
Genome Sequence of the Bacteriophage CL31 and Interaction with the Host Strain Corynebacterium glutamicum ATCC 13032


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids and Growth Conditions
2.2. Recombinant DNA Work and Construction of Chromosomal Insertions or Deletions
2.3. Phage Propagation
2.4. Phage Purification with Cesium Chloride Gradients
2.5. DNA Isolation
2.6. DNA Sequencing and Genome Assembly
2.7. Gene Prediction and Functional Annotation
2.8. Genomic Distance Analysis
2.9. Phage Classification
2.10. Lifestyle Prediction
2.11. Phage Infection Curves and Titer Determination
2.12. Transcriptome Analysis (RNA-seq)
2.12.1. Sample Preparation and Sequencing
2.12.2. Bioinformatic Transcriptome Analysis
2.13. Staining of CL31 for Fluorescence Microscopy
2.14. Scanning Electron Microscopy
2.15. Proteome Analysis
2.15.1. Sample Preparation for Proteome Analyses
2.15.2. In-Gel Digestion
2.15.3. Digestion of Whole Phage Particles
2.16. LC separation and Mass Spectrometry
2.16.1. Shotgun Proteomic Measurement
2.16.2. Trapping Conditions
2.16.3. Separation Conditions
2.16.4. IDA-MS
2.16.5. Data Analysis
3. Results
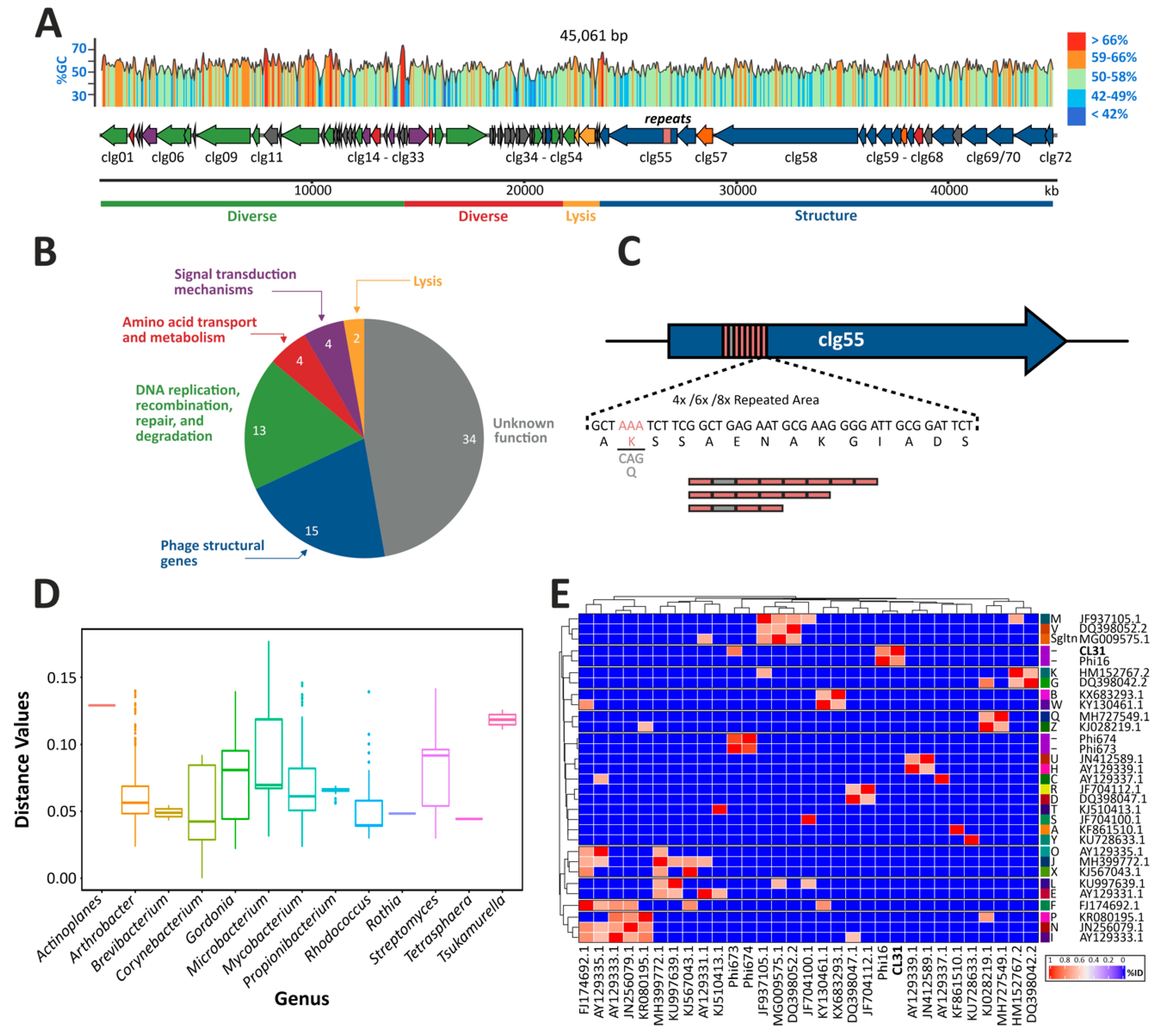
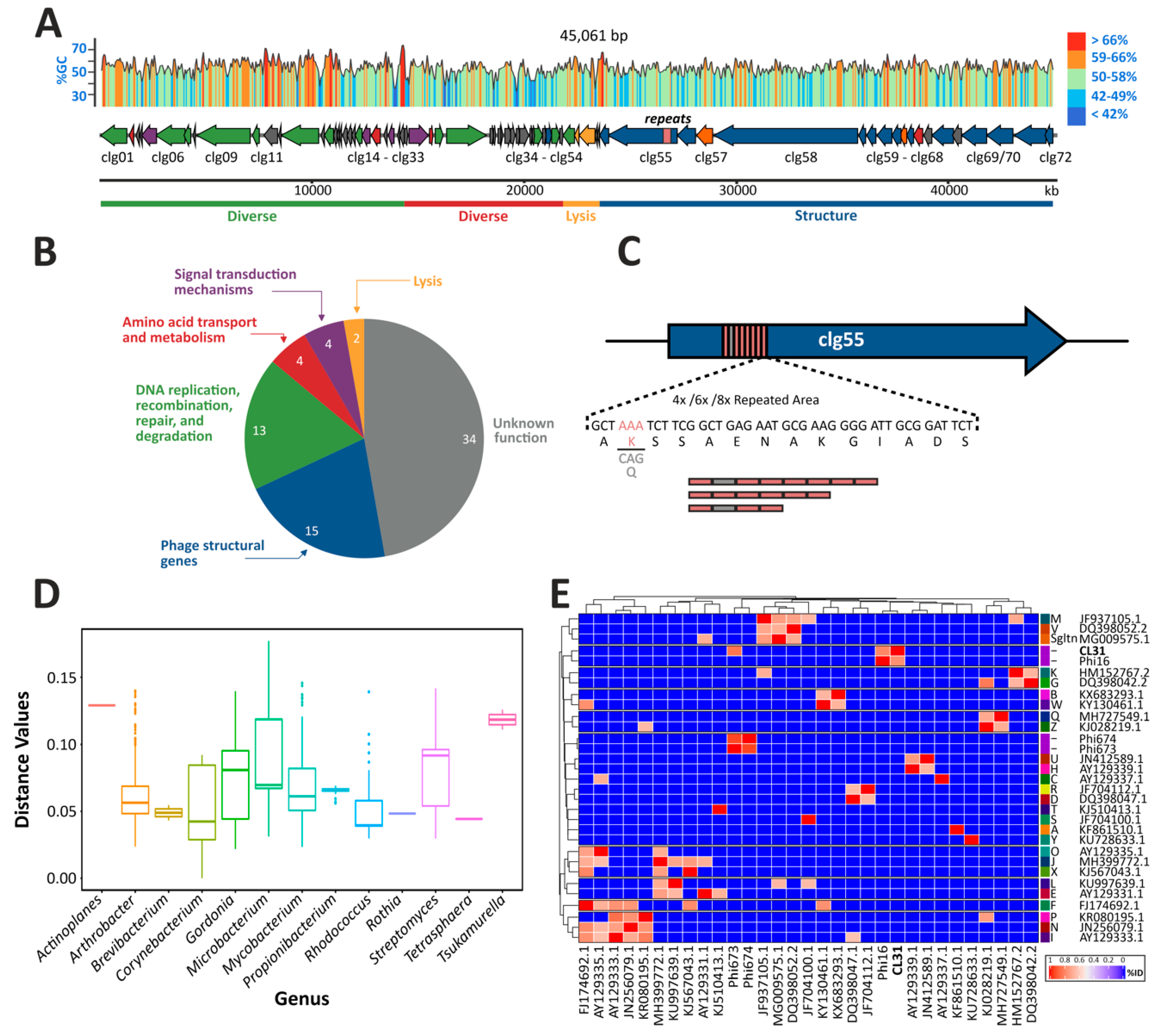
3.1. Genome Assembly, Annotation and Comparisons
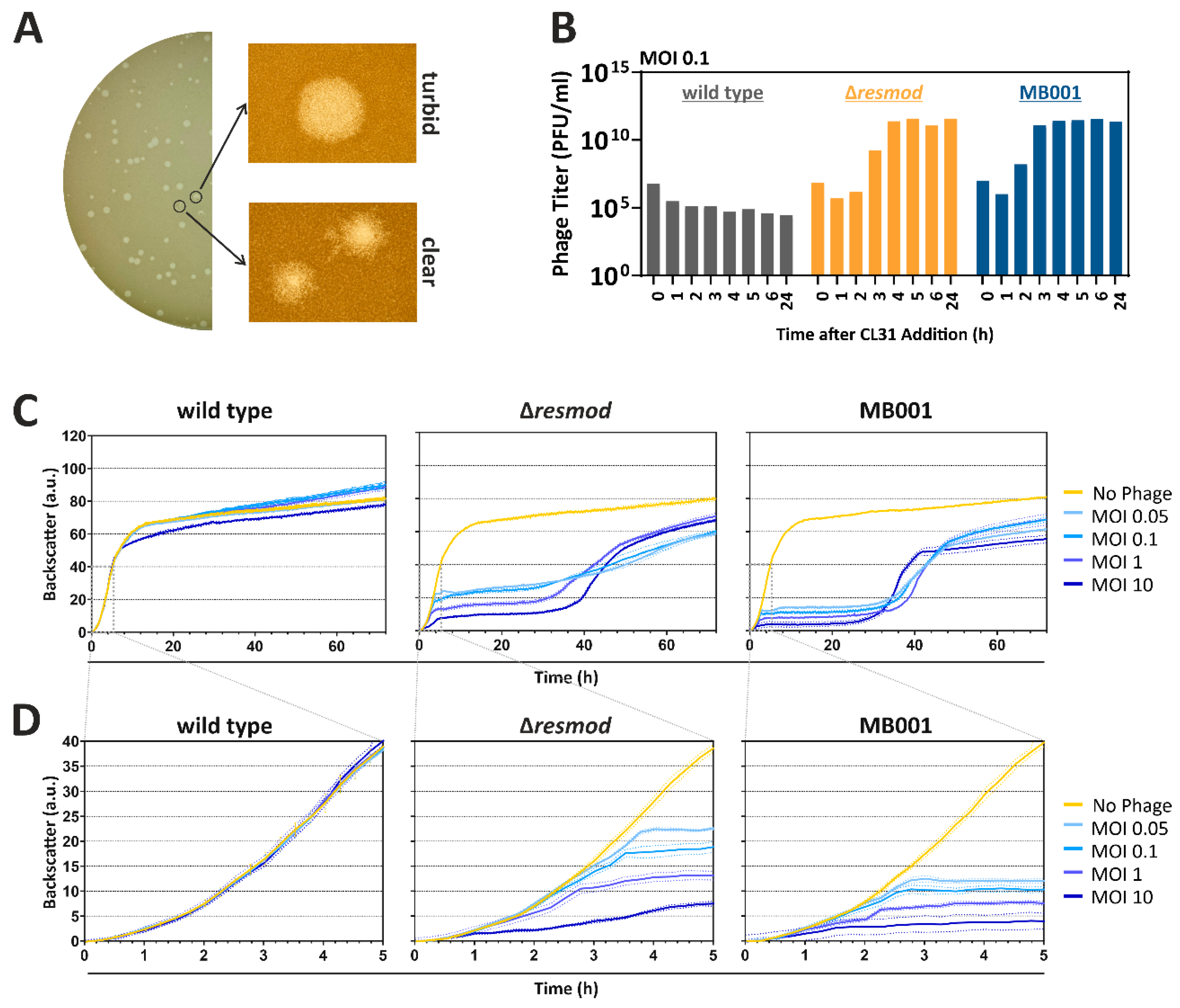
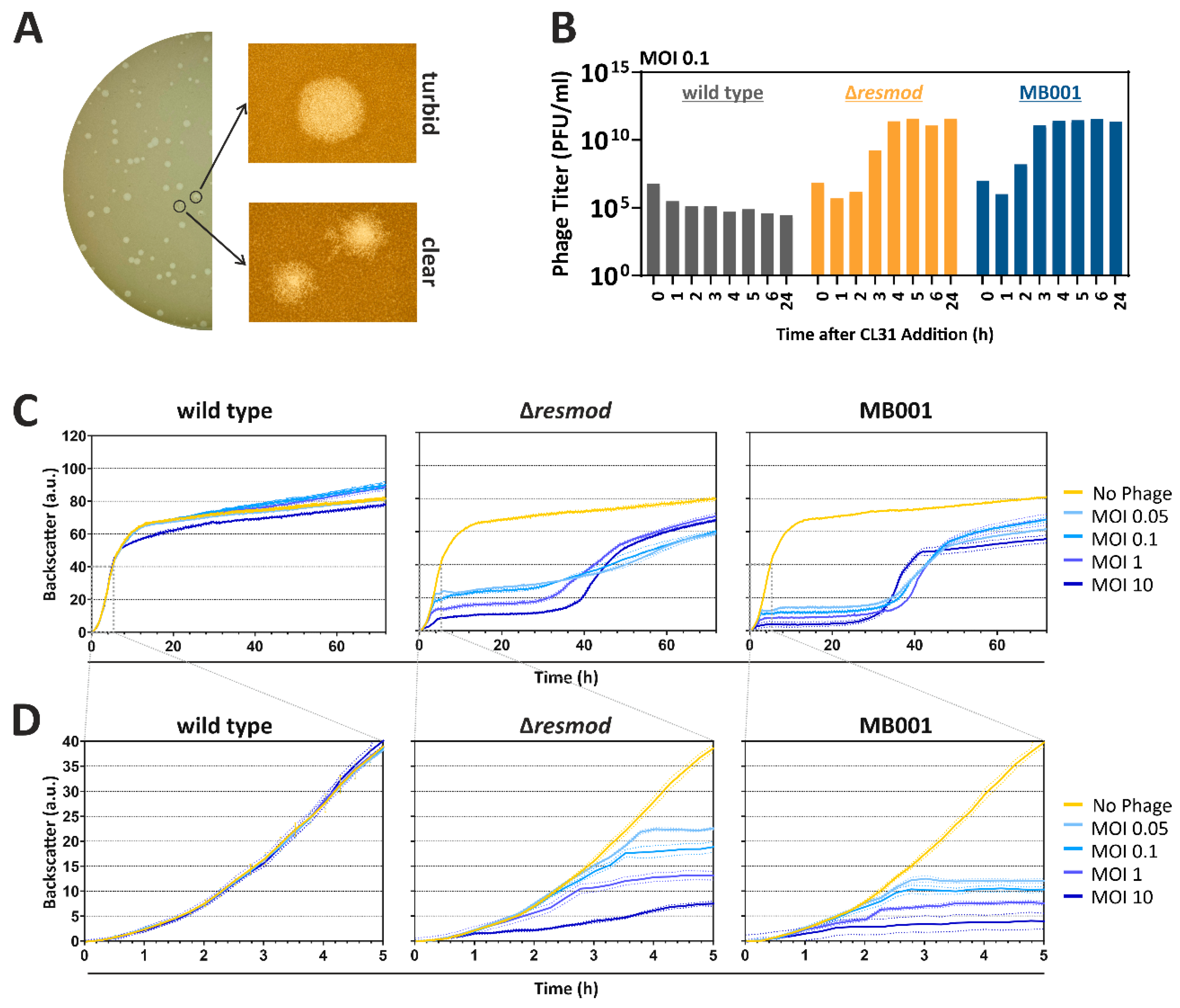
3.2. Plaque Morphologies and Infection Dynamics of CL31
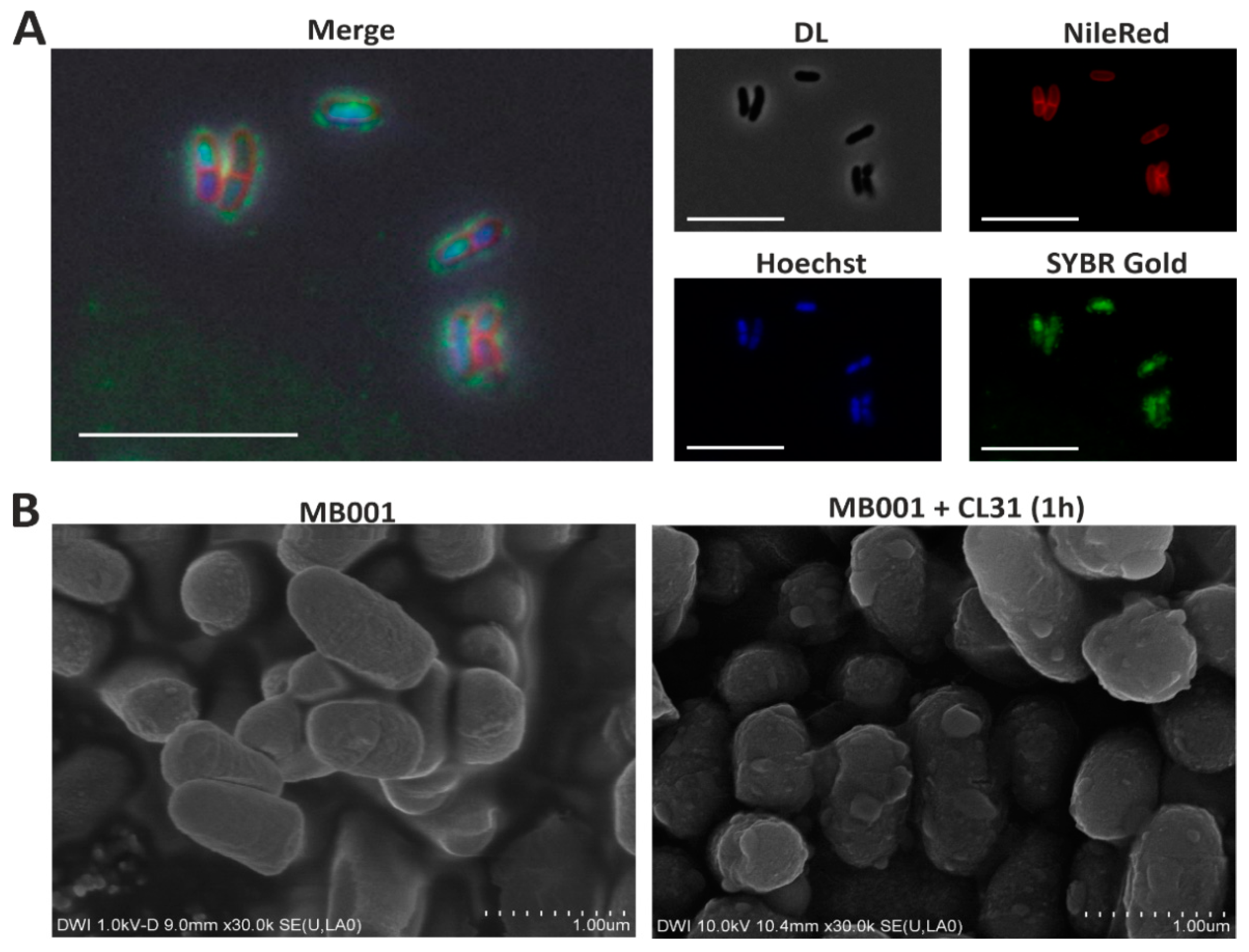
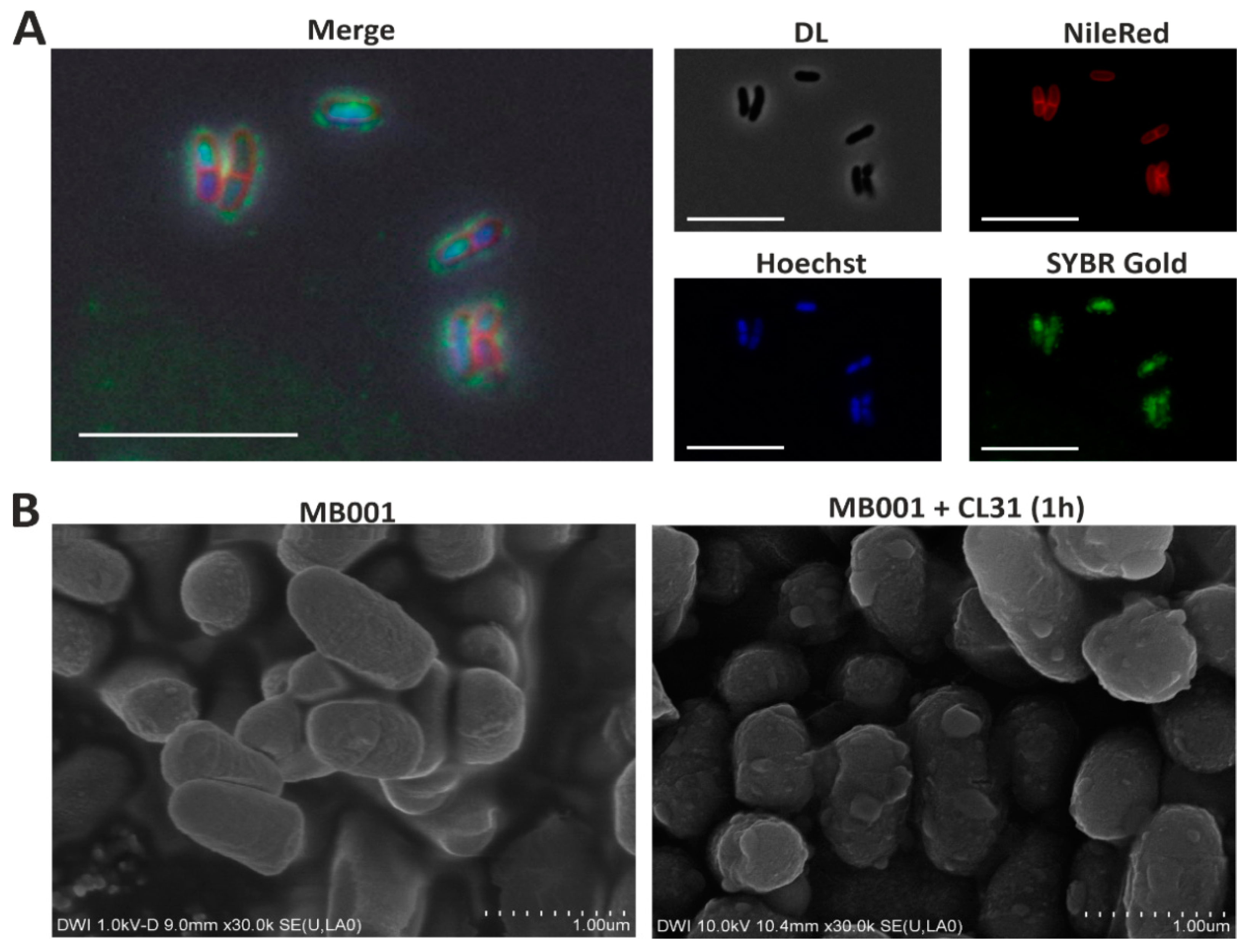
3.3. Visualization of CL31 Attachment and Lysis
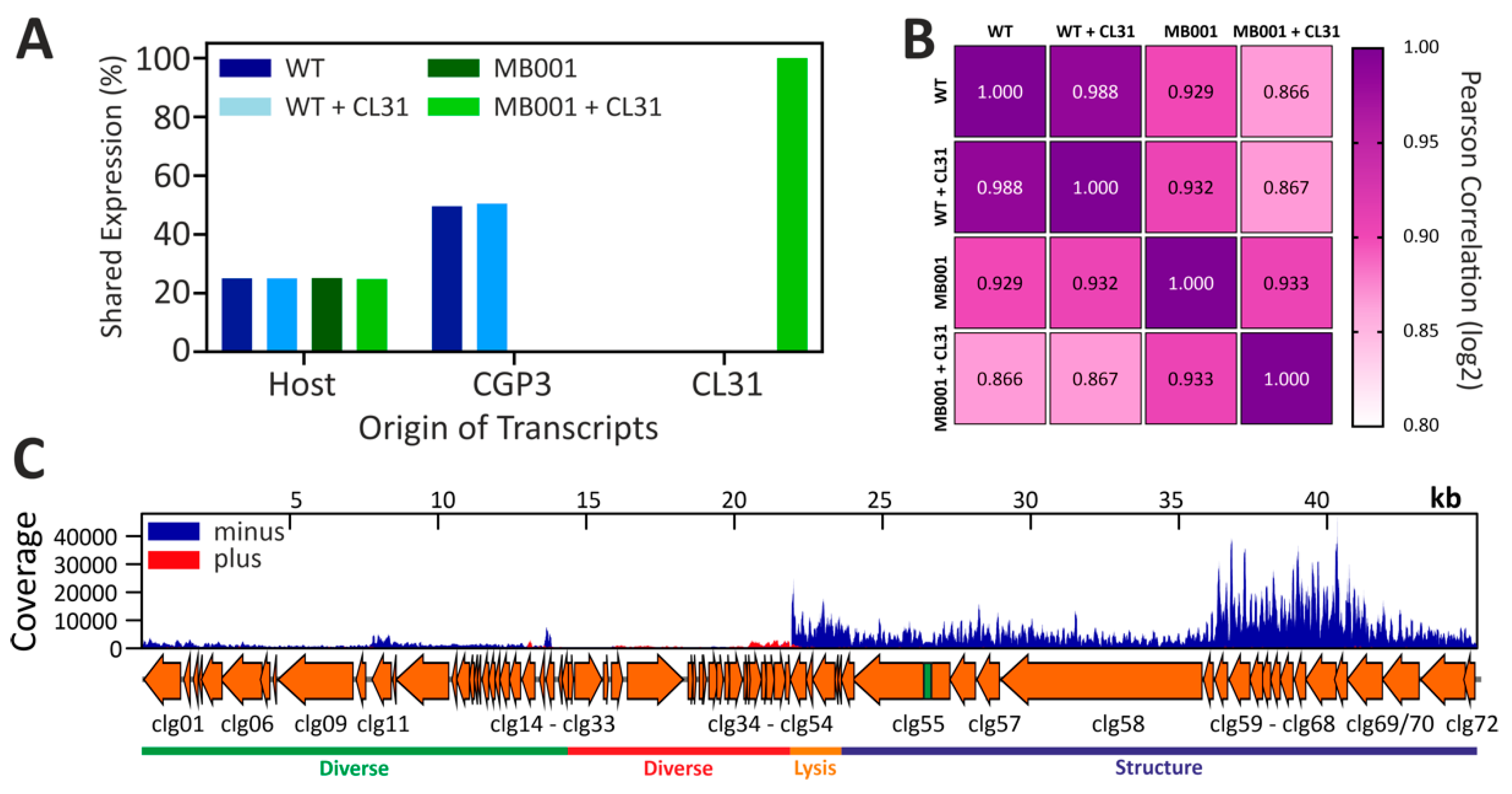
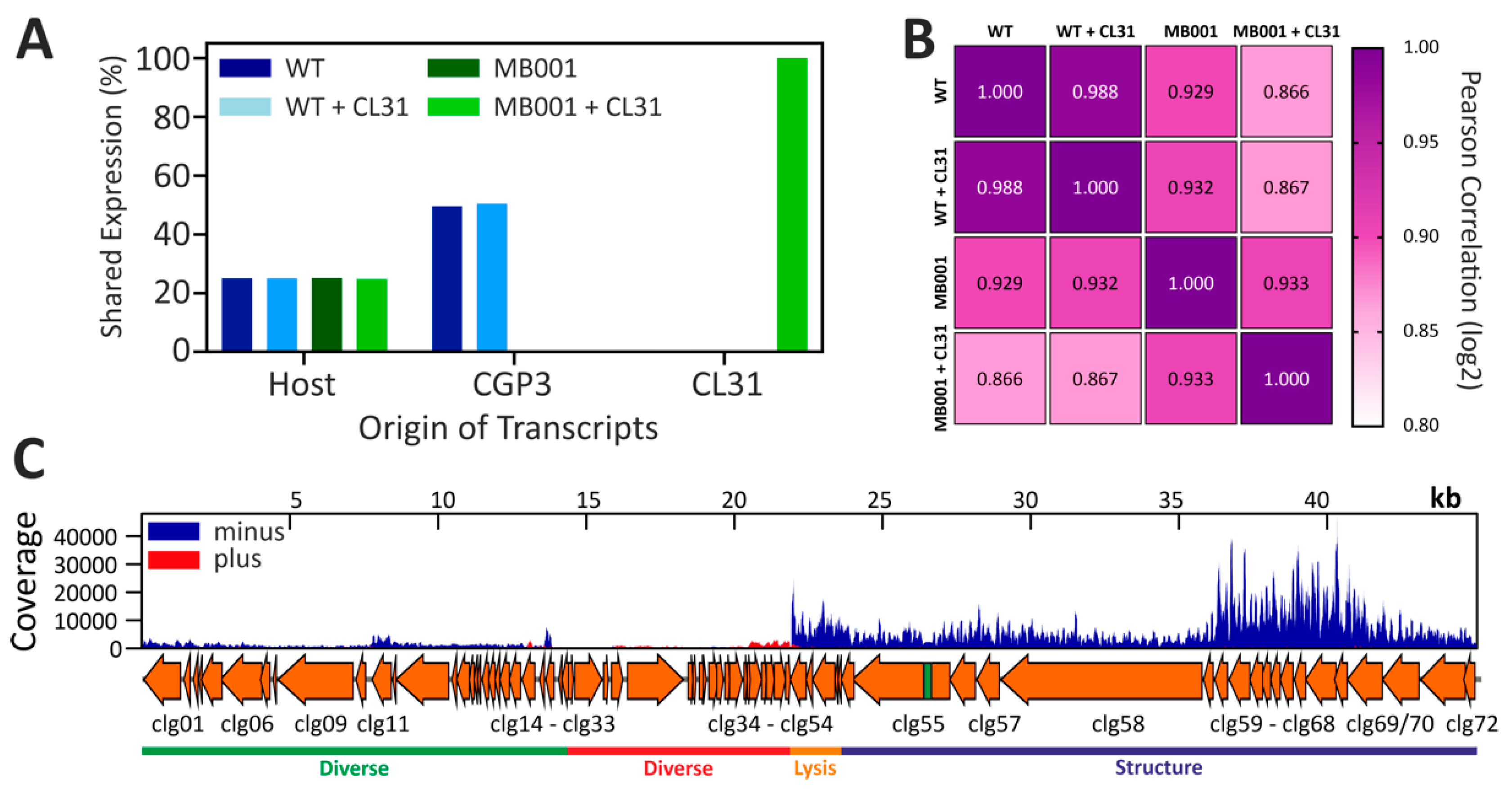
3.4. Global Transcriptome of C. glutamicum during Infection with CL31
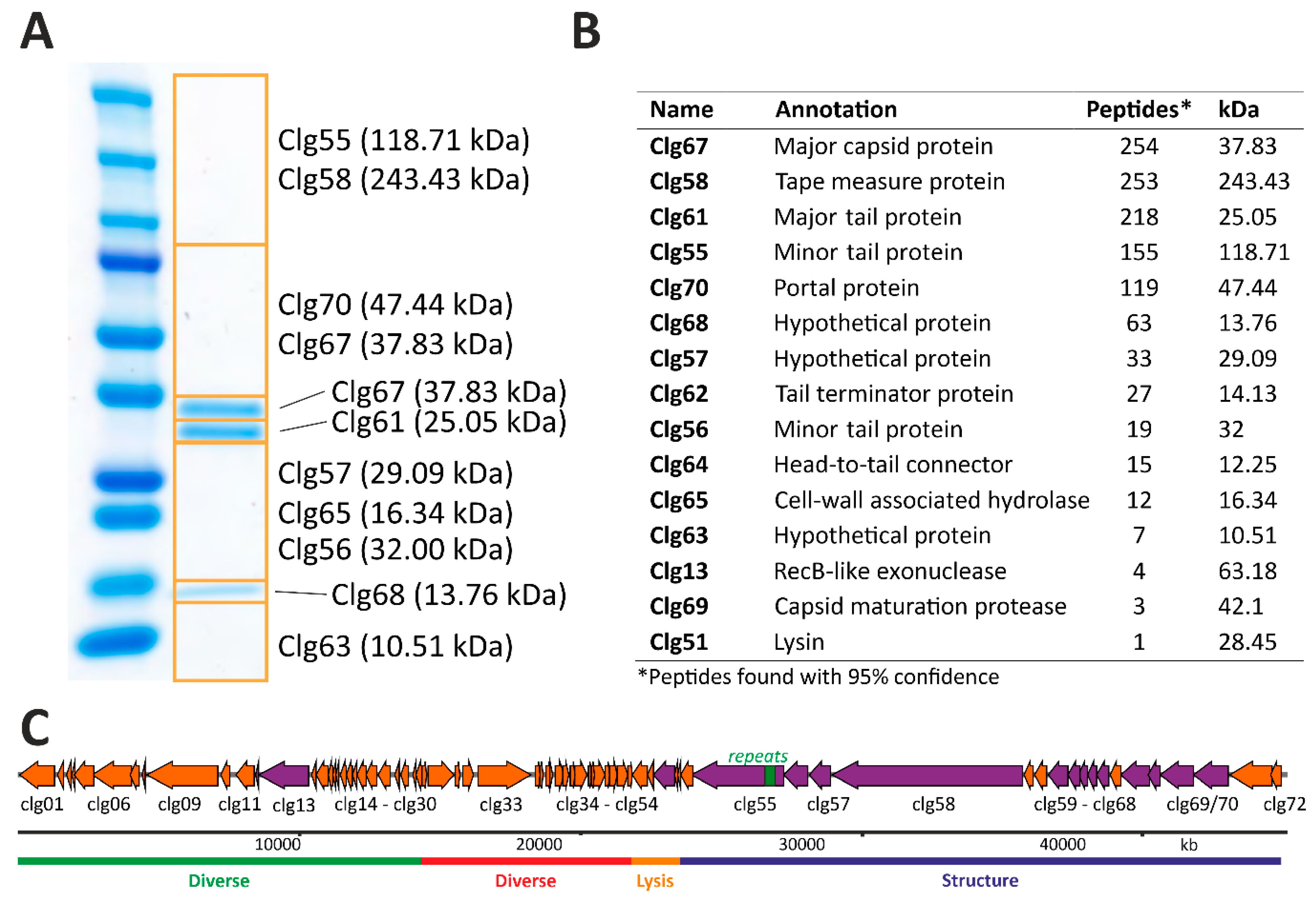
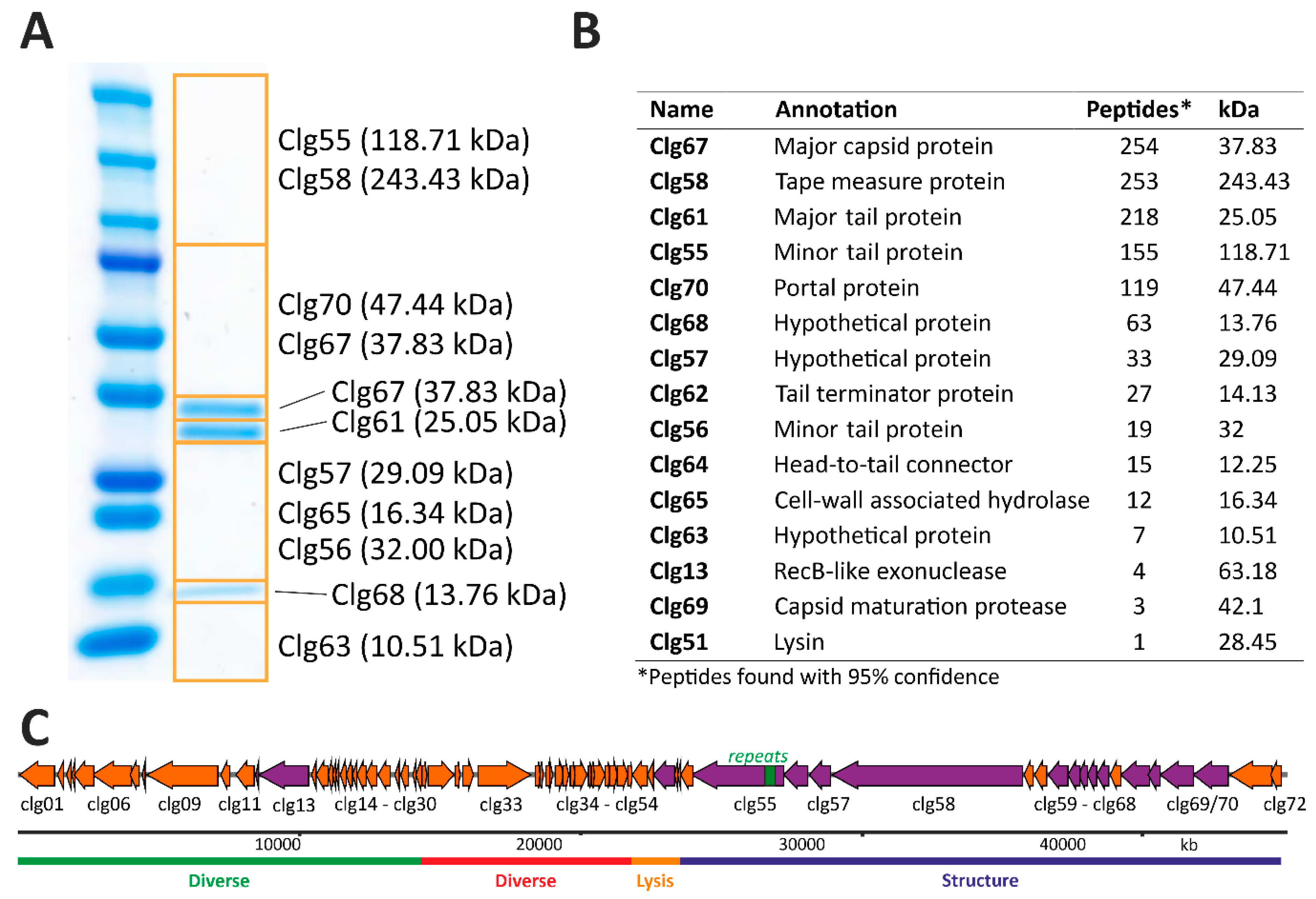
3.5. Structural Proteome Analysis of Purified CL31 Virions
3.6. Mutations in Genes Involved in Mycolic Acid Biosynthesis Lead to Resistance to Infection by CL31
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Becker, J.; Rohles, C.M.; Wittmann, C. Metabolically engineered Corynebacterium glutamicum for bio-based production of chemicals, fuels, materials, and healthcare products. Metab. Eng. 2018, 50, 122–141. [Google Scholar] [CrossRef]
- Kalinowski, J.; Bathe, B.; Bartels, D.; Bischoff, N.; Bott, M.; Burkovski, A.; Dusch, N.; Eggeling, L.; Eikmanns, B.J.; Gaigalat, L.; et al. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J. Biotechnol. 2003, 104, 5–25. [Google Scholar] [CrossRef]
- Frunzke, J.; Bramkamp, M.; Schweitzer, J.E.; Bott, M. Population Heterogeneity in Corynebacterium glutamicum ATCC 13032 caused by prophage CGP3. J. Bacteriol. 2008, 190, 5111–5119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, A.; Tauch, A.; Droste, N.; Pühler, A.; Kalinowski, J. The Corynebacterium glutamicum cglIM gene encoding a 5-cytosine methyltransferase enzyme confers a specific DNA methylation pattern in an McrBC-deficient Escherichia coli strain. Gene 1997, 203, 95–101. [Google Scholar] [CrossRef]
- Pfeifer, E.; Hünnefeld, M.; Popa, O.; Polen, T.; Kohlheyer, D.; Baumgart, M.; Frunzke, J. Silencing of cryptic prophages in Corynebacterium glutamicum. Nucleic Acids Res. 2016, 44, 10117–10131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgart, M.; Unthan, S.; Rückert, C.; Sivalingam, J.; Grünberger, A.; Kalinowski, J.; Bott, M.; Noack, S.; Frunzke, J. Construction of a prophage-free variant of Corynebacterium glutamicum ATCC 13032 for use as a platform strain for basic research and industrial biotechnology. Appl. Environ. Microbiol. 2013, 79, 6006–6015. [Google Scholar] [CrossRef] [Green Version]
- Schroven, K.; Aertsen, A.; Lavigne, R. Bacteriophages as drivers of bacterial virulence and their potential for biotechnological exploitation. FEMS Microbiol. Rev. 2021, 45, 1–15. [Google Scholar] [CrossRef]
- Lammens, E.M.; Nikel, P.I.; Lavigne, R. Exploring the synthetic biology potential of bacteriophages for engineering non-model bacteria. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Lobanova, J.S.; Gak, E.R.; Andreeva, I.G.; Rybak, K.V.; Krylov, A.A.; Mashko, S.V. Complete nucleotide sequence and annotation of the temperate corynephage ϕ16 genome. Arch. Virol. 2017, 162, 2489–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, S.; Blanco, C.; Trautwetter, A. Site-specific integration of corynephage Phi16: Construction of an integration vector. Microbiology 1999, 145, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Moreau, S.; Leret, V.; Le Marrec, C.; Varangot, H.; Ayache, m.; Bonnassie, S.; Blanco, C.; Trautwetter, A. Prophage distribution in coryneform bacteria. Res. Microbiol. 1995, 146, 493–505. [Google Scholar] [CrossRef]
- Koptides, M.; Barak, I.; Sisova, M.; Baloghova, E.; Ugorcakova, J.; Timko, J. Characterization of bacteriophage BFK20 from Brevibacterium flavum. J. Gen. Microbiol. 1992, 138, 1387–1391. [Google Scholar] [CrossRef] [Green Version]
- Bukovska, G.; Klucar, L.; Vlcek, C.; Adamovic, J.; Turna, J.; Timko, J. Complete nucleotide sequence and genome analysis of bacteriophage BFK20—A lytic phage of the industrial producer Brevibacterium flavum. Virology 2006, 348, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yomantas, Y.A.V.V.; Abalakina, E.G.; Lobanova, J.S.; Mamontov, V.A.; Stoynova, N.V.; Mashko, S.V. Complete nucleotide sequences and annotations of φ673 and φ674, two newly characterised lytic phages of Corynebacterium glutamicum ATCC 13032. Arch. Virol. 2018, 163, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.L.; Pan, T.Y.; Kan, S.C.; Kuan, Y.C.; Hong, L.Y.; Chiu, K.R.; Sheu, C.S.; Yang, J.S.; Hsu, W.H.; Hu, H.Y. Genome sequence of the lytic bacteriophage P1201 from Corynebacterium glutamicum NCHU 87078: Evolutionary relationships to phages from Corynebacterineae. Virology 2008, 378, 226–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majtan, T.; Halgasova, N.; Bukovska, G.; Timko, J. Transcriptional profiling of bacteriophage BFK20: Coexpression interrogated by “guilt-by-association” algorithm. Virology 2007, 359, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Tkacova, A.; Orieskova, M.; Halgasova, N.; Bocanova, L.; Bukovska, G. Identification of Brevibacterium flavum genes related to receptors involved in bacteriophage BFK20 adsorption. Virus Res. 2019, 274, 197775. [Google Scholar] [CrossRef]
- Hashiro, S.; Mitsuhashi, M.; Yasueda, H. Overexpression system for recombinant RNA in Corynebacterium glutamicum using a strong promoter derived from corynephage BFK20. J. Biosci. Bioeng. 2019, 128, 255–263. [Google Scholar] [CrossRef]
- Yeh, P.; Oreglia, J.; Sicard, A.M. Transfection of Corynebacterium lilium Protoplasts. Microbiology 1985, 131, 3179–3183. [Google Scholar] [CrossRef] [Green Version]
- Trautwetter, A.; Blanco, C.; Sicard, A.M. Structural characteristics of the Corynebacterium lilium bacteriophage CL31. J. Virol. 1987, 61, 1540–1545. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.A.; Hatfull, G.F. PhagesDB: The actinobacteriophage database. Bioinformatics 2017, 33, 784–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Joseph, S., David, W., Russell, B., Eds.; Cold Expiring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989; Volume 76, pp. 348–349. [Google Scholar] [CrossRef]
- Kensy, F.; Zang, E.; Faulhammer, C.; Tan, R.-K.K.; Büchs, J.; Buchs, J.; Büchs, J. Validation of a high-throughput fermentation system based on online monitoring of biomass and fluorescence in continuously shaken microtiter plates. Microb. Cell Fact. 2009, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, S.; Udaka, S.; Shimono, M. Studies on the amino acid fermentation. Part 1. Production of L-glutamic acid by various microorganisms. J. Gen. Appl. Microbiol. 2004, 50, 331–343. [Google Scholar]
- Schäfer, A.; Tauch, A.; Jäger, W.; Kalinowski, J.; Thierbach, G.; Pühler, A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: Selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 1994, 145, 69–73. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison 3rd, C.A.; Smith, H.O.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Niebisch, A.; Bott, M. Molecular analysis of the cytochrome bc1-aa3branch of the Corynebacterium glutamicum respiratory chain containing an unusual diheme cytochrome c1. Arch. Microbiol. 2001, 175, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Hardy, A.; Sharma, V.; Kever, L.; Frunzke, J. Genome Sequence and Characterization of Five Bacteriophages Infecting Streptomyces coelicolor and Streptomyces venezuelae: Alderaan, Coruscant, Dagobah, Endor1 and Endor2. Viruses 2020, 12, 1065. [Google Scholar] [CrossRef]
- Mcnair, K.; Zhou, C.; Dinsdale, E.A.; Souza, B.; Edwards, R.A. PHANOTATE: A novel approach to gene identification in phage genomes. Bioinformatics 2019, 35, 4537–4542. [Google Scholar] [CrossRef] [Green Version]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Ecale Zhou, C.L.; Malfatti, S.; Kimbrel, J.; Philipson, C.; McNair, K.; Hamilton, T.; Edwards, R.; Souza, B. MultiPhATE: Bioinformatics pipeline for functional annotation of phage isolates. Bioinformatics 2019, 35, 4402–4404. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Sims, G.E.; Jun, S.R.; Wu, G.A.; Kim, S.H. Alignment-free genome comparison with feature frequency profiles (FFP) and optimal resolutions. Proc. Natl. Acad. Sci. USA 2009, 106, 2677–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Ceyssens, P.J.; Robben, J. Phage proteomics: Applications of mass spectrometry. Methods Mol. Biol. 2009, 502, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Yu, Y.; Yu, Y.; Smith, M.; Pieper, R. A spinnable and automatable StageTip for high throughput peptide desalting and proteomics. Protoc. Exch. 2014, 1. [Google Scholar] [CrossRef]
- Mavrich, T.N.; Hatfull, G.F. Bacteriophage evolution differs by host, lifestyle and genome. Nat. Microbiol. 2017, 2, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Snippy: Rapid Haploid Variant Calling and Core Genome Alignment. Available online: https://github.com/tseemann/snippy (accessed on 11 December 2020).
- Gande, R.; Gibson, K.J.C.; Brown, A.K.; Krumbach, K.; Dover, L.G.; Sahm, H.; Shioyama, S.; Oikawa, T.; Besra, G.S.; Eggeling, L. Acyl-CoA carboxylases (accD2 and accD3), together with a unique polyketide synthase (Cg-pks), are key to mycolic acid biosynthesis in Corynebacterianeae such as Corynebacterium glutamicum and Mycobacterium tuberculosis. J. Biol. Chem. 2004, 279, 44847–44857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, A.; Schwarzer, A.; Kalinowski, J.; Puhler, A. Cloning and characterization of a DNA region encoding a stress-sensitive restriction system from Corynebacterium glutamicum ATCC 13032 and analysis of its role in intergeneric conjugation with Escherichia coli. J. Bacteriol. 1994, 176, 7309–7319. [Google Scholar] [CrossRef] [Green Version]
- Chopin, M.C.; Chopin, A.; Bidnenko, E. Phage abortive infection in lactococci: Variations on a theme. Curr. Opin. Microbiol. 2005, 8, 473–479. [Google Scholar] [CrossRef]
- Halgašová, N.; Majtán, T.; Ugorčáková, J.; Timko, J.; Bukovská, G. Resistance of corynebacterial strains to infection and lysis by corynephage BFK 20. J. Appl. Microbiol. 2005, 98, 184–192. [Google Scholar] [CrossRef]
- Mavrich, T.N.; Hatfull, G.F. Evolution of Superinfection Immunity in Cluster A Mycobacteriophages. MBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Dedrick, R.M.; Jacobs-Sera, D.; Guerrero Bustamante, C.A.; Garlena, R.A.; Mavrich, T.N.; Pope, W.H.; Cervantes Reyes, J.C.; Russell, D.A.; Adair, T.; Alvey, R.; et al. Prophage-mediated defence against viral attack and viral counter-defence. Nat. Microbiol. 2017, 2, 1–13. [Google Scholar] [CrossRef]
- Gentile, G.M.; Wetzel, K.S.; Dedrick, R.M.; Montgomery, M.T.; Garlena, R.A.; Jacobs-Sera, D.; Hatfull, G.F. More Evidence of Collusion: A New Prophage-Mediated Viral Defense System Encoded by Mycobacteriophage Sbash. MBio 2019, 10, e00196-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, E.; Hünnefeld, M.; Popa, O.; Frunzke, J. Impact of Xenogeneic Silencing on Phage–Host Interactions. J. Mol. Biol. 2019, 431, 4670–4683. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, J.; Filipchyk, A.; Hünnefeld, M.; Gätgens, C.; Brehm, J.; Heermann, R.; Frunzke, J. Deciphering the rules underlying xenogeneic silencing and counter-silencing of Lsr2-like proteins using CgpS of Corynebacterium glutamicum as a model. MBio 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Argov, T.; Sapir, S.R.; Pasechnek, A.; Azulay, G.; Stadnyuk, O.; Rabinovich, L.; Sigal, N.; Borovok, I.; Herskovits, A.A. Coordination of cohabiting phage elements supports bacteria–phage cooperation. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groth, A.C.; Calos, M.P. Phage integrases: Biology and applications. J. Mol. Biol. 2004, 335, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Rostøl, J.T.; Marraffini, L. (Ph)ighting Phages: How Bacteria Resist Their Parasites. Cell Host Microbe 2019, 25, 184–194. [Google Scholar] [CrossRef] [Green Version]
- Verheust, C.; Fornelos, N.; Mahillon, J. GIL16, a new gram-positive tectiviral phage related to the Bacillus thuringiensis GIL01 and the Bacillus cereus pBClin15 elements. J. Bacteriol. 2005, 187, 1966–1973. [Google Scholar] [CrossRef] [Green Version]
- Klatt, S.; Brammananth, R.; O’Callaghan, S.; Kouremenos, K.A.; Tull, D.; Crellin, P.K.; Coppel, R.L.; McConville, M.J. Identification of novel lipid modifications and intermembrane dynamics in Corynebacterium glutamicum using high-resolution mass spectrometry. J. Lipid Res. 2018, 59, 1190–1204. [Google Scholar] [CrossRef] [Green Version]
- Houssin, C.; de Sousa d’Auria, C.; Constantinesco, F.; Dietrich, C.; Labarre, C.; Bayan, N. Architecture and Biogenesis of the Cell Envelope of Corynebacterium glutamicum; Springer: Cham, Switzerland, 2020; pp. 25–60. [Google Scholar]
- Gande, R.; Dover, L.G.; Krumbach, K.; Besra, G.S.; Sahm, H.; Oikawa, T.; Eggeling, L. The two carboxylases of Corynebacterium glutamicum essential for fatty acid and mycolic acid synthesis. J. Bacteriol. 2007, 189, 5257–5264. [Google Scholar] [CrossRef] [Green Version]
- Portevin, D.; De Sousa-D’Auria, C.; Houssin, C.; Grimaldi, C.; Chami, M.; Daffe, M.; Guilhot, C.; Daffé, M.; Guilhot, C. A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc. Natl. Acad. Sci. USA 2004, 101, 314–319. [Google Scholar] [CrossRef] [Green Version]
- Di Capua, C.B.; Belardinelli, J.M.; Buchieri, M.V.; Bortolotti, A.; Franceschelli, J.J.; Morbidoni, H.R. Deletion of msmeg_1350 in Mycobacterium smegmatis causes loss of epoxy-mycolic acids, fitness alteration at low temperature and resistance to a set of mycobacteriophages. Microbiology 2018, 164, 1567–1582. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, M.; Fan, X.; Yan, J.; Li, W.; Xie, J. Mycobacteriophage SWU1 gp39 can potentiate multiple antibiotics against Mycobacterium via altering the cell wall permeability. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangalea, M.R.; Duerkop, B.A. Fitness trade-offs resulting from bacteriophage resistance potentiate synergistic antibacterial strategies. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Shi, F.; Tao, G.; Wang, X. Purification and Structure Analysis of Mycolic Acids in Corynebacterium glutamicum. J. Microbiol. 2012, 50, 235–240. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, P.A.; Von Meijenfeldt, F.A.B.; Van Rooijen, L.E.; Brouns, S.J.J.; Dutilh, B.E. Evolution of BACON domain tandem repeats in crassphage and novel gut bacteriophage lineages. Viruses 2019, 11, 1085. [Google Scholar] [CrossRef] [Green Version]
- Mello, L.V.; Chen, X.; Rigden, D.J. Mining metagenomic data for novel domains: BACON, a new carbohydrate-binding module. FEBS Lett. 2010, 584, 2421–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, A.; Chang, H.Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S.; et al. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef] [PubMed]
- De Melo, A.G.; Rousseau, G.M.; Tremblay, D.M.; Labrie, S.J.; Moineau, S. DNA tandem repeats contribute to the genetic diversity of Brevibacterium aurantiacum phages. Environ. Microbiol. 2020, 22, 3413–3428. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain or Plasmid | Characteristics | Reference |
|---|---|---|
| Bacterial strains | ||
| E. coli DH5α | supE44ΔlacU169 (ϕ80lacZDM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | Invitrogen |
| C. glutamicum ATCC 13032 | Biotin-auxotrophic wild type (Accession: BX927147) | [24] |
| C. glutamicum Δresmod | ATCC 13032 with in-frame deletion of restriction-modification system cglIR-cglIIR-cglIM (cg1996-cg1998), located within prophage CGP3 | [6] |
| C. glutamicum MB001 | ATCC 13032 with in-frame deletion of prophages CGP1 (cg1507-cg1524), CGP2 (cg1746-cg1752), and CGP3 (cg1890-cg2071) | [6] |
| C. glutamicum MB001::pks_Tyr886Asp | C. glutamicum MB001 with a nucleotide exchange (T2656G) causing the amino acid exchange Tyr886Asp | This work |
| C. glutamicum MB001::accD2_Gly350Asp | C. glutamicum MB001 with a nucleotide exchange (G1049A) causing the amino acid exchange Gly350Asp | This work |
| C. glutamicum MB001::accD3_Gly341Asp | C. glutamicum MB001 with a nucleotide exchange (G1022A) causing the amino acid exchange Gly341Asp | This work |
| Plasmids | ||
| pK19mobsacB | KanR; plasmid for allelic exchange in C. glutamicum; (pK18 oriVE.c., sacB, lacZα) | [25] |
| pK19mobsacB -pks_ T2656G | KanR; Variant of pK19 mob sacB; plasmid used for the integration of the nucleotide exchange causing the missense mutation Tyr886Asp in pks gene | This work |
| pK19mobsacB -accD2_ G1049A | KanR; Variant of pK19 mob sacB; plasmid used for the integration of the nucleotide exchange causing the missense mutation Gly350Asp in accD2 gene | This work |
| pK19mobsacB -accD3_ G1022A | KanR; Variant of pK19 mob sacB; plasmid used for the integration of the nucleotide exchange causing the missense mutation Gly341Asp in accD3 gene | This work |
| Strain | Reference | Gene | Locus | Base | Amino Acid | Position | Frequency | Mutation |
|---|---|---|---|---|---|---|---|---|
| ∆resmod | BX927147 | dtsR2 (accD2) | Cg0811 | C -> T | Gly350Asp | 728777 | 97.7% | missense |
| MB001 | CP005959 | pccB (accD3) | Cg3177 | C -> T | Gly341Asp | 2831759 | 95.4% | missense |
| WT | BX927147 | pks | Cg3178 | A -> T | Tyr886Asp | 3038431 | 100% | missense |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hünnefeld, M.; Viets, U.; Sharma, V.; Wirtz, A.; Hardy, A.; Frunzke, J. Genome Sequence of the Bacteriophage CL31 and Interaction with the Host Strain Corynebacterium glutamicum ATCC 13032. Viruses 2021, 13, 495. https://doi.org/10.3390/v13030495
Hünnefeld M, Viets U, Sharma V, Wirtz A, Hardy A, Frunzke J. Genome Sequence of the Bacteriophage CL31 and Interaction with the Host Strain Corynebacterium glutamicum ATCC 13032. Viruses. 2021; 13(3):495. https://doi.org/10.3390/v13030495
Chicago/Turabian StyleHünnefeld, Max, Ulrike Viets, Vikas Sharma, Astrid Wirtz, Aël Hardy, and Julia Frunzke. 2021. "Genome Sequence of the Bacteriophage CL31 and Interaction with the Host Strain Corynebacterium glutamicum ATCC 13032" Viruses 13, no. 3: 495. https://doi.org/10.3390/v13030495
APA StyleHünnefeld, M., Viets, U., Sharma, V., Wirtz, A., Hardy, A., & Frunzke, J. (2021). Genome Sequence of the Bacteriophage CL31 and Interaction with the Host Strain Corynebacterium glutamicum ATCC 13032. Viruses, 13(3), 495. https://doi.org/10.3390/v13030495