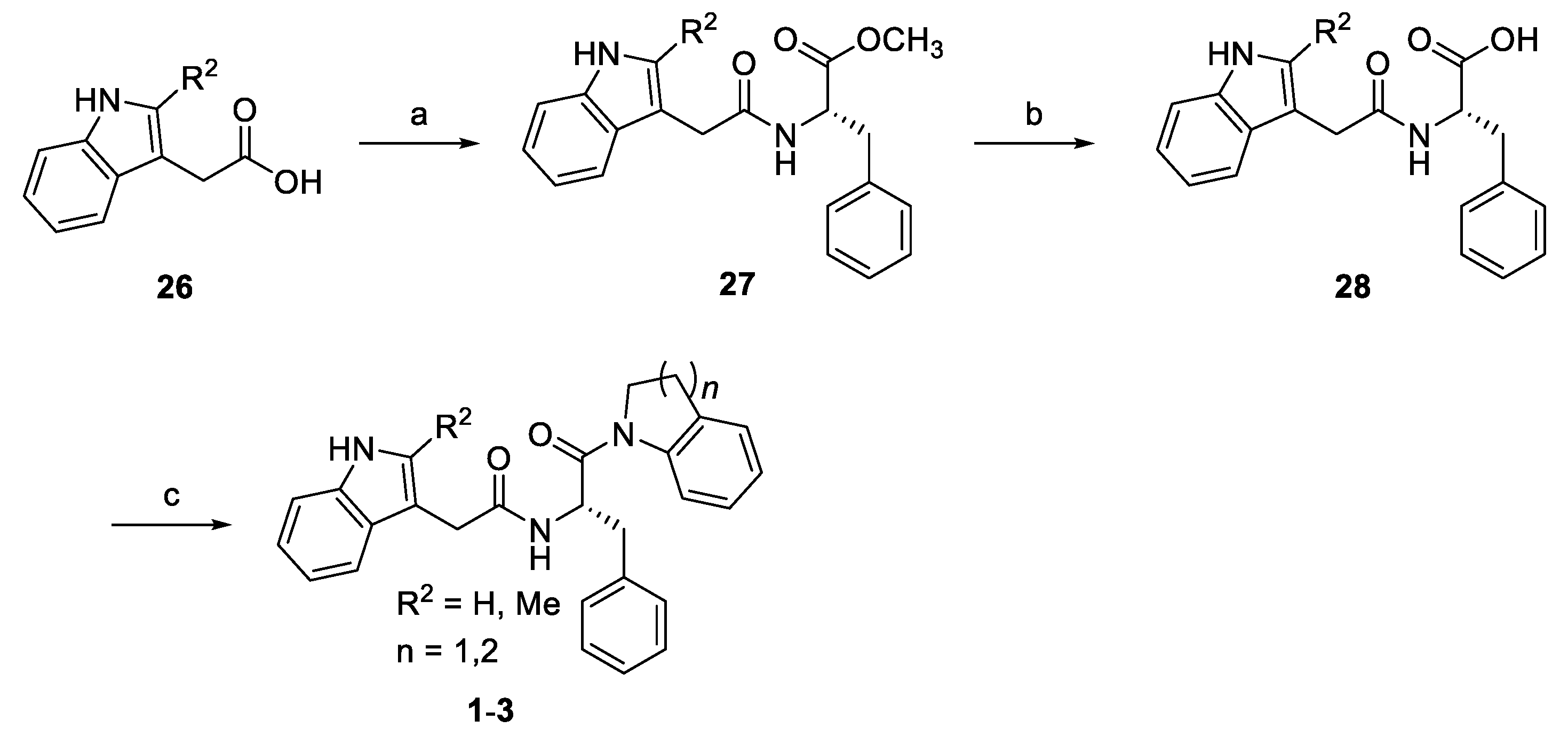
2.1. Chemistry
2.2. Cells
2.3. Method Details
2.3.1. Thermal Shift Assays (TSAs) to Screen Compounds for Effect on HIV-1 CA Hexamer Stability
2.3.2. Virus Production
2.3.3. Anti-HIV-1 and Cytotoxicity Assays
2.4. Metabolic Stability Assay
2.5. Molecular Modeling
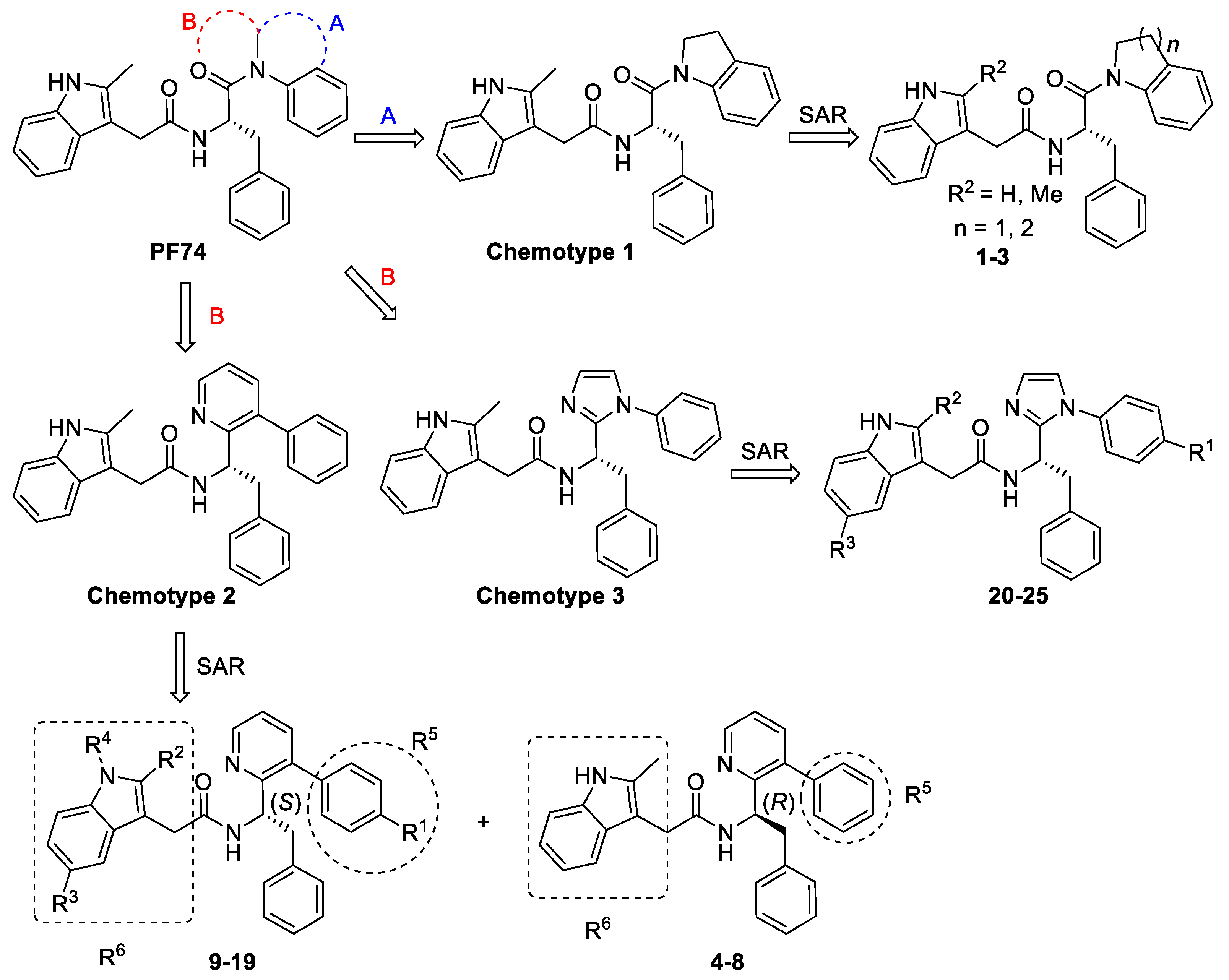
3. Results
3.1. The Indoline and Quinoline Analogs (Chemotype 1)
3.2. SAR of the Pyridine Series (Chemotype 2)
3.3. SAR of the Imidazole Series (Chemotype 3)
3.4. Metabolic Stability
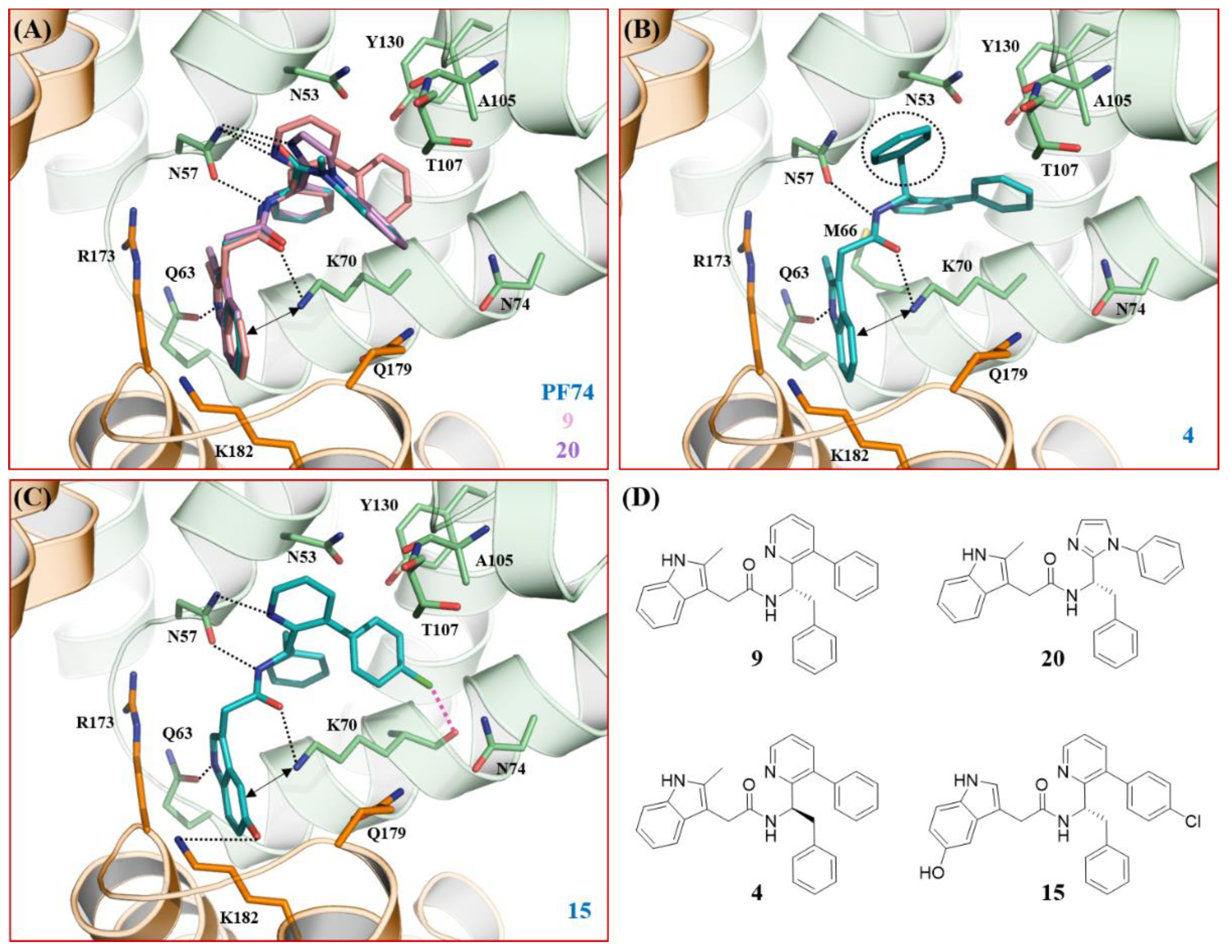
3.5. Molecular Modeling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Novikova, M.; Zhang, Y.; Freed, E.O.; Peng, K. Multiple Roles of HIV-1 Capsid during the Virus Replication Cycle. Virol. Sin. 2019, 34, 119–134. [Google Scholar] [CrossRef] [Green Version]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef]
- Buffone, C.; Martinez-Lopez, A.; Fricke, T.; Opp, S.; Severgnini, M.; Cifola, I.; Petiti, L.; Frabetti, S.; Skorupka, K.; Zadrozny, K.K.; et al. Nup153 Unlocks the Nuclear Pore Complex for HIV-1 Nuclear Translocation in Nondividing Cells. J. Virol. 2018, 92, e00648-18. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.R.; Perreira, J.M.; Savidis, G.; Portmann, J.M.; Aker, A.M.; Feeley, E.M.; Smith, M.C.; Brass, A.L. Direct Visualization of HIV-1 Replication Intermediates Shows that Capsid and CPSF6 Modulate HIV-1 Intra-nuclear Invasion and Integration. Cell Rep. 2015, 13, 1717–1731. [Google Scholar] [CrossRef] [Green Version]
- Bejarano, D.A.; Peng, K.; Laketa, V.; Börner, K.; Jost, K.L.; Lucic, B.; Glass, B.; Lusic, M.; Müller, B.; Kräusslich, H.-G. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the nuclear pore complex. eLife 2019, 8, e41800. [Google Scholar] [CrossRef]
- Sowd, G.A.; Serrao, E.; Wang, H.; Wang, W.; Fadel, H.J.; Poeschla, E.M.; Engelman, A.N. A critical role for alternative polyadenylation factor CPSF6 in targeting HIV-1 integration to transcriptionally active chromatin. Proc. Natl. Acad. Sci. USA 2016, 113, E1054–E1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S.; et al. Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration. Cell Host Microbe 2018, 24, 392–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Dauphin, A.; Komurlu, S.; McCauley, S.M.; Yurkovetskiy, L.; Carbone, C.; Diehl, W.E.; Strambio-De-Castillia, C.; Campbell, E.M.; Luban, J. Cyclophilin A protects HIV-1 from restriction by human TRIM5α. Nat. Microbiol. 2019, 4, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Carnes, S.K.; Sheehan, J.H.; Aiken, C. Inhibitors of the HIV-1 capsid, a target of opportunity. Curr. Opin. HIV AIDS 2018, 13, 359–365. [Google Scholar] [CrossRef]
- Lamorte, L.; Titolo, S.; Lemke, C.T.; Goudreau, N.; Mercier, J.-F.; Wardrop, E.; Shah, V.B.; von Schwedler, U.K.; Langelier, C.; Banik, S.S.R.; et al. Discovery of Novel Small-Molecule HIV-1 Replication Inhibitors That Stabilize Capsid Complexes. Antimicrob. Agents Chemother. 2013, 57, 4622–4631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, C.T.; Titolo, S.; von Schwedler, U.; Goudreau, N.; Mercier, J.-F.; Wardrop, E.; Faucher, A.-M.; Coulombe, R.; Banik, S.S.R.; Fader, L.; et al. Distinct Effects of Two HIV-1 Capsid Assembly Inhibitor Families That Bind the Same Site within the N-Terminal Domain of the Viral CA Protein. J. Virol. 2012, 86, 6643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, B.N.; Kyere, S.; Kinde, I.; Tang, C.; Howard, B.R.; Robinson, H.; Sundquist, W.I.; Summers, M.F.; Hill, C.P. Structure of the Antiviral Assembly Inhibitor CAP-1 Complex with the HIV-1 CA Protein. J. Mol. Biol. 2007, 373, 355–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, W.S.; Pickford, C.; Irving, S.L.; Brown, D.G.; Anderson, M.; Bazin, R.; Cao, J.; Ciaramella, G.; Isaacson, J.; Jackson, L.; et al. HIV Capsid is a Tractable Target for Small Molecule Therapeutic Intervention. PLoS Pathog. 2010, 6, e1001220. [Google Scholar] [CrossRef] [Green Version]
- Link, J.O.; Rhee, M.S.; Tse, W.C.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D.; et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Bester, S.M.; Wei, G.; Zhao, H.; Adu-Ampratwum, D.; Iqbal, N.; Courouble, V.V.; Francis, A.C.; Annamalai, A.S.; Singh, P.K.; Shkriabai, N.; et al. Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science 2020, 370, 360. [Google Scholar] [CrossRef]
- Pornillos, O.; Ganser-Pornillos, B.K.; Yeager, M. Atomic-level modelling of the HIV capsid. Nature 2011, 469, 424–427. [Google Scholar] [CrossRef]
- Gres, A.T.; Kirby, K.A.; KewalRamani, V.N.; Tanner, J.J.; Pornillos, O.; Sarafianos, S.G. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 2015, 349, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, A.J.; Jacques, D.A.; McEwan, W.A.; Fletcher, A.J.; Essig, S.; Chin, J.W.; Halambage, U.D.; Aiken, C.; James, L.C. Host Cofactors and Pharmacologic Ligands Share an Essential Interface in HIV-1 Capsid That Is Lost upon Disassembly. PLoS Pathog. 2014, 10, e1004459. [Google Scholar] [CrossRef]
- Kleinpeter, A.B.; Freed, E.O. HIV-1 Maturation: Lessons Learned from Inhibitors. Viruses 2020, 12, 940. [Google Scholar] [CrossRef]
- Wang, L.; Casey, M.C.; Vernekar, S.K.V.; Do, H.T.; Sahani, R.L.; Kirby, K.A.; Du, H.; Hachiya, A.; Zhang, H.; Tedbury, P.R.; et al. Chemical profiling of HIV-1 capsid-targeting antiviral PF74. Eur. J. Med. Chem. 2020, 200, 112427. [Google Scholar] [CrossRef]
- Vernekar, S.K.V.; Sahani, R.L.; Casey, M.C.; Kankanala, J.; Wang, L.; Kirby, K.A.; Du, H.; Zhang, H.; Tedbury, P.R.; Xie, J.; et al. Toward Structurally Novel and Metabolically Stable HIV-1 Capsid-Targeting Small Molecules. Viruses 2020, 12, 452. [Google Scholar] [CrossRef]
- Wang, L.; Casey, M.C.; Vernekar, S.K.V.; Sahani, R.L.; Kankanala, J.; Kirby, K.A.; Du, H.; Hachiya, A.; Zhang, H.; Tedbury, P.R.; et al. Novel HIV-1 capsid-targeting small molecules of the PF74 binding site. Eur. J. Med. Chem. 2020, 204, 112626. [Google Scholar] [CrossRef]
- Wang, L.; Casey, M.C.; Vernekar, S.K.V.; Sahani, R.L.; Kirby, K.A.; Du, H.; Zhang, H.; Tedbury, P.R.; Xie, J.; Sarafianos, S.G.; et al. Novel PF74-like small molecules targeting the HIV-1 capsid protein: Balance of potency and metabolic stability. Acta Pharm. Sin. B 2020. [Google Scholar] [CrossRef]
- Chang, C.E.A.; Chen, W.; Gilson, M.K. Ligand configurational entropy and protein binding. Proc. Natl. Acad. Sci. USA 2007, 104, 1534. [Google Scholar] [CrossRef] [Green Version]
- Lawson, A.D.G.; MacCoss, M.; Heer, J.P. Importance of Rigidity in Designing Small Molecule Drugs To Tackle Protein–Protein Interactions (PPIs) through Stabilization of Desired Conformers. J. Med. Chem. 2018, 61, 4283–4289. [Google Scholar] [CrossRef] [PubMed]
- Borsari, C.; Rageot, D.; Dall’Asen, A.; Bohnacker, T.; Melone, A.; Sele, A.M.; Jackson, E.; Langlois, J.-B.; Beaufils, F.; Hebeisen, P.; et al. A Conformational Restriction Strategy for the Identification of a Highly Selective Pyrimido-pyrrolo-oxazine mTOR Inhibitor. J. Med. Chem. 2019, 62, 8609–8630. [Google Scholar] [CrossRef]
- Kedderis, G.L.; Hollenberg, P.F. Peroxidase-catalyzed N-demethylation reactions: Deuterium solvent isotope effects. Biochemistry 1985, 24, 6158–6163. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, P.F.; Miwa, G.T.; Walsh, J.S.; Dwyer, L.A.; Rickert, D.E.; Kedderis, G.L. Mechanisms of N-demethylation reactions catalyzed by cytochrome P-450 and peroxidases. Drug Metab. Dispos. 1985, 13, 272. [Google Scholar] [PubMed]
- Bondy, S.S.; Cannizzaro, C.E.; Chou, C.-H.; Halcomb, R.L.; Hu, E.Y.; Link, J.O.; Liu, Q.; Schroeder, S.D.; Tse, W.C.; Zhang, J.R. Compounds for the treatment of HIV. Patent WO 2013/006738 A1, 1 October 2013. [Google Scholar]
- Belema, M.; Bender, J.A.; Beno, B.; Gentles, R.G.; Li, G.; Meanwell, N.A.; Pendri, A.; Yang, Z.; Zhu, S. Inhibitors of Human Immunodeficiency Virus Replication. U.S. Patent 2017/0304239 A1, 27 November 2017. [Google Scholar]
- Lange, M.J.; Lyddon, T.D.; Johnson, M.C. Diphtheria Toxin A-Resistant Cell Lines Enable Robust Production and Evaluation of DTA-Encoding Lentiviruses. Sci. Rep. 2019, 9, 8985. [Google Scholar] [CrossRef] [Green Version]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Pornillos, O.; Ganser-Pornillos, B.K.; Kelly, B.N.; Hua, Y.; Whitby, F.G.; Stout, C.D.; Sundquist, W.I.; Hill, C.P.; Yeager, M. X-Ray Structures of the Hexameric Building Block of the HIV Capsid. Cell 2009, 137, 1282–1292. [Google Scholar] [CrossRef] [Green Version]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986, 59, 284. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger. Schrödinger Small-Molecule Drug Discovery Suite 2019-4; Schrödinger, LLC: New York, NY, USA, 2019. [Google Scholar]
- Schrödinger. Schrödinger Release 2019-4: Maestro; Schrödinger, LLC: New York, NY, USA, 2019. [Google Scholar]
- Schrödinger. The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC: New York, NY, USA, 2019. [Google Scholar]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Gallazzi, F.; Hill, K.J.; Burke, D.H.; Lange, M.J.; Quinn, T.P.; Neogi, U.; Sönnerborg, A. GS-CA Compounds: First-In-Class HIV-1 Capsid Inhibitors Covering Multiple Grounds. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Xu, J.P.; Francis, A.C.; Meuser, M.E.; Mankowski, M.; Ptak, R.G.; Rashad, A.A.; Melikyan, G.B.; Cocklin, S. Exploring Modifications of an HIV-1 Capsid Inhibitor: Design, Synthesis, and Mechanism of Action. J. Drug Des. Res. 2018, 5, 1070. [Google Scholar] [PubMed]
- Sun, L.; Huang, T.; Dick, A.; Meuser, M.E.; Zalloum, W.A.; Chen, C.-H.; Ding, X.; Gao, P.; Cocklin, S.; Lee, K.-H.; et al. Design, synthesis and structure-activity relationships of 4-phenyl-1H-1,2,3-triazole phenylalanine derivatives as novel HIV-1 capsid inhibitors with promising antiviral activities. Eur. J. Med. Chem. 2020, 190, 112085. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zalloum, W.A.; Meuser, M.E.; Jing, L.; Kang, D.; Chen, C.-H.; Tian, Y.; Zhang, F.; Cocklin, S.; Lee, K.-H.; et al. Discovery of phenylalanine derivatives as potent HIV-1 capsid inhibitors from click chemistry-based compound library. Eur. J. Med. Chem. 2018, 158, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Wacher, V.J.; Silverman, J.A.; Zhang, Y.; Benet, L.Z. Role of P-Glycoprotein and Cytochrome P450 3A in Limiting Oral Absorption of Peptides and Peptidomimetics. J. Pharm. Sci. 1998, 87, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, T.; Vidova, V.; Bienertova-Vasku, J.; Janku, P.; Almasi, M.; Klanova, J.; Spacil, Z. Urinary intermediates of tryptophan as indicators of the gut microbial metabolism. Anal. Chim. Acta 2017, 987, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Höglund, E.; Øverli, Ø.; Winberg, S. Tryptophan Metabolic Pathways and Brain Serotonergic Activity: A Comparative Review. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Hesse, L.M.; Venkatakrishnan, K.; von Moltke, L.L.; Shader, R.I.; Greenblatt, D.J. CYP3A4 Is the Major CYP Isoform Mediating the in Vitro Hydroxylation and Demethylation of Flunitrazepam. Drug Metab. Dispos. 2001, 29, 133. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | Structure | EC50 (μM) a | CC50 (μM) b | TSA ∆Tm (°C) c |
|---|---|---|---|---|
| PF74 |  | 0.70 ± 0.07 | 76 ± 9 | 6.9 ± 0.5 |
| 1 |  | >20 | >100 | −0.5 |
| 2 |  | >20 | >100 | −1.0 |
| 3 |  | >20 | >50 | 1.8 ± 1.2 |
| Cpd d | Structure | EC50 (μM) a | CC50 (μM) b | TSA ∆Tm (°C) c |
|---|---|---|---|---|
| PF74 |  | 0.70 ± 0.07 | 76 ± 9 | 6.9 ± 0.5 |
| 4 |  | >20 | <50 | 0.1 ± 0.1 |
| 5 |  | >20 | <50 | 0.1 ± 0.07 |
| 6 |  | >15 | >50 | 0.3 ± 0.1 |
| 7 |  | >20 | >50 | 0 ± 0 |
| 8 |  | >20 | >50 | 0.1 ± 0.1 |
| 9 |  | 2.6 ± 0.4 | <50 | 4.5 ± 0.0 |
| 10 |  | 8.8 ± 0.4 | <50 | 1.0 ± 0.07 |
| 11 |  | 5.1 ± 0.7 | >50 | 1.1 ± 0.07 |
| 12 |  | >20 | >50 | −0.2 ± 0.07 |
| 13 |  | 0.79 ± 0.25 | 22 ± 2 | 7.7 ± 0.2 |
| 14 |  | 1.4 ± 0.7 | 39 ± 0.2 | 7.8 ± 0.3 |
| 15 d |  | 0.31 ± 0.07 | 44 ± 1 | 8.7 ± 0.3 |
| 16 |  | 2.7 ± 0.2 | 41 ± 0.1 | 6.6 ± 0.2 |
| 17 |  | 3.6 ± 0.2 | 48 ± 6 | 5.9 ± 0.2 |
| 18 |  | 2.1 ± 0.8 | 40 ± 4 | 6.3 ± 0.6 |
| 19 |  | 3.2 ± 0.3 | 41 ± 2 | 6.3 ± 0.1 |
| Cpd | Structure | EC50 (μM) a | CC50 (μM) b | TSA ∆Tm (°C) c |
|---|---|---|---|---|
| PF74 |  | 0.70 ± 0.07 | 76 ± 9 | 6.9 ± 0.5 |
| 20 |  | 3.7 ± 0.8 | >50 | 3.9 ± 0.4 |
| 21 |  | 6.6 ± 0.4 | >50 | 2.2 ± 0.0 |
| 22 |  | >20 | >50 | −0.3 ± 0.1 |
| 23 |  | 3.3 ± 0.03 | 93 ± 2 | 4.0 ± 0.3 |
| 24 |  | 3.5 ± 0.3 | >100 | 2.8 ± 0.3 |
| 25 |  | 3.3 ± 0.2 | >100 | 2.7 ± 0.1 |
| Cpd | HLMs a | MLMs b | Metabolite (s) |
|---|---|---|---|
| PF74 | 0.7 c | 0.6 c | -- |
| 9 | 1.2 ± 0.04 | 1.0 ± 0.01 | -- |
| 14 | 1.7 ± 0.02 | 0.9 ± 0.004 | 15 |
| 15 | 27.2 ± 0.5 | 3.1 ± 0.3 | -- |
| 16 | 1.4 ± 0.02 | 1.4 ± 0.02 | 15 |
| 17 | 1.3 ± 0.01 | 1.3 ± 0.005 | 15 and 14 |
| 20 | 0.9 ± 0.004 | 0.7 ± 0.01 | -- |
| 22 | 1.3 ± 0.01 | 0.9 ± 0.01 | -- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahani, R.L.; Diana-Rivero, R.; Vernekar, S.K.V.; Wang, L.; Du, H.; Zhang, H.; Castaner, A.E.; Casey, M.C.; Kirby, K.A.; Tedbury, P.R.; et al. Design, Synthesis and Characterization of HIV-1 CA-Targeting Small Molecules: Conformational Restriction of PF74. Viruses 2021, 13, 479. https://doi.org/10.3390/v13030479
Sahani RL, Diana-Rivero R, Vernekar SKV, Wang L, Du H, Zhang H, Castaner AE, Casey MC, Kirby KA, Tedbury PR, et al. Design, Synthesis and Characterization of HIV-1 CA-Targeting Small Molecules: Conformational Restriction of PF74. Viruses. 2021; 13(3):479. https://doi.org/10.3390/v13030479
Chicago/Turabian StyleSahani, Rajkumar Lalji, Raquel Diana-Rivero, Sanjeev Kumar V. Vernekar, Lei Wang, Haijuan Du, Huanchun Zhang, Andres Emanuelli Castaner, Mary C. Casey, Karen A. Kirby, Philip R. Tedbury, and et al. 2021. "Design, Synthesis and Characterization of HIV-1 CA-Targeting Small Molecules: Conformational Restriction of PF74" Viruses 13, no. 3: 479. https://doi.org/10.3390/v13030479
APA StyleSahani, R. L., Diana-Rivero, R., Vernekar, S. K. V., Wang, L., Du, H., Zhang, H., Castaner, A. E., Casey, M. C., Kirby, K. A., Tedbury, P. R., Xie, J., Sarafianos, S. G., & Wang, Z. (2021). Design, Synthesis and Characterization of HIV-1 CA-Targeting Small Molecules: Conformational Restriction of PF74. Viruses, 13(3), 479. https://doi.org/10.3390/v13030479