
Detection of H3K4me3 Identifies NeuroHIV Signatures, Genomic Effects of Methamphetamine and Addiction Pathways in Postmortem HIV+ Brain Specimens that Are Not Amenable to Transcriptome Analysis


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Mouse Postmortem Specimens and ChIP-qPCR
2.2. Human Postmortem Specimens and ChIP-qPCR
2.3. ChIP-qPCR Statistical Analysis
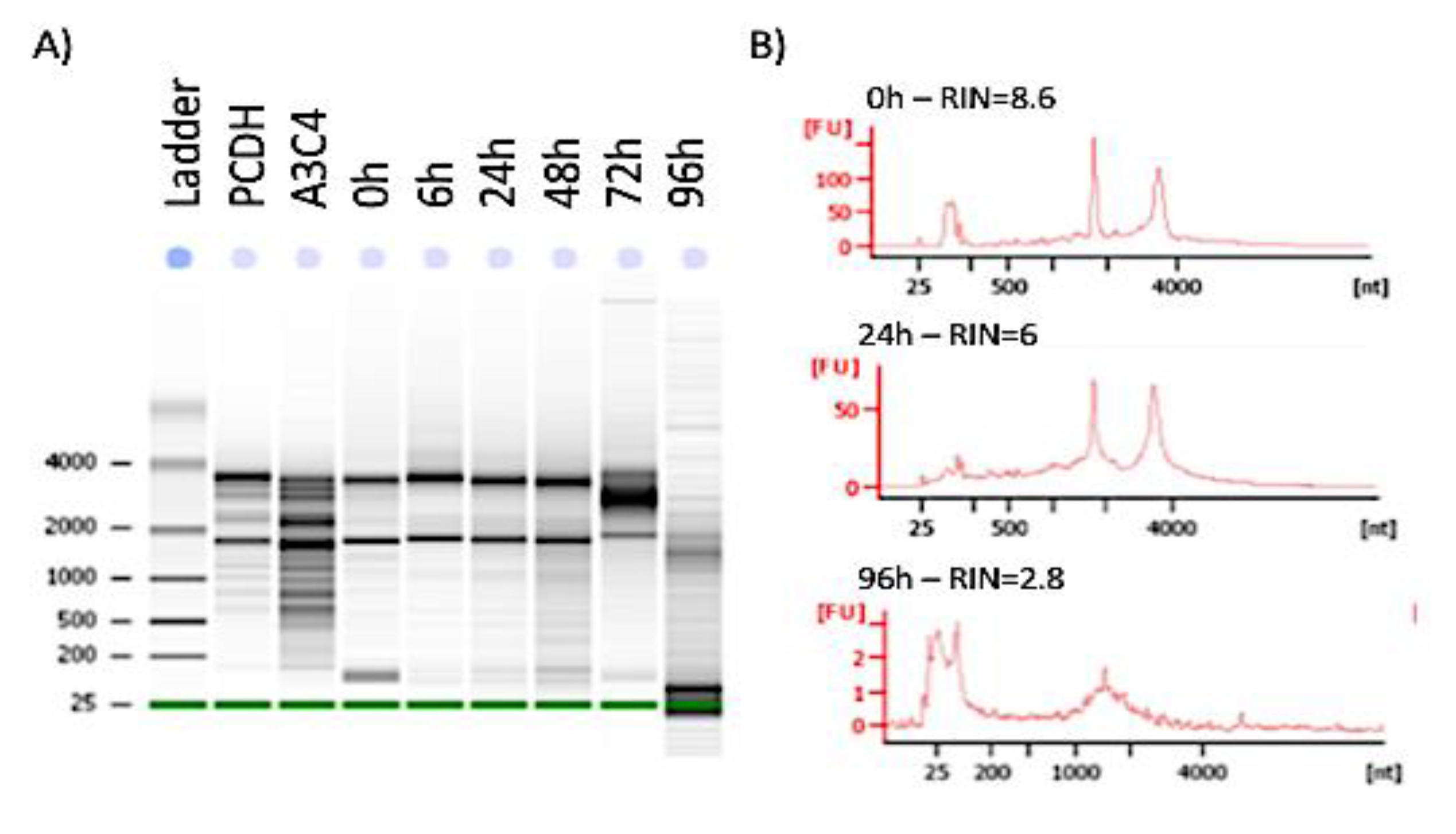
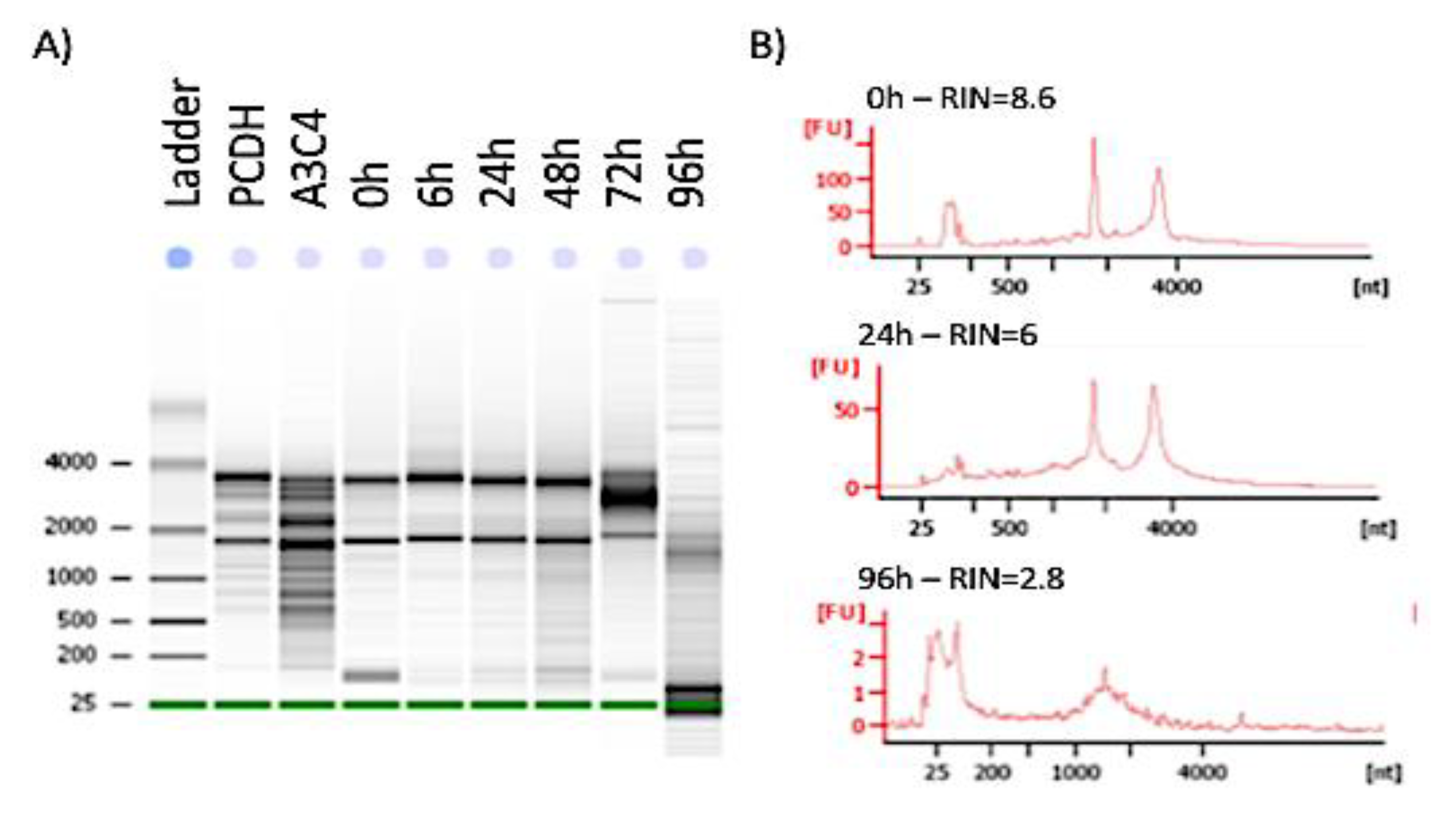
2.4. RNA Extraction and Quality Assessment
2.5. Chromatin Preparation
2.6. ChIP-Seq
2.7. ChIP Analysis
2.8. Systems Analysis
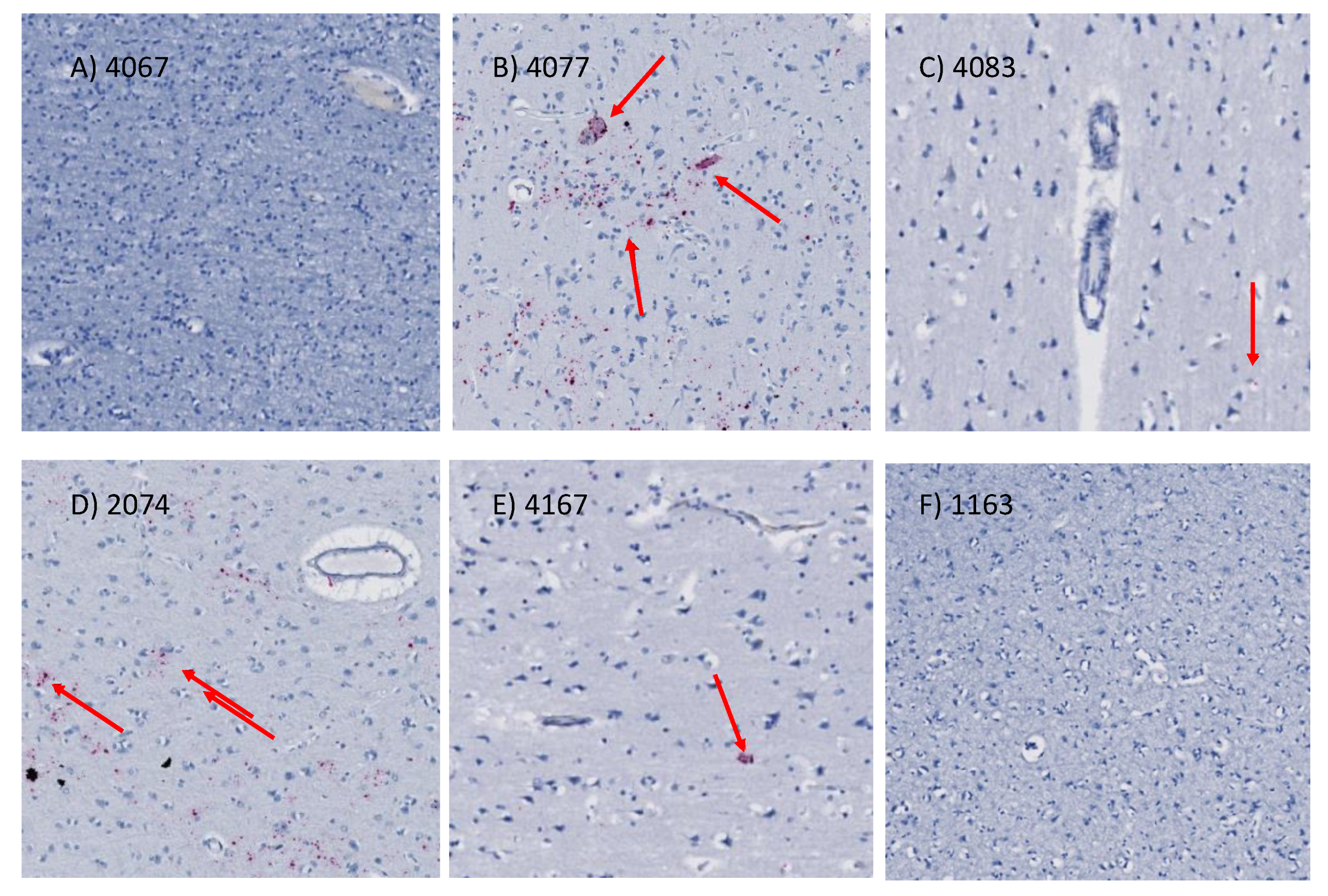
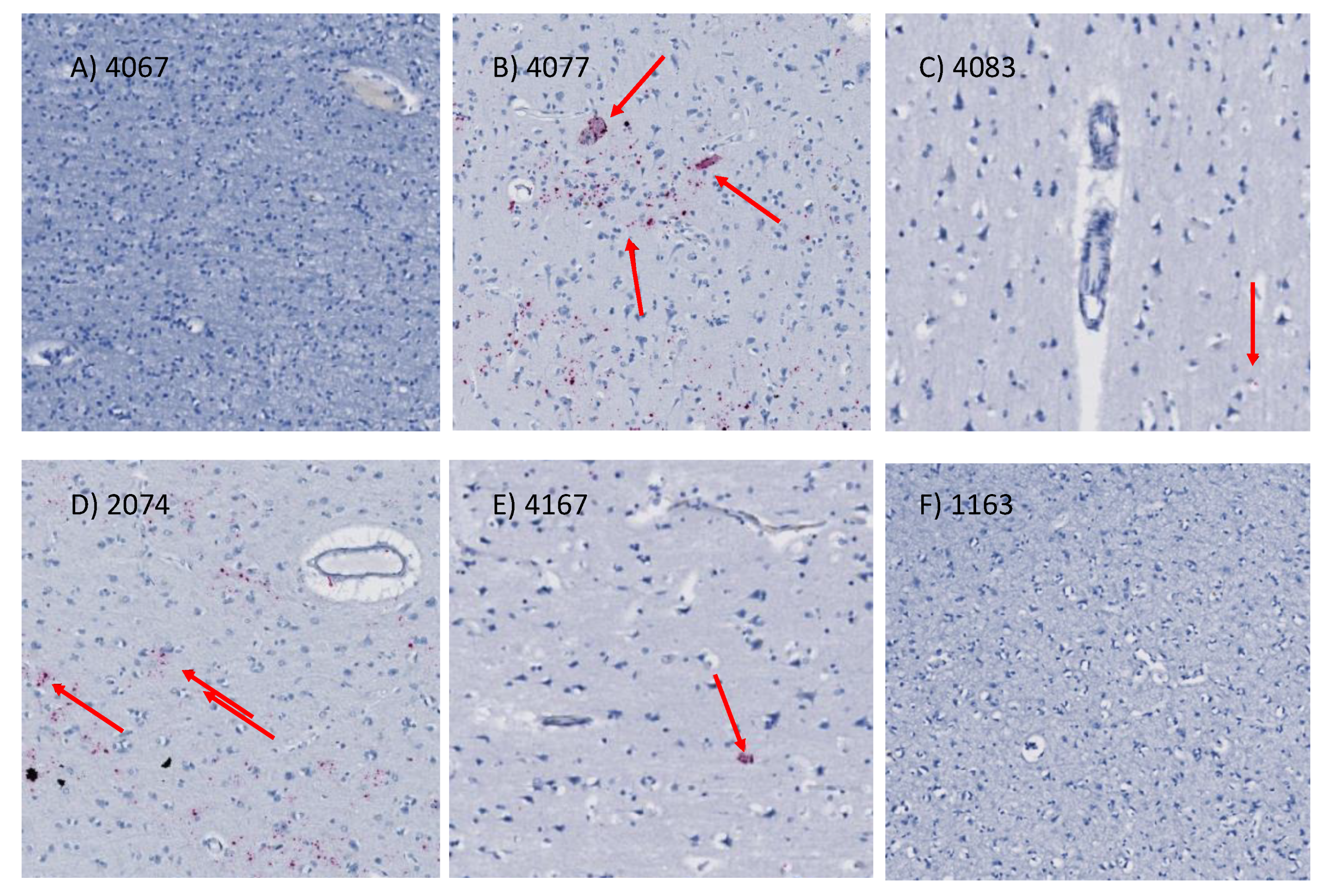
2.9. In Situ Hybridization for HIV (vRNA) Detection
3. Results
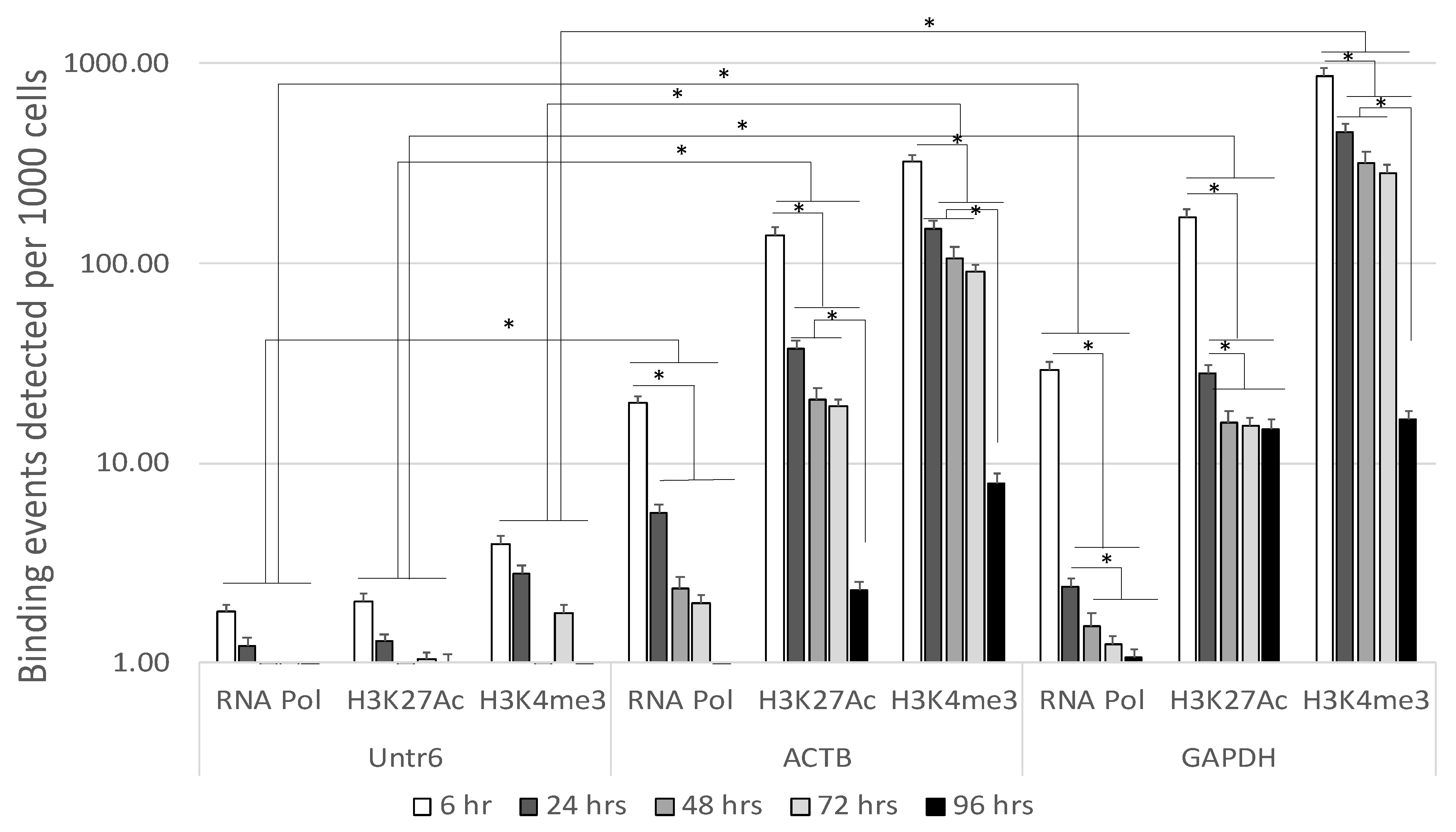
3.1. Stability of Enhancer Epigenetic Marks in Mouse Postmortem Brains Overtime
3.2. Stability of Functional Genomic Marks in Human Postmortem Prefrontal Cortex Specimens with RIN < 7.2
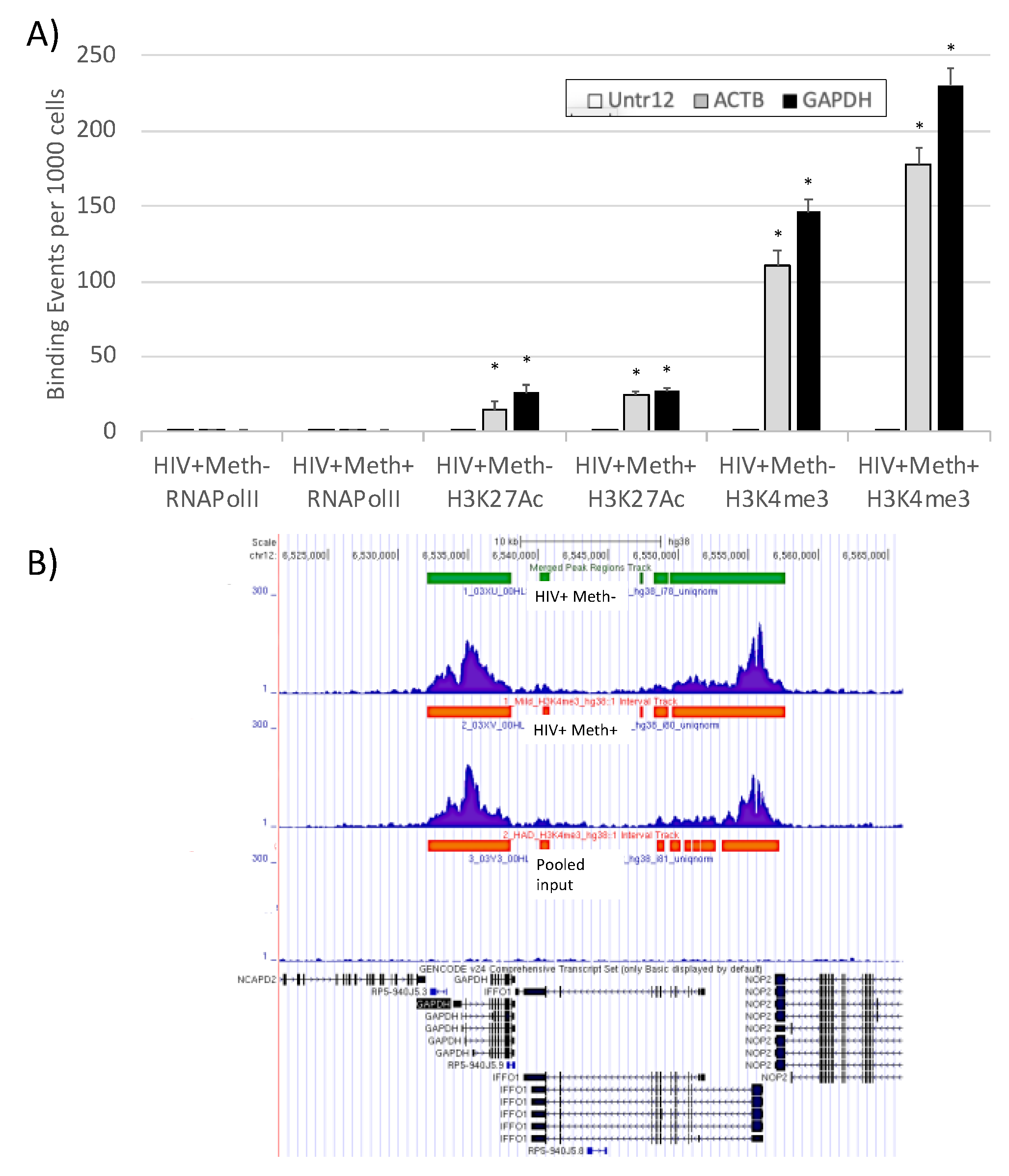
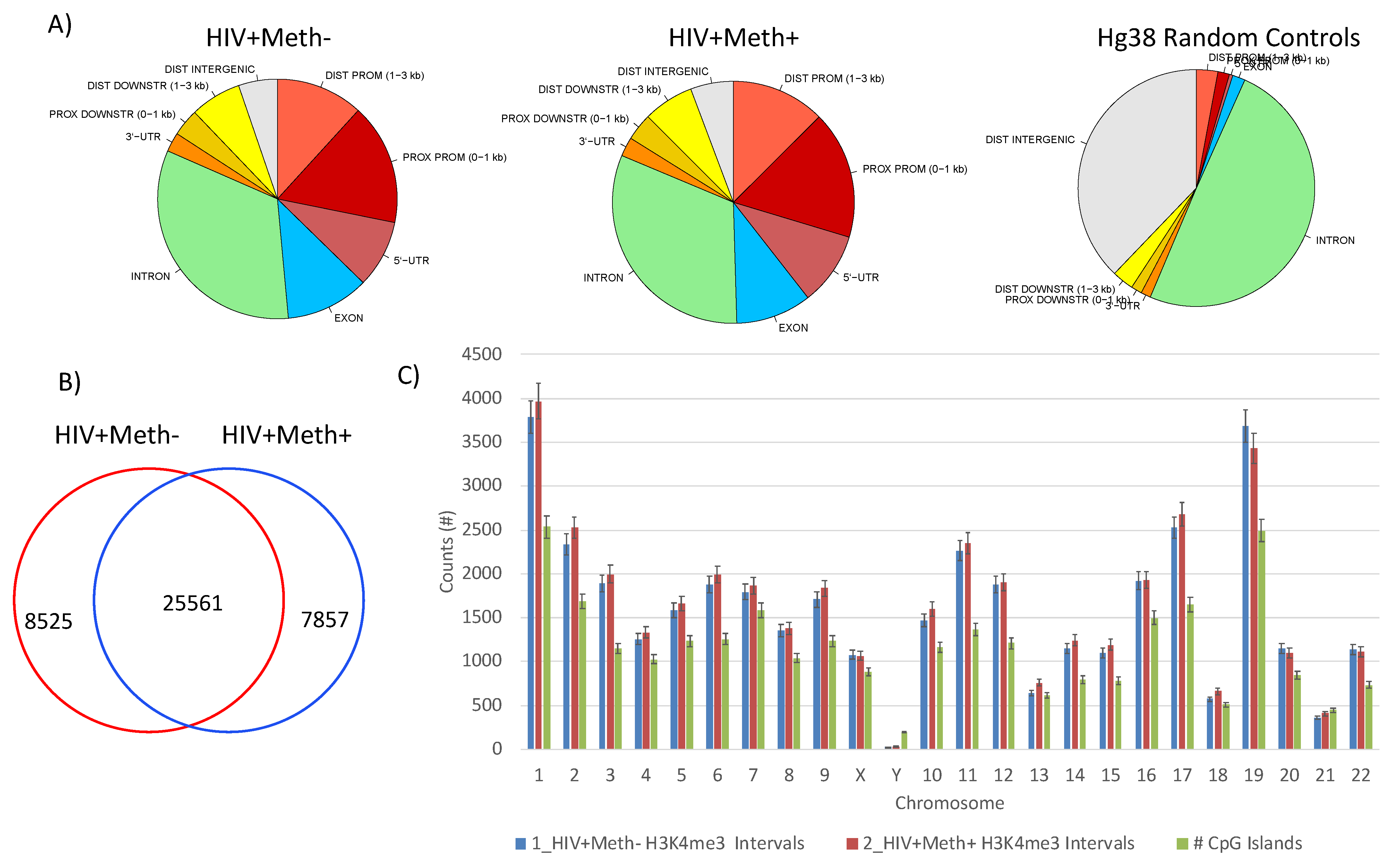
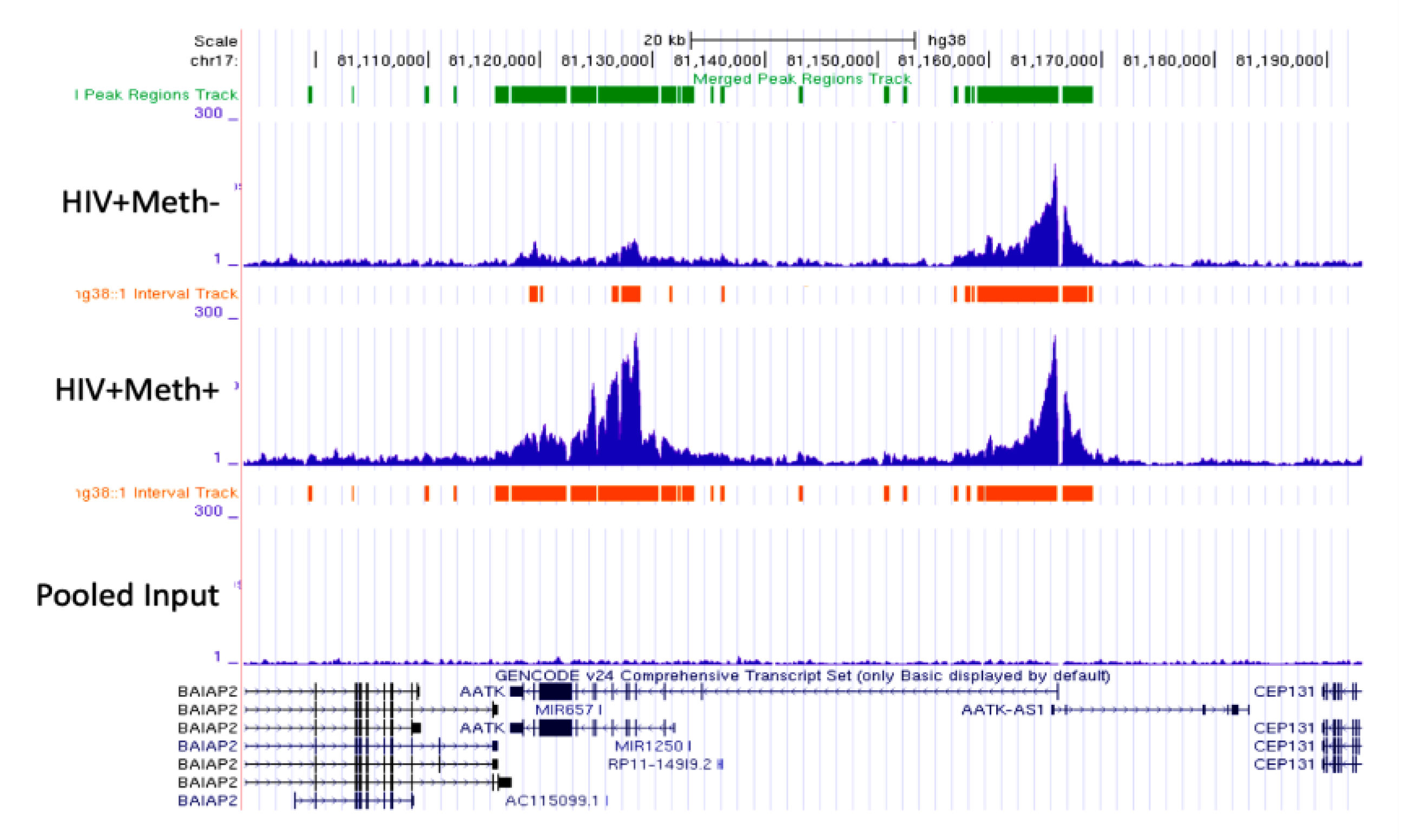
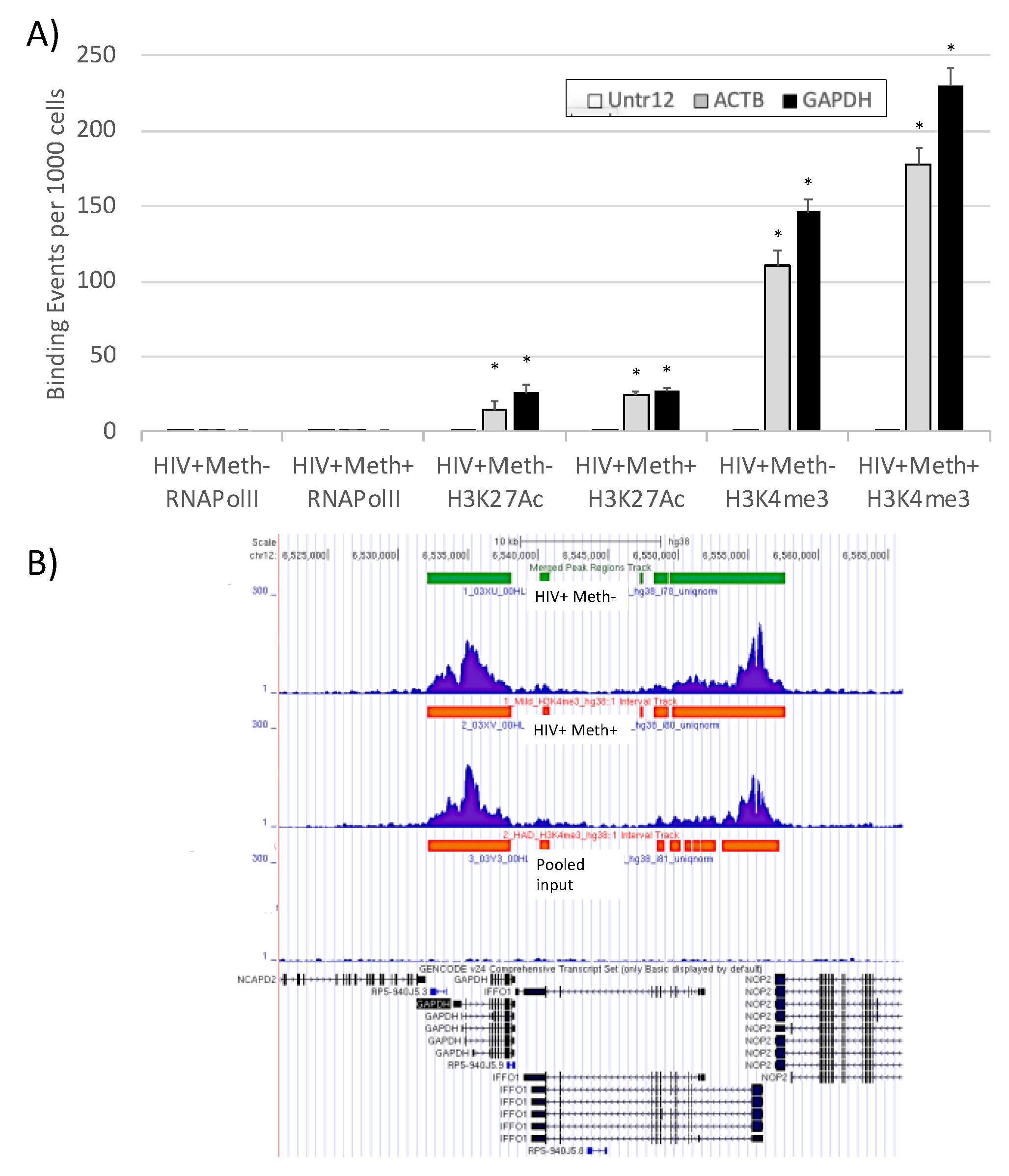
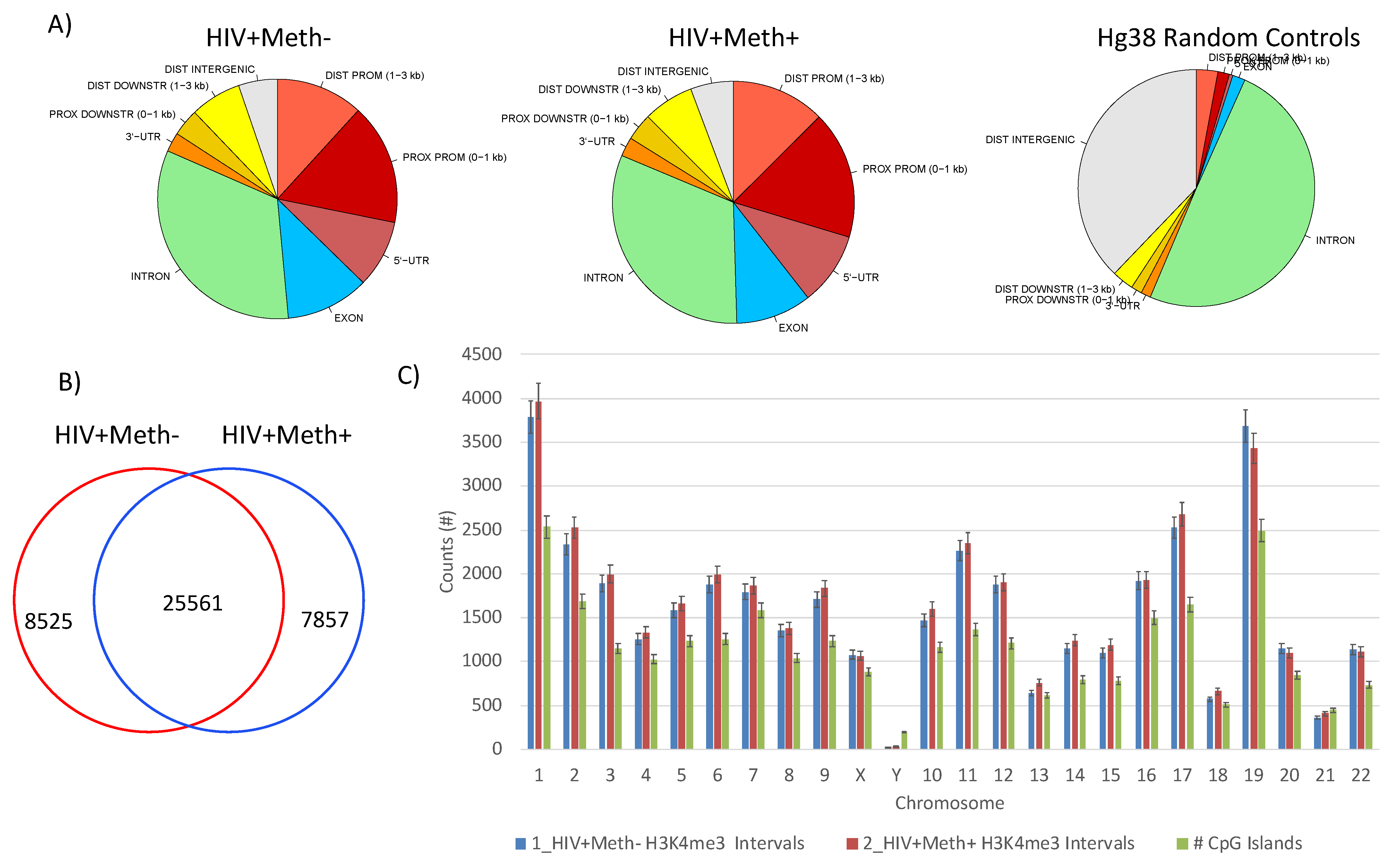
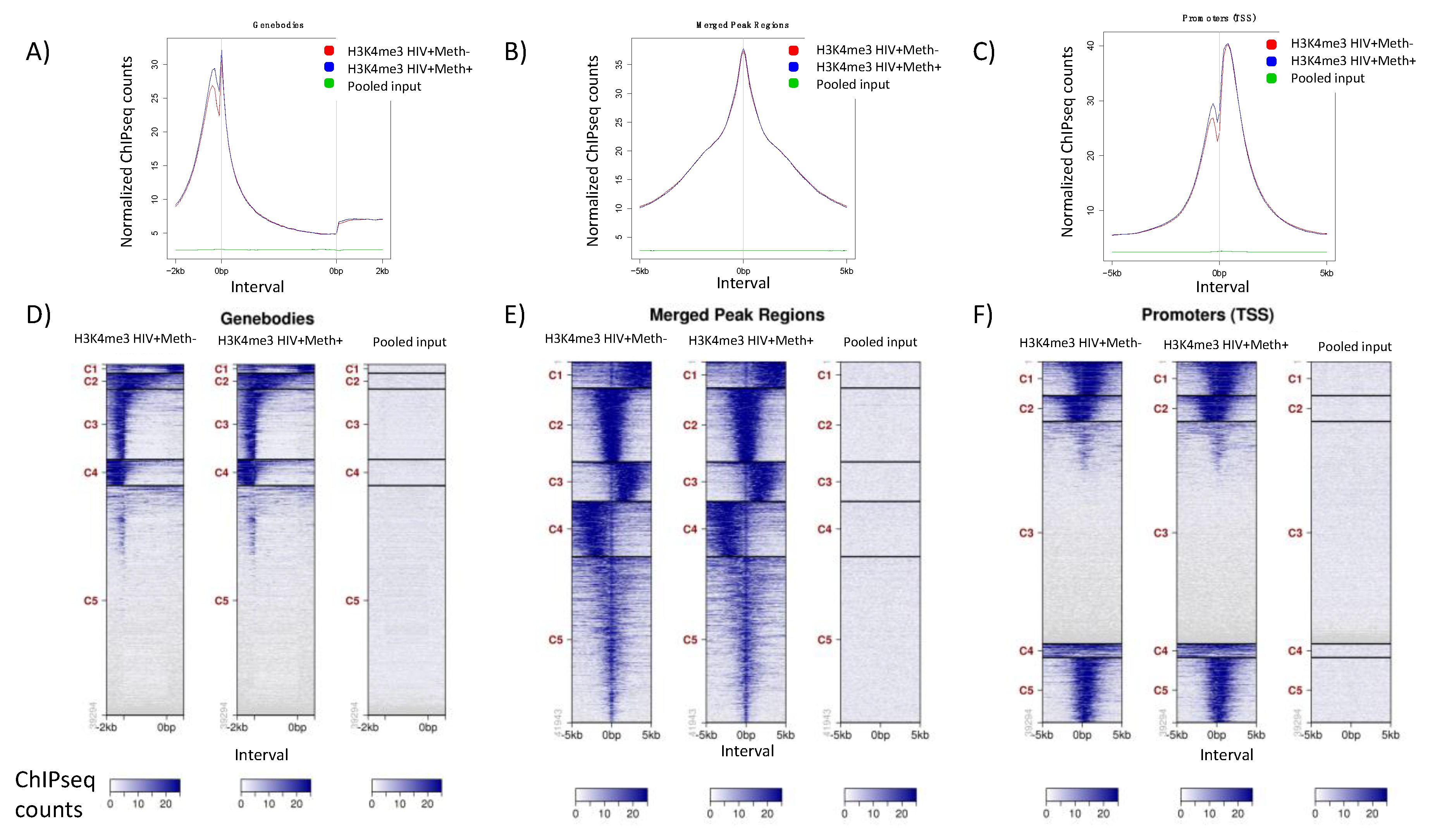
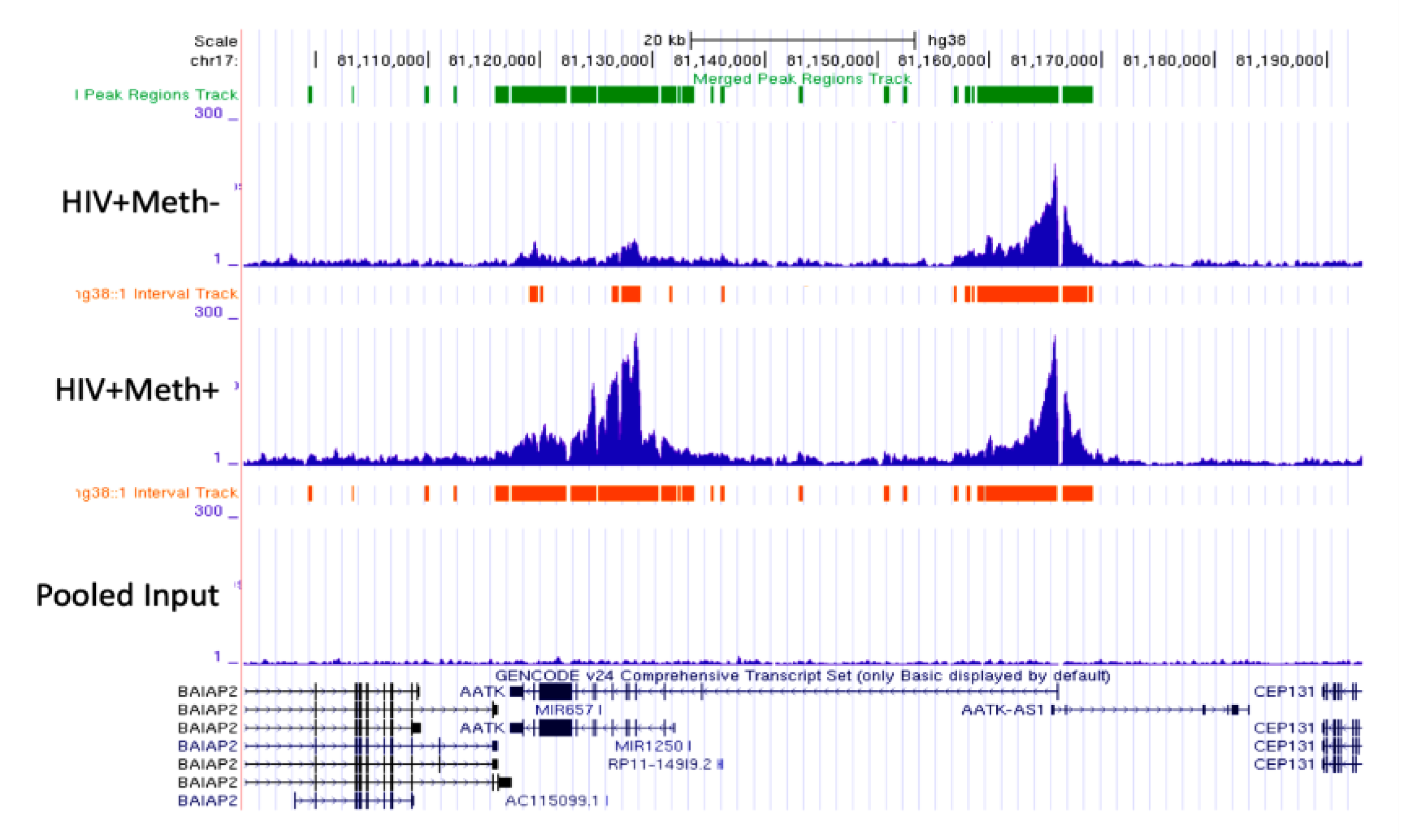
3.3. Quality Controls in Genome-Wide Peak Data Confirms H3K4me3 Stability in Postmortem Human Specimens
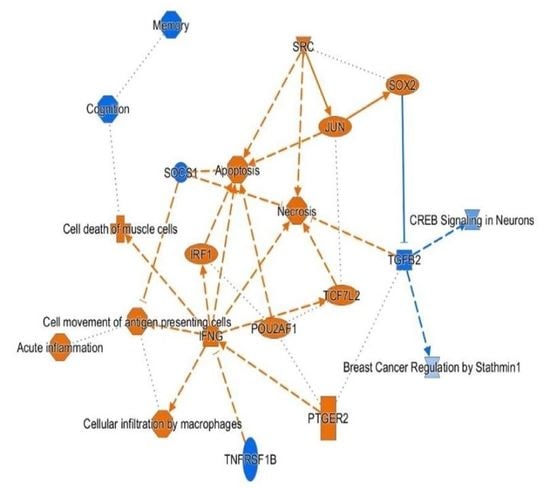
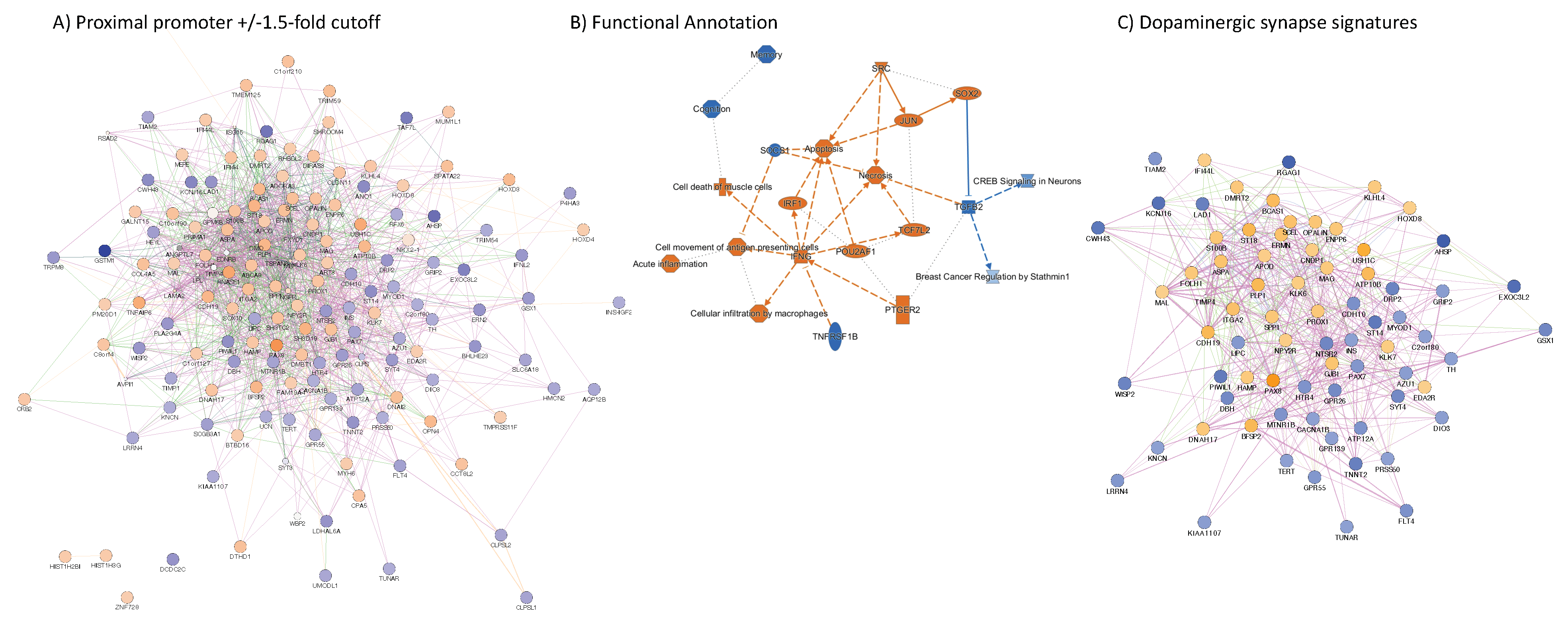
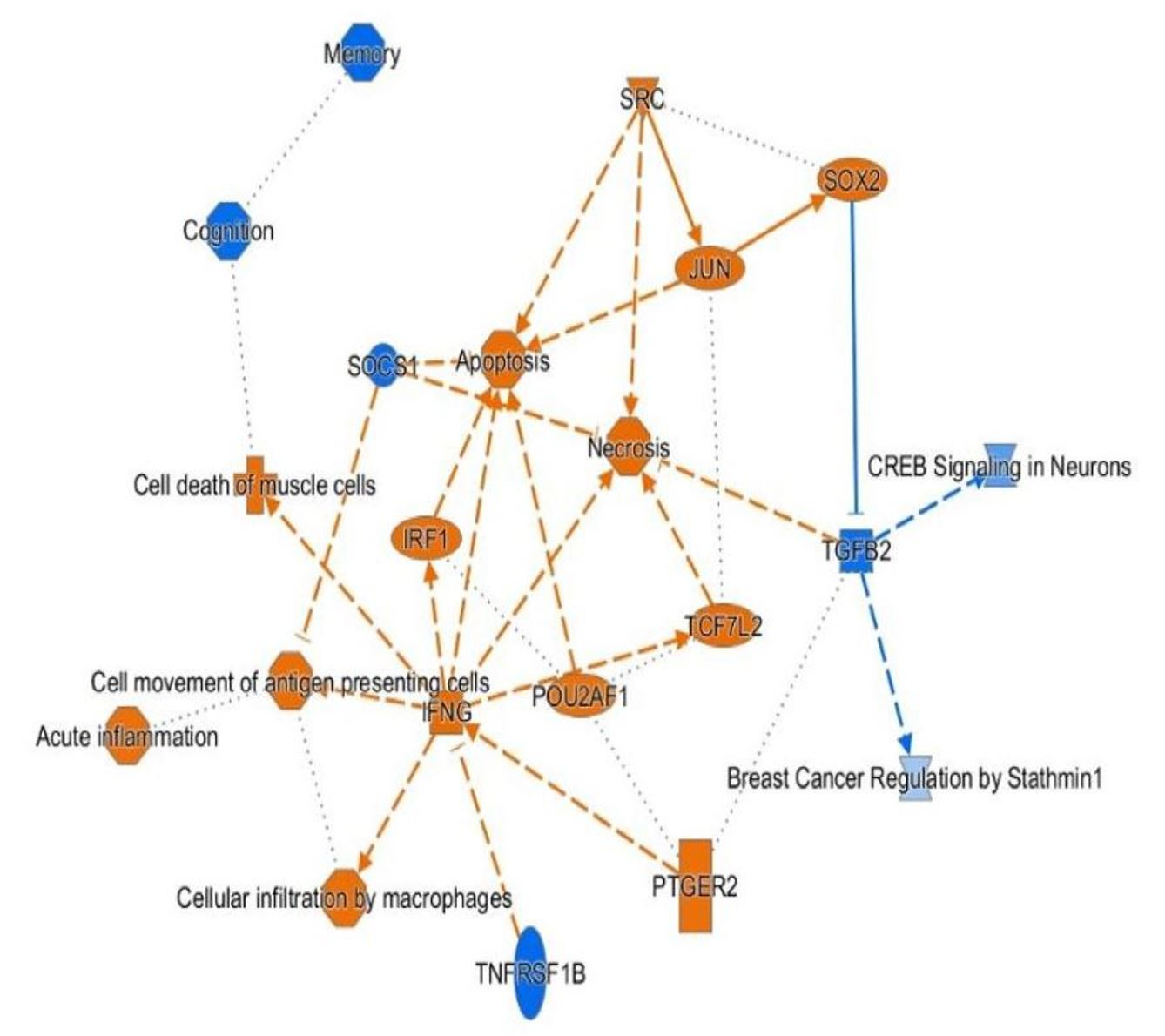
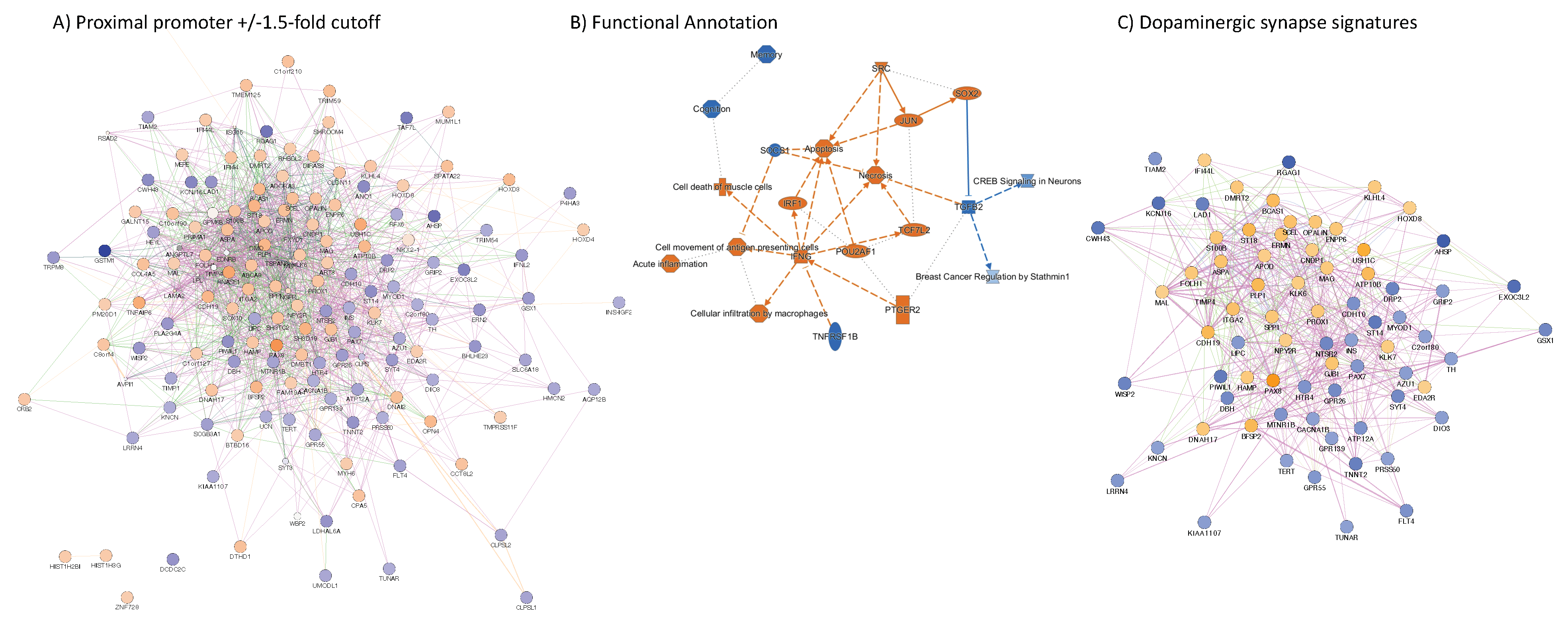
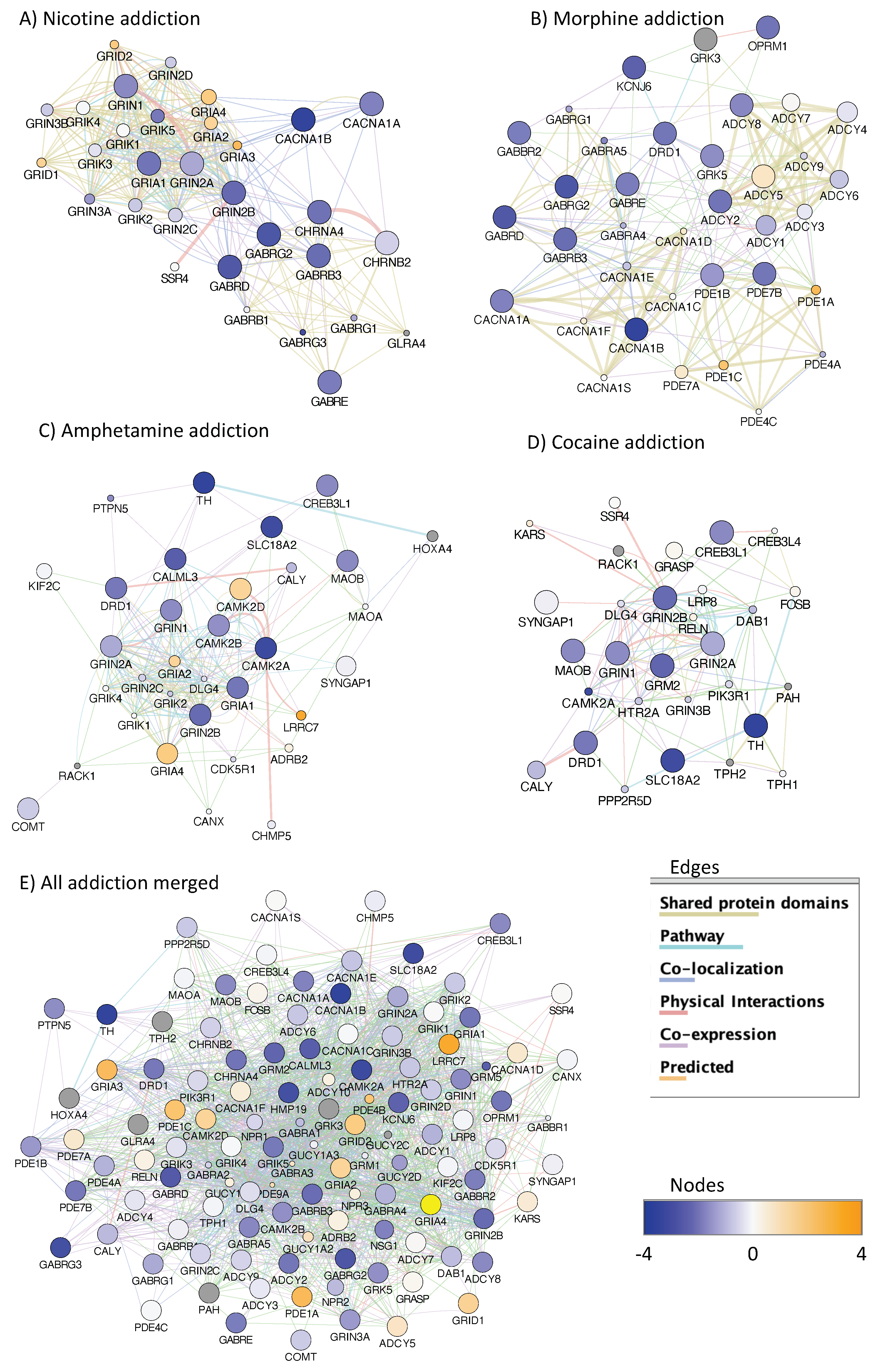
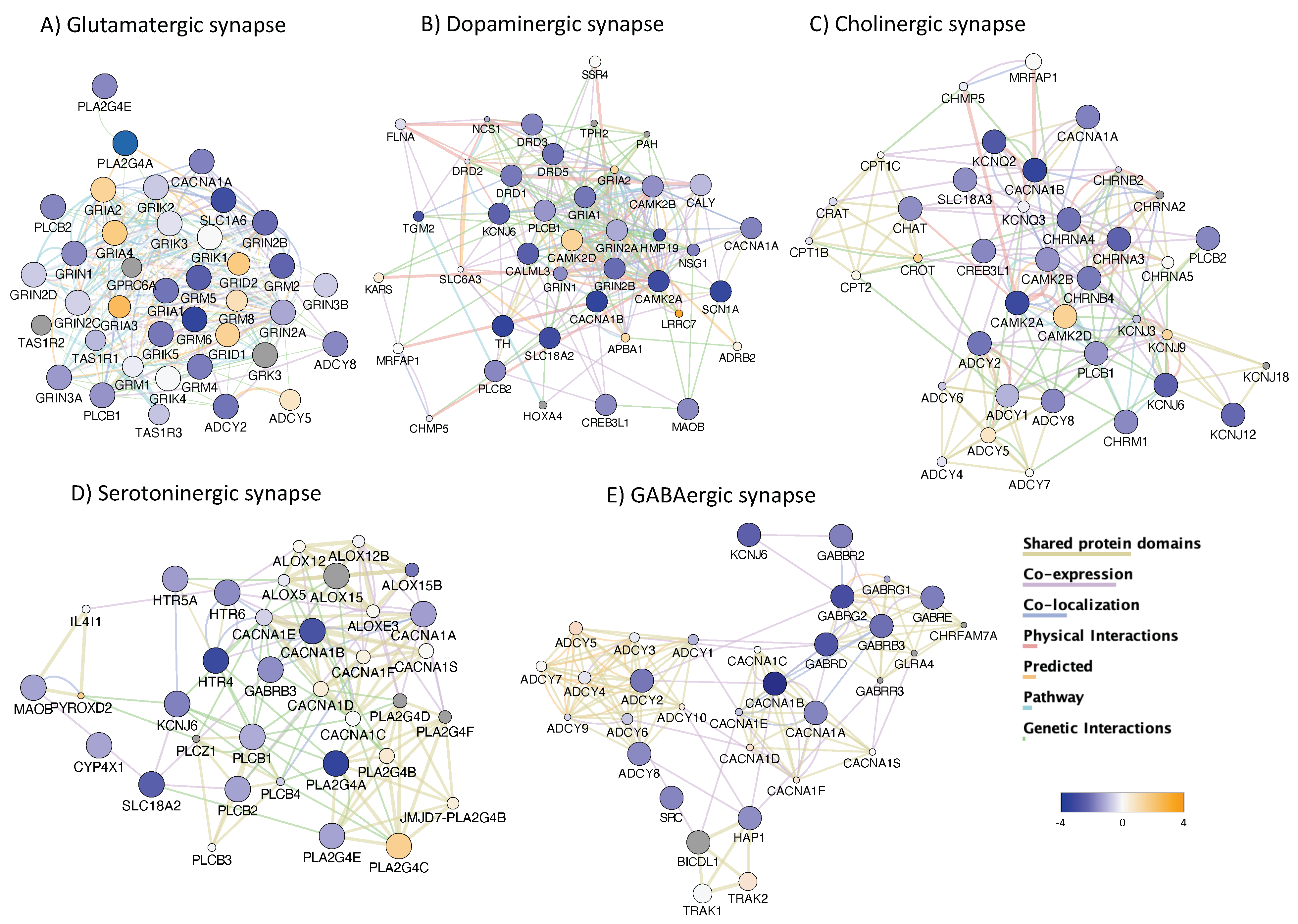
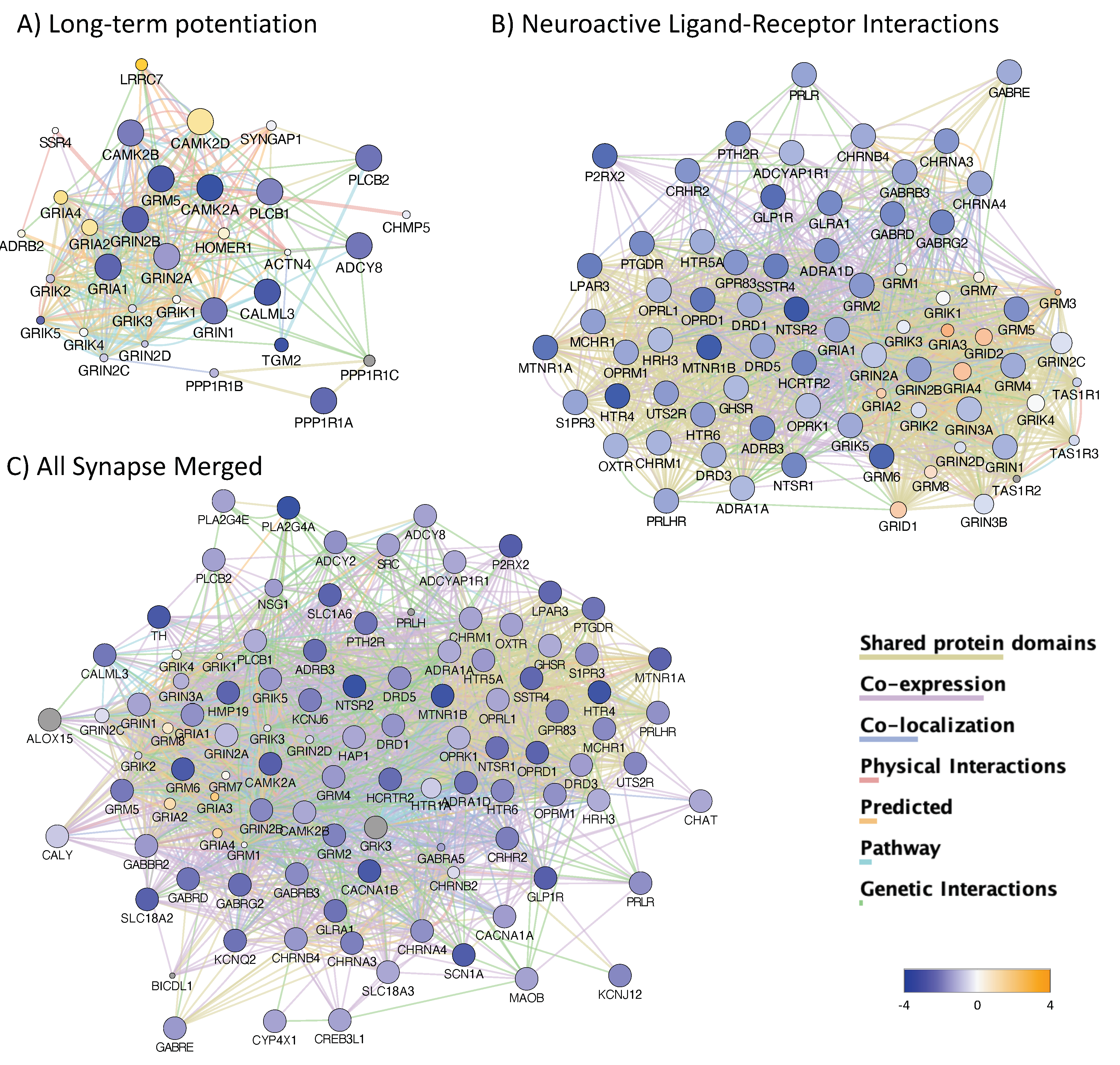
3.4. H3K4me3 Can Provide Insights into Biological Processes and Pathway Usage in Specimens with Limited Value in Global Transcriptome Studies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fan, J.; Khanin, R.; Sakamoto, H.; Zhong, Y.; Michael, C.; Pena, D.; Javier, B.; Wood, L.D.; Iacobuzio-Donahue, C.A. Quantification of nucleic acid quality in postmortem tissues from a cancer research autopsy program. Oncotarget 2016, 7, 66906–66921. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, L.; Yin, Y.; Yang, E. Systematic analysis of gene expression patterns associated with postmortem interval in human tissues. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Tolliver, S.S.; Hearn, W.L.; Furton, K.G. Evaluating the Relationship Between Postmortem and Antemortem Morphine and Codeine Concentrations in Whole Blood. J. Anal. Toxicol. 2010, 34, 491–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gates, L.A.; Foulds, C.E.; O’Malley, B.W. Histone Marks in the ‘Driver’s Seat’: Functional Roles in Steering the Transcription Cycle. Trends Biochem. Sci. 2017, 42, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Karlić, R.; Chung, H.-R.; Lasserre, J.; Vlahoviček, K.; Vingron, M. Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2926–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huebert, D.J.; Kamal, M.; O’Donovan, A.; Bernstein, B.E. Genome-wide analysis of histone modifications by ChIP-on-chip. Methods 2006, 40, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Kamal, M.; Lindblad-Toh, K.; Bekiranov, S.; Bailey, D.K.; Huebert, D.J.; McMahon, S.; Karlsson, E.K.; Kulbokas, E.J.; Gingeras, T.R.; et al. Genomic Maps and Comparative Analysis of Histone Modifications in Human and Mouse. Cell 2005, 120, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, B.E.; Humphrey, E.L.; Liu, C.L.; Schreiber, S.L. The use of Chromatin Immunoprecipitation Assays in Genome-Wide Analyses of Histone Modifications. Methods Enzym. 2003, 376, 349–360. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Humphrey, E.L.; Erlich, R.L.; Schneider, R.; Bouman, P.; Liu, J.S.; Kouzarides, T.; Schreiber, S.L. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. USA 2002, 99, 8695–8700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hightower, G.K.; Wong, J.K.; Letendre, S.L.; Umlauf, A.A.; Ellis, R.J.; Ignacio, C.C.; Heaton, R.K.; Collier, A.C.; Marra, C.M.; Clifford, D.B.; et al. Higher HIV-1 genetic diversity is associated with AIDS and neuropsychological impairment. Virology 2012, 433, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Everall, I.; Vaida, F.; Khanlou, N.; Lazzaretto, D.; Achim, C.; Letendre, S.; Moore, D.; Ellis, R.; Cherner, M.; Gelman, B.; et al. Cliniconeuropathologic correlates of human immunodeficiency virus in the era of antiretroviral therapy. J. Neurovirol. 2009, 15, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Gelman, B.B.; Chen, T.; Lisinicchia, J.G.; Soukup, V.M.; Carmical, J.R.; Starkey, J.M.; Masliah, E.; Commins, D.L.; Brandt, D.; Grant, I.; et al. The National Neuro AIDS Tissue Consortium Brain Gene Array: Two Types of HIV-Associated Neurocognitive Impairment. PLoS ONE 2012, 7, e46178. [Google Scholar] [CrossRef] [PubMed]
- May, P.E.; Heithoff, A.J.; Wichman, C.S.; Phatak, V.S.; Moore, D.J.; Heaton, R.K.; Fox, H.S. Assessing Cognitive Functioning in People Living with HIV (PLWH): Factor Analytic Results From CHARTER and NNTC Cohorts. JAIDS J. Acquir. Immune Defic. Syndr. 2020, 83, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Basova, L.; Najera, J.A.; Bortell, N.; Wang, D.; Moya, R.; Lindsey, A.; Semenova, S.; Ellis, R.J.; Marcondes, M.C.G. Dopamine and its receptors play a role in the modulation of CCR5 expression in innate immune cells following exposure to Methamphetamine: Implications to HIV infection. PLoS ONE 2018, 13, e0199861. [Google Scholar] [CrossRef] [PubMed]
- Tjitro, R.; Campbell, L.A.; Basova, L.; Johnson, J.; Najera, J.A.; Lindsey, A.; Marcondes, M.C.G. Modeling the Function of TATA Box Binding Protein in Transcriptional Changes Induced by HIV-1 Tat in Innate Immune Cells and the Effect of Methamphetamine Exposure. Front. Immunol. 2019, 9, 3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, T.A.; Meyer, C.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nussbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, C.; Schones, D.E.; Zeng, C.; Cui, K.; Zhao, K.; Peng, W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 2009, 25, 1952–1958. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda, J.L. Using R and Bioconductor in Clinical Genomics and Transcriptomics. J. Mol. Diagn. 2020, 22, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Montojo, J.; Zuberi, K.; Rodriguez, H.; Bader, G.D.; Morris, Q. GeneMANIA: Fast gene network construction and function prediction for Cytoscape. F1000Research 2014, 3, 153. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Franz, M.; Rodriguez, H.; Lopes, C.; Zuberi, K.; Montojo, J.; Bader, G.D.; Morris, Q. GeneMANIA update 2018. Nucleic Acids Res. 2018, 46, W60–W64. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2013, 13, 2498–2504. [Google Scholar] [CrossRef]
- Stein, L.D. Using the Reactome Database. Curr. Protoc. Bioinform. 2004, 7. [Google Scholar] [CrossRef]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Oughtred, R.; Stark, C.; Breitkreutz, B.-J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef] [Green Version]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [Green Version]
- Matys, V. Transfac (R) and its module TransCompel (R): Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [Green Version]
- Wingender, E.; Chen, X.; Fricke, E.; Geffers, R.; Hehl, R.; Liebich, I.; Krull, M.; Matys, V.; Michael, H.; Ohnhäuser, R.; et al. The Transfac system on gene expression regulation. Nucleic Acids Res. 2001, 29, 281–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingender, E. Transfac: An integrated system for gene expression regulation. Nucleic Acids Res. 2000, 28, 316–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornes, O.A.; Castro-Mondragon, J.; Khan, A.; Van Der Lee, R.; Zhang, X.A.; Richmond, P.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. Jaspar 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2019, 48, D87–D92. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.-C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.-T.; Ma, X.-J.; Luo, Y. RNAscope. J. Mol. Diagn. 2012, 14, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brands, C.; Morcock, D.; Estes, J.; Deleage, C. Next-generation Viral RNA/DNA in situ Hybridization Applications in Human Immunodeficiency Virus/Simian Immunodeficiency Virus Research. J. Vis. Exp. 2020, 160. [Google Scholar] [CrossRef] [PubMed]
- Deleage, C.; Chan, C.N.; Busman-Sahay, K.; Estes, J.D. Next-generation in situ hybridization approaches to define and quantify HIV and SIV reservoirs in tissue microenvironments. Retrovirology 2018, 15, 1–10. [Google Scholar] [CrossRef]
- Mendoza-Parra, M.A.; Saleem, M.-A.M.; Blum, M.; Cholley, P.-E.; Gronemeyer, H. NGS-QC Generator: A Quality Control System for ChIP-Seq and Related Deep Sequencing-Generated Datasets. Adv. Struct. Saf. Stud. 2016, 1418, 243–265. [Google Scholar] [CrossRef]
- Andersson, E.; Tryggvason, U.; Deng, Q.; Friling, S.; Alekseenko, Z.; Robert, B.; Perlmann, T.; Ericson, J. Identification of Intrinsic Determinants of Midbrain Dopamine Neurons. Cell 2006, 124, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Koppelkamm, A.; Vennemann, B.; Lutz-Bonengel, S.; Fracasso, T.; Vennemann, M. RNA integrity in post-mortem samples: Influencing parameters and implications on RT-qPCR assays. Int. J. Leg. Med. 2011, 125, 573–580. [Google Scholar] [CrossRef]
- Ferrer, I.; Martinez, A.; Boluda, S.; Parchi, P.; Barrachina, M. Brain banks: Benefits, limitations and cautions concerning the use of post-mortem brain tissue for molecular studies. Cell Tissue Bank. 2008, 9, 181–194. [Google Scholar] [CrossRef]
- Reiman, M.; Laan, M.; Rull, K.; Sõber, S. Effects of RNA integrity on transcript quantification by total RNA sequencing of clinically collected human placental samples. FASEB J. 2017, 31, 3298–3308. [Google Scholar] [CrossRef] [Green Version]
- White, K.; Yang, P.; Li, L.; Farshori, A.; Medina, A.E.; Zielke, H.R. Effect of Postmortem Interval and Years in Storage on RNA Quality of Tissue at a Repository of the NIH NeuroBioBank. Biopreserv. Biobank. 2018, 16, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlner, J.; Holmgren, A.; Jones, A.W. Demographics and post-mortem toxicology findings in deaths among people arrested multiple times for use of illicit drugs and/or impaired driving. Forensic Sci. Int. 2016, 265, 138–143. [Google Scholar] [CrossRef]
- Jarmasz, J.S.; Stirton, H.; Davie, J.R.; Del Bigio, M.R. DNA methylation and histone post-translational modification stability in post-mortem brain tissue. Clin. Epigenet. 2019, 11, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almouzni, G.; Cedar, H. Maintenance of Epigenetic Information. Cold Spring Harb. Perspect. Biol. 2016, 8, a019372. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chen, Z.; Wu, D.; Zhang, L.; Lin, X.; Su, J.; Rodriguez, B.; Xi, Y.; Xia, Z.; Chen, X.; et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat. Genet. 2015, 47, 1149–1157. [Google Scholar] [CrossRef]
- Sato, Y.; Hilbert, L.; Oda, H.; Wan, Y.; Heddleston, J.M.; Chew, T.-L.; Zaburdaev, V.; Keller, P.; Lionnet, T.; Vastenhouw, N.; et al. Histone H3K27 acetylation precedes active transcription during zebrafish zygotic genome activation as revealed by live-cell analysis. Development 2019, 146, dev179127. [Google Scholar] [CrossRef] [Green Version]
- Nikolov, D.B.; Burley, S.K. RNA polymerase II transcription initiation: A structural view. Proc. Natl. Acad. Sci. USA 1997, 94, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klar, A.J.S. The chromosome 1;11 translocation provides the best evidence supporting genetic etiology for schizophrenia and bipolar affective disorders. Genetics 2002, 160, 1745–1747. [Google Scholar] [PubMed]
- Lacour, A.; Espinosa, A.; Louwersheimer, E.; Heilmann, S.; Hernández, I.; Wolfsgruber, S.; Fernández, V.; Wagner, H.; Rosende-Roca, M.; Mauleón, A.; et al. Genome-wide significant risk factors for Alzheimer’s disease: Role in progression to dementia due to Alzheimer’s disease among subjects with mild cognitive impairment. Mol. Psychiatry 2017, 22, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Grau, S.M.-G.; Hernández, I.; Heilmann-Heimbach, S.; Ruiz, S.; Rosende-Roca, M.; Mauleón, A.; Vargas, L.; Gómez, O.R.; Alegret, M.; Espinosa, A.; et al. Genome-wide significant risk factors on chromosome 19 and the APOE locus. Oncotarget 2018, 9, 24590–24600. [Google Scholar] [CrossRef] [Green Version]
- Joutel, A.; Corpechot, C.; Ducros, A.; Vahedi, K.; Chabriat, H.; Mouton, P.; Alamowitch, S.; Domenga, V.; Cécillion, M.; Maréchal, E.; et al. Notch3 Mutations in Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL), a Mendelian Condition Causing Stroke and Vascular Dementia. Ann. N. Y. Acad. Sci. 1997, 826, 213–217. [Google Scholar] [CrossRef]
- Joutel, A.; Corpechot, C.; Ducros, A.; Vahedi, K.; Chabriat, H.; Mouton, P.; Alamowitch, S.; Domenga, V.; Cécillion, M.; Maréchal, E.; et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nat. Cell Biol. 1996, 383, 707–710. [Google Scholar] [CrossRef]
- Canet, G.; Dias, C.; Gabelle, A.; Simonin, Y.; Gosselet, F.; Marchi, N.; Makinson, A.; Tuaillon, E.; Van De Perre, P.; Givalois, L.; et al. HIV Neuroinfection and Alzheimer’s Disease: Similarities and Potential Links? Front. Cell. Neurosci. 2018, 12, 307. [Google Scholar] [CrossRef] [PubMed]
- Okitsu, C.Y.; Hsieh, J.C.F.; Hsieh, C.-L. Transcriptional Activity Affects the H3K4me3 Level and Distribution in the Coding Region. Mol. Cell. Biol. 2010, 30, 2933–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkers, R.C.; Van Heeringen, S.J.; Jacobi, U.G.; Janssen-Megens, E.M.; Françoijs, K.-J.; Stunnenberg, H.G.; Veenstra, G.J.C. A Hierarchy of H3K4me3 and H3K27me3 Acquisition in Spatial Gene Regulation in Xenopus Embryos. Dev. Cell 2009, 17, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 2011, 508, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Najera, J.A.; Bustamante, E.A.; Bortell, N.; Morsey, B.; Fox, H.S.; Ravasi, T.; Marcondes, M.C.G. Methamphetamine abuse affects gene expression in brain-derived microglia of SIV-infected macaques to enhance inflammation and promote virus targets. BMC Immunol. 2016, 17, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Bortell, N.; Morsey, B.; Basova, L.; Fox, H.S.; Marcondes, M.C.G. Phenotypic changes in the brain of SIV-infected macaques exposed to methamphetamine parallel macrophage activation patterns induced by the common gamma-chain cytokine system. Front. Microbiol. 2015, 6, 900. [Google Scholar] [CrossRef] [Green Version]
- Kesby, J.P.; Najera, J.A.; Romoli, B.; Fang, Y.; Basova, L.; Birmingham, A.; Marcondes, M.C.G.; Dulcis, D.; Semenova, S. HIV-1 TAT protein enhances sensitization to methamphetamine by affecting dopaminergic function. Brain Behav. Immun. 2017, 65, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Mediouni, S.; Marcondes, M.C.G.; Miller, C.A.; McLaughlin, J.P.; Valente, S.T. The cross-talk of HIV-1 Tat and methamphetamine in HIV-associated neurocognitive disorders. Front. Microbiol. 2015, 6, 1164. [Google Scholar] [CrossRef] [Green Version]
- Ellis, C.; Hoffman, W.; Jaehnert, S.; Plagge, J.; Loftis, J.M.; Schwartz, D.; Huckans, M. Everyday Problems with Executive Dysfunction and Impulsivity in Adults Recovering from Methamphetamine Addiction. Addict. Disord. Their Treat. 2016, 15, 1–5. [Google Scholar] [CrossRef]
- Kohno, M.; Loftis, J.M.; Huckans, M.; Dennis, L.E.; McCready, H.; Hoffman, W.F. The relationship between interleukin-6 and functional connectivity in methamphetamine users. Neurosci. Lett. 2018, 677, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Loftis, J.M.; Janowsky, A. Neuroimmune Basis of Methamphetamine Toxicity. Int. Rev. Neurobiol. 2014, 118, 165–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basova, L.V.; Kesby, J.P.; Kaul, M.; Semenova, S.; Marcondes, M.C.G. Systems Biology Analysis of the Antagonizing Effects of HIV-1 Tat Expression in the Brain over Transcriptional Changes Caused by Methamphetamine Sensitization. Viruses 2020, 12, 426. [Google Scholar] [CrossRef] [Green Version]
- Baek, E.J.; Kim, H.; Basova, L.A.; Rosander, A.; Kesby, J.P.; Semenova, S.; Marcondes, M.C.G. Sex differences and Tat expression affect dopaminergic receptor expression and response to antioxidant treatment in methamphetamine-sensitized HIV Tat transgenic mice. Neuropharmacology 2020, 178, 108245. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, K.; Iudicello, J.E.; Morgan, E.E.; Weber, E.; Moore, D.J.; Franklin, D.R.; Ellis, R.J.; Grant, I.; Woods, S.P. Human Immunodeficiency Virus Infection Heightens Concurrent Risk of Functional Dependence in Persons with Long-Term Methamphetamine use. J. Addict. Med. 2013, 7, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koob, G.F.; Volkow, N.D. Neurocircuitry of Addiction. Neuropsychopharmacology 2009, 35, 217–238. [Google Scholar] [CrossRef] [Green Version]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773. [Google Scholar] [CrossRef]
- Volkow, N.D.; Koob, G.; Baler, R. Biomarkers in Substance Use Disorders. ACS Chem. Neurosci. 2015, 6, 522–525. [Google Scholar] [CrossRef]
- Nickoloff-Bybel, E.A.; Calderon, T.M.; Gaskill, P.J.; Berman, J.W. HIV Neuropathogenesis in the Presence of a Disrupted Dopamine System. J. Neuroimmune Pharmacol. 2020, 15, 729–742. [Google Scholar] [CrossRef]
- Gonçalves, J.; Baptista, S.; Martins, T.; Milhazes, N.; Borges, F.; Ribeiro, C.F.; Malva, J.O.; Silva, A.P. Methamphetamine-induced neuroinflammation and neuronal dysfunction in the mice hippocampus: Preventive effect of indomethacin. Eur. J. Neurosci. 2010, 31, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Ott, T.; Nieder, A. Dopamine and Cognitive Control in Prefrontal Cortex. Trends Cogn. Sci. 2019, 23, 213–234. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.M. The Prefrontal Cortex—An Update. Neuron 2001, 30, 319–333. [Google Scholar] [CrossRef] [Green Version]
- Fuster, J.M. Memory networks in the prefrontal cortex. Prog. Brain Res. 2000, 122, 309–316. [Google Scholar] [CrossRef]
- Fuster, J.M. Prefrontal Cortex and the Bridging of Temporal Gaps in the Perception-Action Cycle. Ann. N. Y. Acad. Sci. 1990, 608, 318–336. [Google Scholar] [CrossRef]
- Goldman-Rakic, P.S. Architecture of the Prefrontal Cortex and the Central Executive. Ann. N. Y. Acad. Sci. 1995, 769, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J.E.; Fellous, J.-M.; Wang, X.-J. A role for NMDA-receptor channels in working memory. Nat. Neurosci. 1998, 1, 273–275. [Google Scholar] [CrossRef]
- Tegnér, J.; Compte, A.; Wang, X.-J. The dynamical stability of reverberatory neural circuits. Biol. Cybern. 2002, 87, 471–481. [Google Scholar] [CrossRef]
- Rolls, E.T.; Loh, M.; Deco, G.; Winterer, G. Computational models of schizophrenia and dopamine modulation in the prefrontal cortex. Nat. Rev. Neurosci. 2008, 9, 696–709. [Google Scholar] [CrossRef]
- Wearne, T.A.; Cornish, J.L. Inhibitory regulation of the prefrontal cortex following behavioral sensitization to amphetamine and/or methamphetamine psychostimulants: A review of GABAergic mechanisms. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 95, 109681. [Google Scholar] [CrossRef] [PubMed]
- Gelman, B.B.; Lisinicchia, J.G.; Chen, T.; Johnson, K.M.; Jennings, K.; Freeman, D.H.; Soukup, V.M. Prefrontal Dopaminergic and Enkephalinergic Synaptic Accommodation in HIV-associated Neurocognitive Disorders and Encephalitis. J. Neuroimmune Pharmacol. 2012, 7, 686–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelman, B.B.; Spencer, J.A.; Holzer, C.E.; Soukup, V.M.; Iii, C.E.H. Abnormal Striatal Dopaminergic Synapses in National NeuroAIDS Tissue Consortium Subjects with HIV Encephalitis. J. Neuroimmune Pharmacol. 2006, 1, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Skowronska, M.; McDonald, M.; Velichkovska, M.; Leda, A.R.; Park, M.; Toborek, M. Methamphetamine increases HIV infectivity in neural progenitor cells. J. Biol. Chem. 2018, 293, 296–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, H.; Cadet, J.L. p53-knockout mice are protected against the long-term effects of methamphetamine on dopaminergic terminals and cell bodies. J. Neurochem. 2002, 69, 780–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagoulias, I.; Karagiannis, F.; Aggeletopoulou, I.; Georgakopoulos, T.; Argyropoulos, C.P.; Akinosoglou, K.; Gogos, C.; Skoutelis, A.; Mouzaki, A. Ets-2 Acts as a Transcriptional Repressor of the Human Immunodeficiency Virus Type 1 through Binding to a Repressor-Activator Target Sequence of 5′-LTR. Front. Immunol. 2018, 8, 1924. [Google Scholar] [CrossRef]
- Panagoulias, I.; Georgakopoulos, T.; Aggeletopoulou, I.; Agelopoulos, M.; Thanos, D.; Mouzaki, A. Transcription Factor Ets-2 Acts as a Preinduction Repressor of Interleukin-2 (IL-2) Transcription in Naive T Helper Lymphocytes. J. Biol. Chem. 2016, 291, 26707–26721. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Average HIV+Meth- H3K4me3 | Average HIV+Meth+ H3K4me3 | Pooled Input hg38 | |
|---|---|---|---|
| Total number of reads | 39,365,646 | 40,840,923 | 42,982,011 |
| Total number of alignments (hg38) | 35,790,215 | 38,438,156 | 39,813,903 |
| Unique alignments (-q 25) | 33,267,427 | 35,878,237 | 34,930,380 |
| Unique alignments (without duplicates) | 23,200,850 | 26,581,519 | 32,071,577 |
| Group | Case# | Plasma VL at CD4 nadir | ART History | Frontal Cortex RNA RIN |
|---|---|---|---|---|
| HIV+Meth- | 4067 | 5.12 | ZDV + 3TC, LPV + RTV, TFV | 6.6 |
| HIV+Meth- | 4077 | 2.6 | ATV, FTC, RTV, TFV, TFV + FTC | 5.7 |
| HIV+Meth- | 4083 | 3.35 | ZDV + 3TC, RFV, ATV, RTV, TFV/FTC | 6.8 |
| HIV+Meth+ | 1163 | 5.57 | DRV, RTV, TFV + FTC | 4.6 |
| HIV+Meth+ | 2074 | 4.57 | 3TC, D4T, IDV, NVP, RTV, ABC, LPV + RTV, NFV, DDI, TFV | 5.2 |
| HIV+Meth+ | 4167 | 6.28 | LPV + RTV, TFV + FTC | 6.3 |
| Matrix | Factor | Consensus Sequence | Classification | Average #Sites Interval Sequence |
|---|---|---|---|---|
| V$SP1_13 | Sp1 | gcggctctgcggGGCGGggcgggg | ZFC2H2 | 4.33 |
| V$P53_03 | P53 | cgACATGGacacacatgggt | P53 | 3.33 |
| V$ETS2_06 | c-Ets-2 | ggCCGGAgaggctgcccctt | ETS | 2.85 |
| V$JUNDFRA2_01 | JUND:FRA2 | aaTGACTcaa | BZIP | 2.5 |
| V$ASCL1_03 | MASH-1 | cgCAGCTgcc | BHLH | 2.5 |
| V$NR3C1_13 | GR | aacacaataTGTACa | ZFC4-NR | 2.33 |
| V$CREBP1_01 | ATF-2 | tgaCGTCA | BZIP | 2.29 |
| V$AP2ALPHA_03 | AP2aA | cgCGCCCccggctct | BHSH | 2.27 |
| V$MYCMAX_03 | cMyc:Max | agttatgcACGTGtgtacca | BHLH | 2.15 |
| V$HOXA5_03 | HOXA5 | AATTAgtg | HOX | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basova, L.; Lindsey, A.; McGovern, A.M.; Ellis, R.J.; Marcondes, M.C.G. Detection of H3K4me3 Identifies NeuroHIV Signatures, Genomic Effects of Methamphetamine and Addiction Pathways in Postmortem HIV+ Brain Specimens that Are Not Amenable to Transcriptome Analysis. Viruses 2021, 13, 544. https://doi.org/10.3390/v13040544
Basova L, Lindsey A, McGovern AM, Ellis RJ, Marcondes MCG. Detection of H3K4me3 Identifies NeuroHIV Signatures, Genomic Effects of Methamphetamine and Addiction Pathways in Postmortem HIV+ Brain Specimens that Are Not Amenable to Transcriptome Analysis. Viruses. 2021; 13(4):544. https://doi.org/10.3390/v13040544
Chicago/Turabian StyleBasova, Liana, Alexander Lindsey, Anne Marie McGovern, Ronald J. Ellis, and Maria Cecilia Garibaldi Marcondes. 2021. "Detection of H3K4me3 Identifies NeuroHIV Signatures, Genomic Effects of Methamphetamine and Addiction Pathways in Postmortem HIV+ Brain Specimens that Are Not Amenable to Transcriptome Analysis" Viruses 13, no. 4: 544. https://doi.org/10.3390/v13040544
APA StyleBasova, L., Lindsey, A., McGovern, A. M., Ellis, R. J., & Marcondes, M. C. G. (2021). Detection of H3K4me3 Identifies NeuroHIV Signatures, Genomic Effects of Methamphetamine and Addiction Pathways in Postmortem HIV+ Brain Specimens that Are Not Amenable to Transcriptome Analysis. Viruses, 13(4), 544. https://doi.org/10.3390/v13040544