
Combined Short and Long-Read Sequencing Reveals a Complex Transcriptomic Architecture of African Swine Fever Virus



Abstract
:1. Introduction
2. Materials and Methods
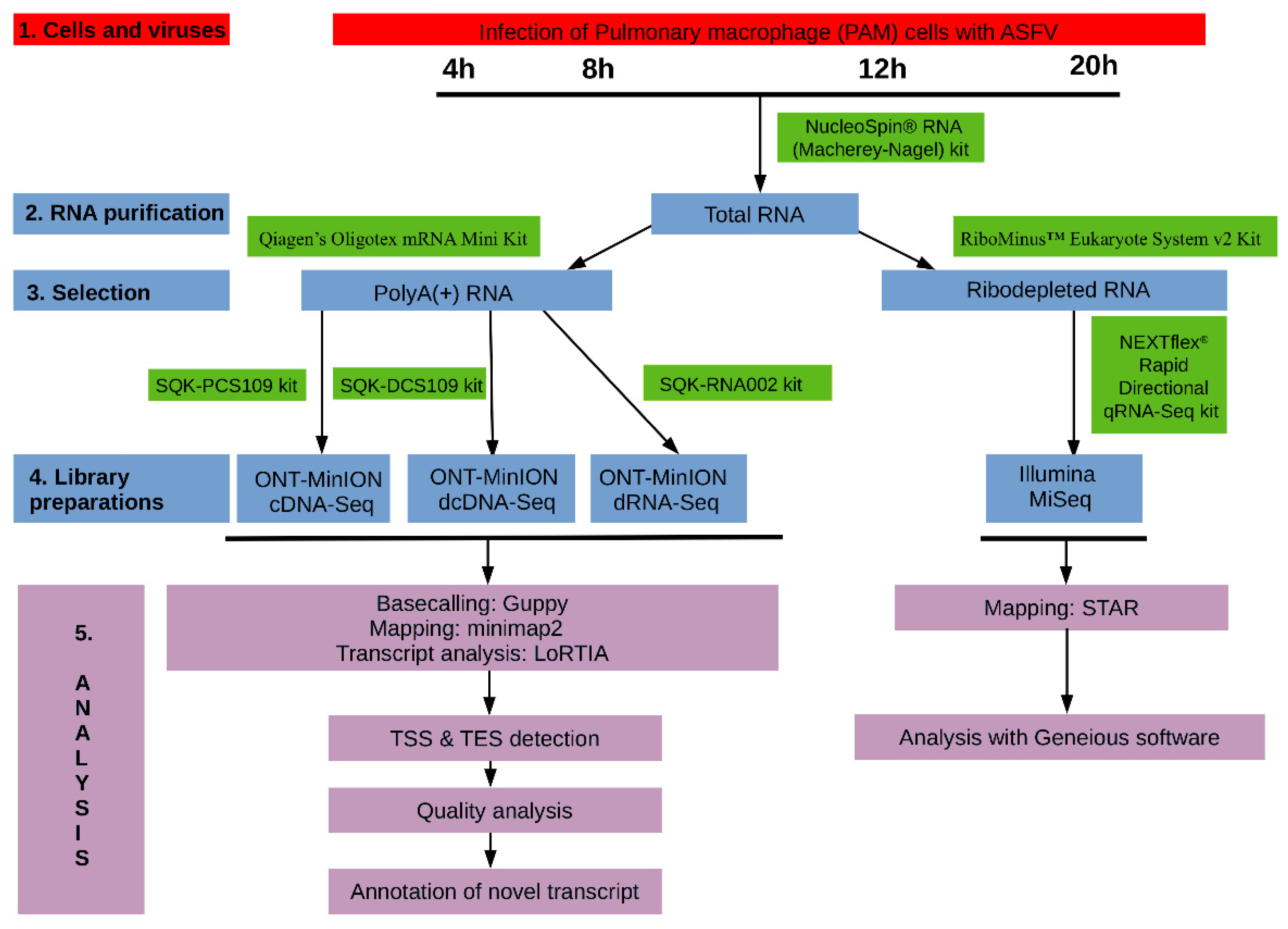
2.1. Cells, Viruses and Infection
2.2. Infection Efficiency
2.3. RNA Purification
2.3.1. Extraction of Total RNA
2.3.2. Purification of Polyadenylated RNAs
2.3.3. Removal of the Ribosomal RNAs
2.4. Library Preparations
2.4.1. Direct RNA Sequencing on MinION SpotON Flow Cells
2.4.2. Direct cDNA Sequencing on MinION Flow Cells
2.4.3. Amplified cDNA-Sequencing Using MinION Device
2.4.4. Amplified cDNA Sequencing Using Illumina MiSeq Sequencer
2.5. Determination of the Quantity and Quality of RNA Samples and Sequencing Ready Libraries
2.6. Data Processing and Analysis
2.6.1. ONT Sequencing
- For dRNA sequencing and dcDNA sequencing reads: −5 TGCCATTAGGCCGGG --five_score 16 -- check_in_soft 15 −3 AAAAAAAAAAAAAAA --three_score 16 s Poisson–f true.
- For o(dT)-primed cDNA reads: −5 GCTGATATTGCTGGG -- five_score 16 --check_in_soft 15 −3 AAAAAAAAAAAAAAA --three_score 16 s Poisson–f true.
2.6.2. Illumina Sequencing
3. Results
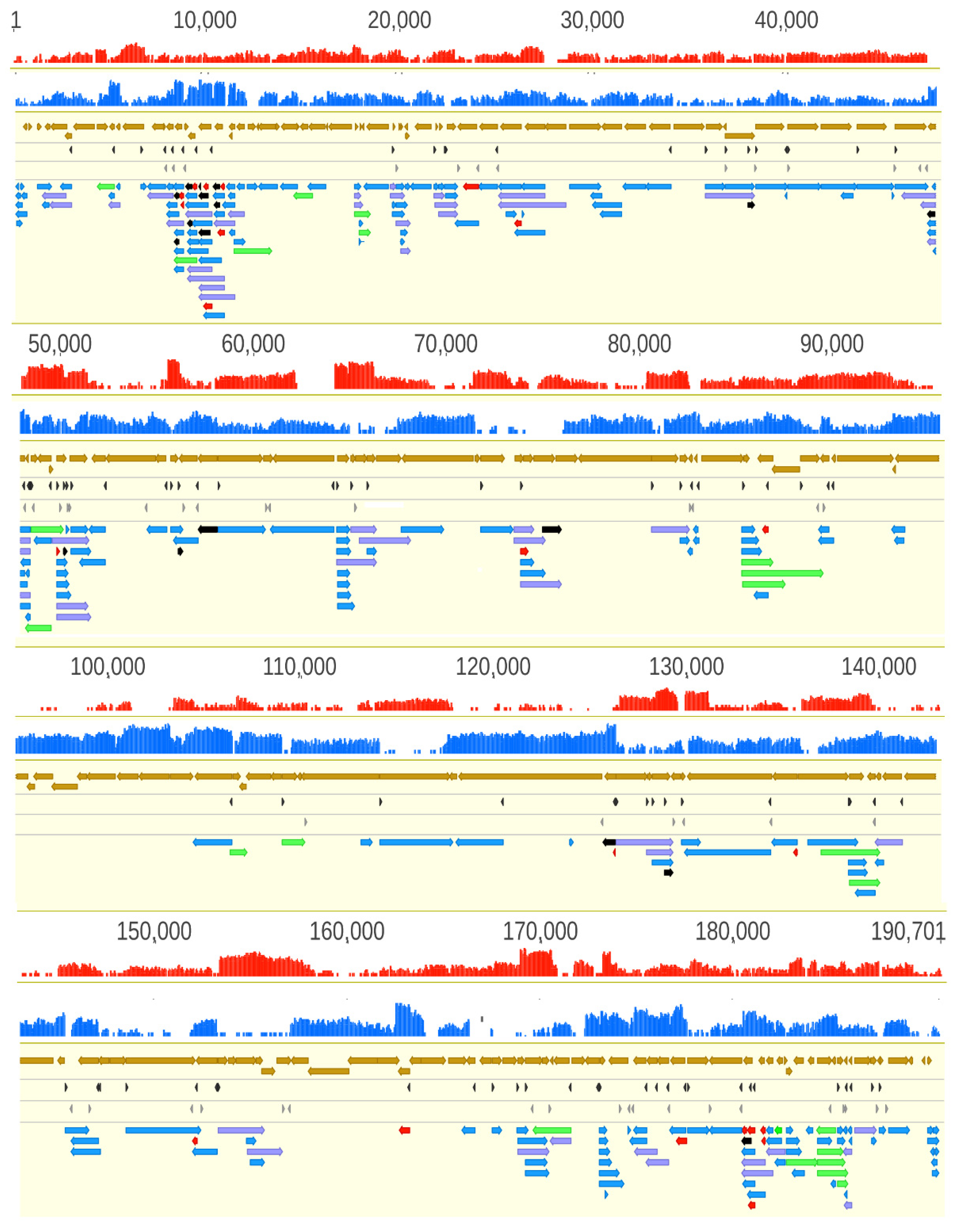
3.1. Comparing the Genomes of Strains ASFV_HU_2018 and Ba71V of ASFV
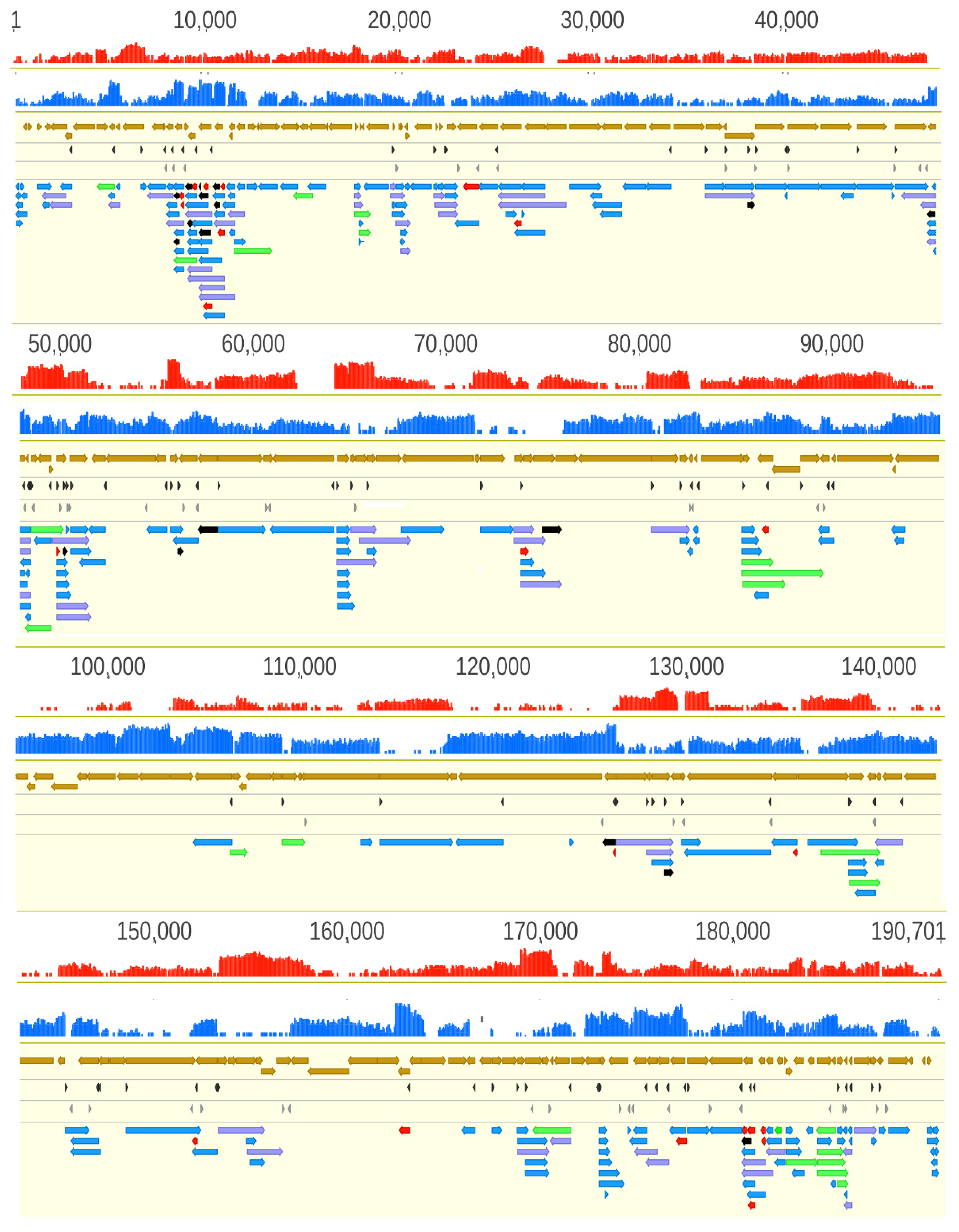
3.2. Analysis of the ASFP Transcriptome with a Dual Sequencing Approach
3.3. Novel Putative Protein-Coding Genes
3.4. Upstream ORF-Containing mRNAs
3.5. Putative Non-Coding Transcripts
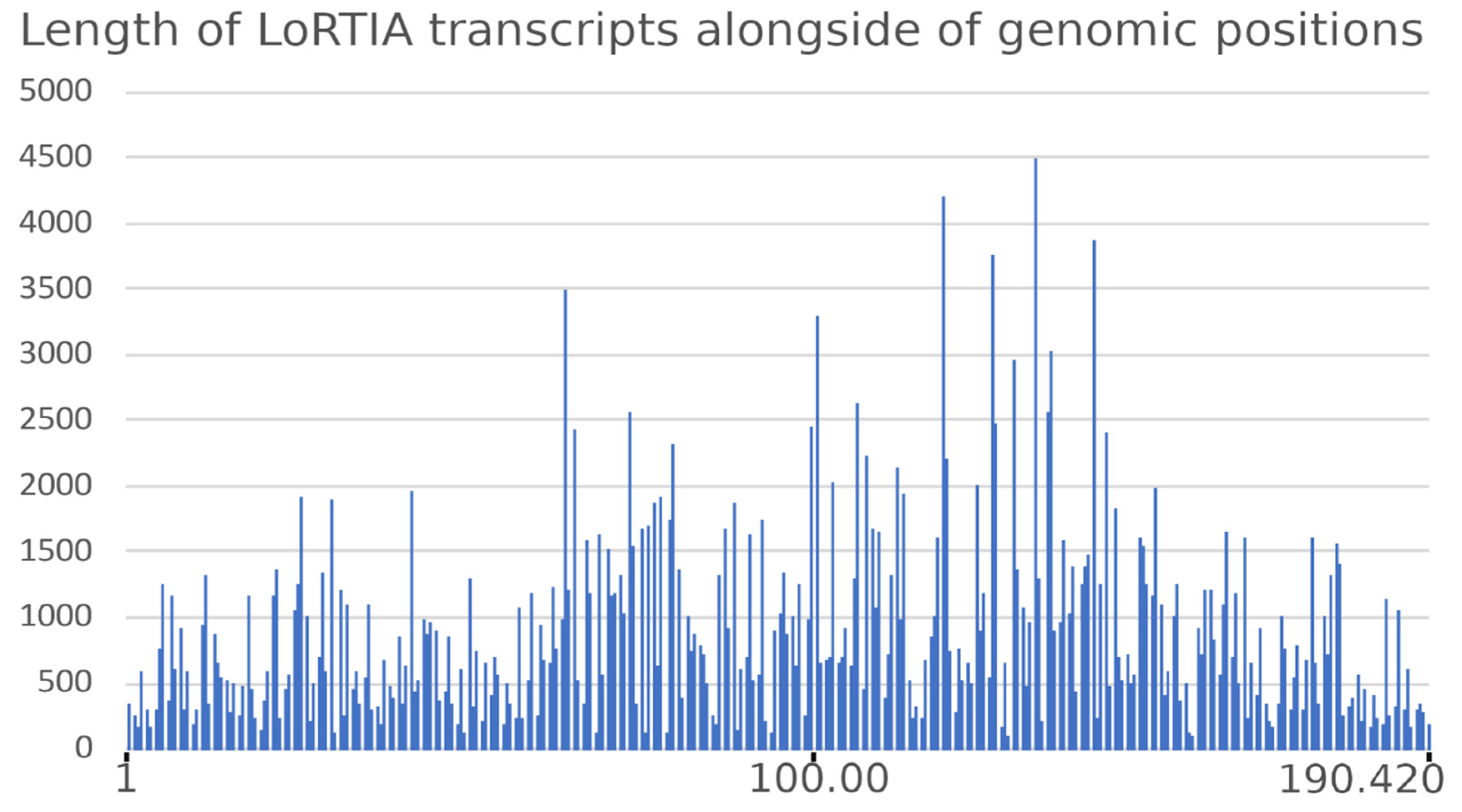
3.6. Transcript Isoforms
3.7. Polycistronic Transcripts
3.8. Complex Transcripts
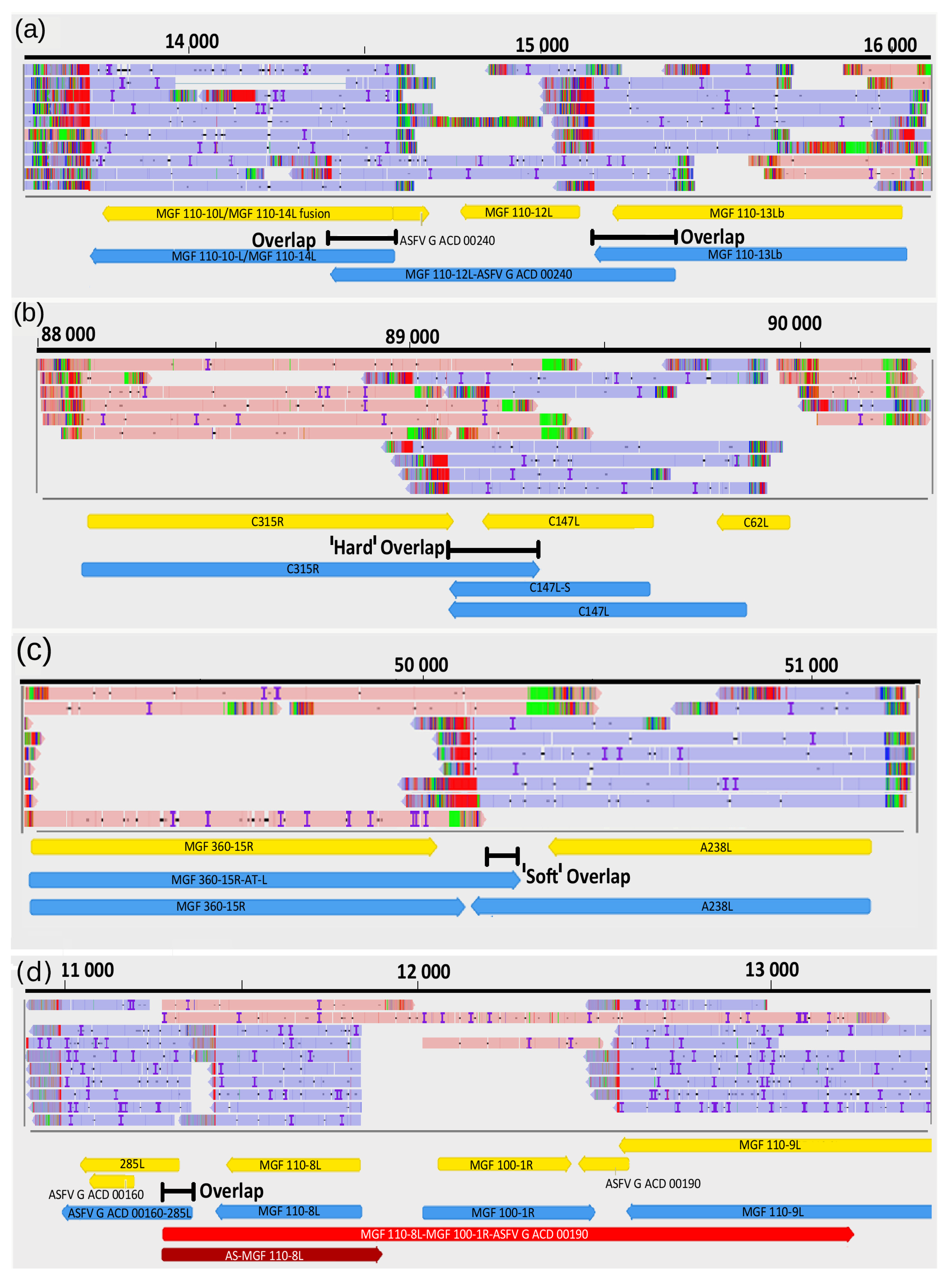
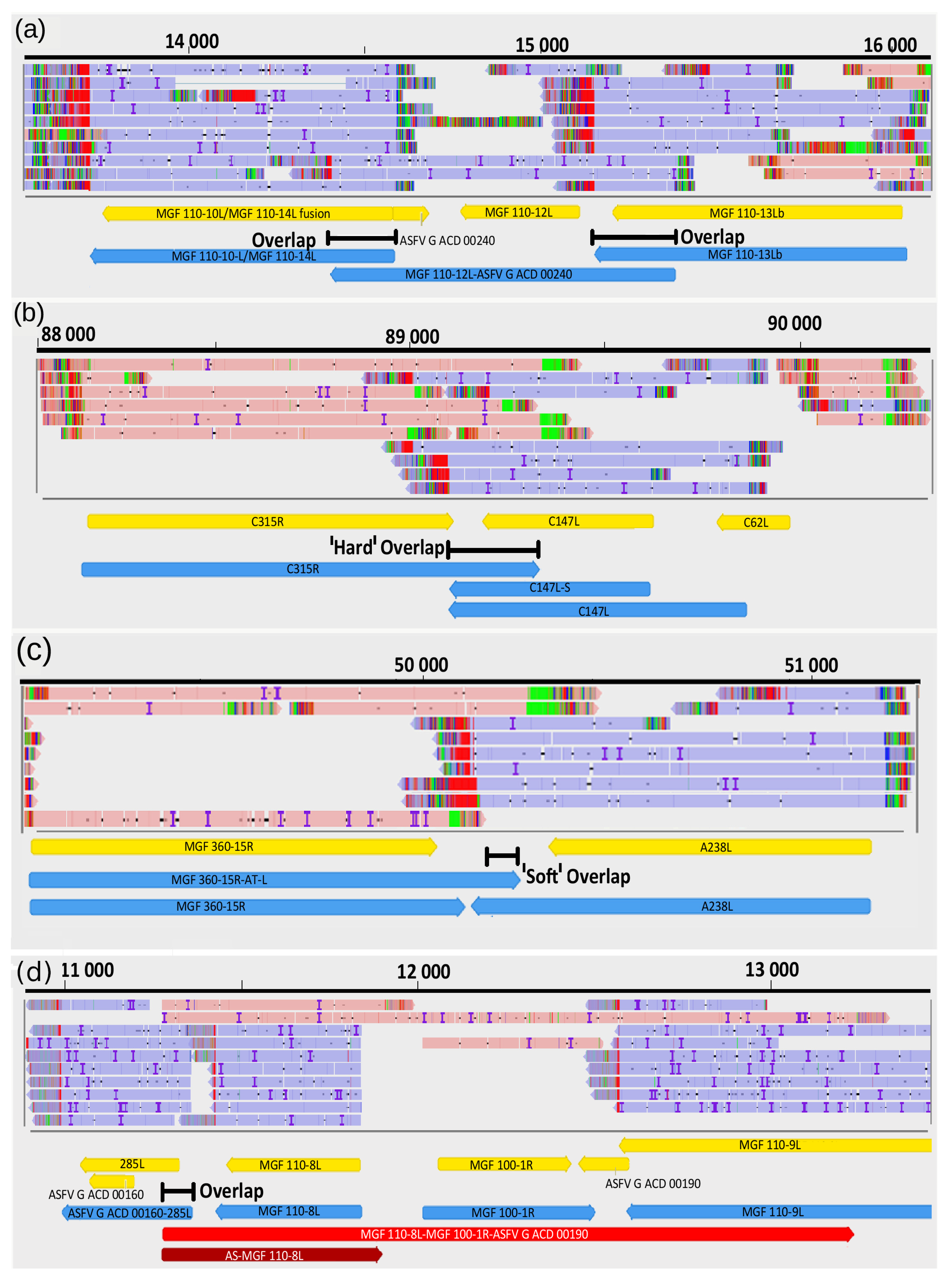
3.9. Transcriptional Overlaps
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Balázs, Z.; Tombácz, D.; Csabai, Z.; Moldován, N.; Snyder, M.; Boldogkoi, Z. Template-switching artifacts resemble alternative polyadenylation. BMC Genom. 2019, 20, 1–10. [Google Scholar] [CrossRef]
- Tombácz, D.; Csabai, Z.; Oláh, P.; Balázs, Z.; Likó, I.; Zsigmond, L.; Sharon, D.; Snyder, M.; Boldogkői, Z. Full-Length Isoform Sequencing Reveals Novel Transcripts and Substantial Transcriptional Overlaps in a Herpesvirus. PLoS ONE 2016, 11, e0162868. [Google Scholar] [CrossRef]
- Moldován, N.; Tombácz, D.; Szűcs, A.; Csabai, Z.; Snyder, M.; Boldogkői, Z. Multi-Platform Sequencing Approach Reveals a Novel Transcriptome Profile in Pseudorabies Virus. Front. Microbiol. 2017, 8, 2708. [Google Scholar] [CrossRef]
- Mazur-Panasiuk, N.; Żmudzki, J.; Woźniakowski, G. African swine fever virus—persistence in different environmental conditions and the possibility of its indirect transmission. J. Vet. Res. 2019, 63, 303–310. [Google Scholar] [CrossRef]
- Reis, A.L.; Netherton, C.; Dixon, L.K. Unraveling the Armor of a Killer: Evasion of Host Defenses by African Swine Fever Virus. J. Virol. 2017, 91, 6–11. [Google Scholar] [CrossRef]
- Achenbach, J.E.; Gallardo, C.; Nieto-Pelegrín, E.; Rivera-Arroyo, B.; Degefa-Negi, T.; Arias, M.; Jenberie, S.; Mulisa, D.D.; Gizaw, D.; Gelaye, E.; et al. Identification of a New Genotype of African Swine Fever Virus in Domestic Pigs from Ethiopia. Transbound. Emerg. Dis. 2017, 64, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.; Martins, C.; Ferreira, F. Early intranuclear replication of African swine fever virus genome modifies the landscape of the host cell nucleus. Virus Res. 2015, 210, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Chapman, D.A.G.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.M.; Salas, M.L. African swine fever virus transcription. Virus Res. 2013, 173, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Almazán, F.; Rodríguez, J.M.; Angulo, A.; Viñuela, E.; Rodriguez, J.F. Transcriptional mapping of a late gene coding for the p12 attachment protein of African swine fever virus. J. Virol. 1993, 67, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Tombácz, D.; Csabai, Z.; Szucs, A.; Balázs, Z.; Moldován, N.; Sharon, D.; Snyder, M.; Boldogkoi, Z. Long-read isoform sequencing reveals a hidden complexity of the transcriptional landscape of herpes simplex virus type 1. Front. Microbiol. 2017, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Cackett, G.; Matelska, D.; Sýkora, M.; Portugal, R.; Malecki, M.; Bähler, J.; Dixon, L.; Werner, F. The African Swine Fever Virus Transcriptome. J. Virol. 2020, 94, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Martens, C.A.; Bruno, D.P.; Porcella, S.F.; Moss, B. Pervasive initiation and 3′-end formation of poxvirus postreplicative RNAs. J. Biol. Chem. 2012, 287, 31050–31060. [Google Scholar] [CrossRef]
- Balázs, Z.; Tombácz, D.; Szűcs, A.; Snyder, M.; Boldogkői, Z. Long-read sequencing of the human cytomegalovirus transcriptome with the Pacific Biosciences RSII platform. Sci. Data 2017, 4, 170194. [Google Scholar] [CrossRef] [PubMed]
- Moldován, N.; Tombácz, D.; Szűcs, A.; Csabai, Z.; Balázs, Z.; Kis, E.; Molnár, J.; Boldogkői, Z. Third-generation Sequencing Reveals Extensive Polycistronism and Transcriptional Overlapping in a Baculovirus. Sci. Rep. 2018, 8, 8604. [Google Scholar] [CrossRef]
- Boldogkői, Z.; Moldován, N.; Balázs, Z.; Snyder, M.; Tombácz, D. Long-Read Sequencing—A Powerful Tool in Viral Transcriptome Research. Trends Microbiol. 2019, 27, 578–592. [Google Scholar] [CrossRef]
- Tombácz, D.; Moldován, N.; Balázs, Z.; Gulyás, G.; Csabai, Z.; Boldogkői, M.; Snyder, M.; Boldogkői, Z. Multiple Long-Read Sequencing Survey of Herpes Simplex Virus Dynamic Transcriptome. Front. Genet. 2019, 10, 1–20. [Google Scholar] [CrossRef]
- Yáñez, R.J.; Rodríguez, J.M.; Nogal, M.L.; Yuste, L.; Enríquez, C.; Rodriguez, J.F.; Viñuela, E. Analysis of the Complete Nucleotide Sequence of African Swine Fever Virus. Virology 1995, 208, 249–278. [Google Scholar] [CrossRef]
- Portugal, R.S.; Bauer, A.; Keil, G.M. Selection of differently temporally regulated African swine fever virus promoters with variable expression activities and their application for transient and recombinant virus mediated gene expression. Virology 2017, 508, 70–80. [Google Scholar] [CrossRef]
- García-Escudero, R.; Viñuela, E. Structure of African Swine Fever Virus Late Promoters: Requirement of a TATA Sequence at the Initiation Region. J. Virol. 2000, 74, 8176–8182. [Google Scholar] [CrossRef]
- Sánchez, E.G.; Riera, E.; Nogal, M.; Gallardo, C.; Fernández, P.; Bello-Morales, R.; López-Guerrero, J.A.; Chitko-Mckown, C.G.; Richt, J.A.; Revilla, Y. Phenotyping and susceptibility of established porcine cells lines to African Swine Fever Virus infection and viral production. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Olasz, F.; Mészáros, I.; Marton, S.; Kaján, G.L.; Tamás, V.; Locsmándi, G.; Magyar, T.; Bálint, Á.; Bányai, K.; Zádori, Z. A simple method for sample preparation to facilitate efficient whole-genome sequencing of African swine fever virus. Viruses 2019, 11, 1129. [Google Scholar] [CrossRef]
- Broyles, S.S. Vaccinia virus transcription. J. Gen. Virol. 2003, 84, 2293–2303. [Google Scholar] [CrossRef]
- Salas, M.L.; Rey-Campos, J.; Almendral, J.M.; Talavera, A.; Viñuela, E. Transcription and translation maps of african swine fever virus. Virology 1986, 152, 228–240. [Google Scholar] [CrossRef]
- Salas, M.L.; Kuznar, J.; Viñuela, E. Polyadenylation, methylation, and capping of the RNA synthesized in vitro by African swine fever virus. Virology 1981, 113, 484–491. [Google Scholar] [CrossRef]
- Jaing, C.; Rowland, R.R.R.; Allen, J.E.; Certoma, A.; Thissen, J.B.; Bingham, J.; Rowe, B.; White, J.R.; Wynne, J.W.; Johnson, D.; et al. Gene expression analysis of whole blood RNA from pigs infected with low and high pathogenic African swine fever viruses. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, 6th ed.; Office International des Epizooties: Paris, France, 2008.
- Zsak, L.; Neilan, J.G. Regulation of apoptosis in African swine fever virus-infected macrophages. Sci. World J. 2002, 2, 1186–1195. [Google Scholar] [CrossRef]
- Hurtado, C.; Bustos, M.J.; Carrascosa, A.L. The use of COS-1 cells for studies of field and laboratory African swine fever virus samples. J. Virol. Methods 2010, 164, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa, A.L.; Bustos, M.J.; de Leon, P. Methods for growing and titrating african swine fever virus: Field and laboratory samples. Curr. Protoc. Cell Biol. 2011, 53, 26.14.1–26.14.25. [Google Scholar] [CrossRef]
- Bustos, M.J.; Nogal, M.L.; Revilla, Y.; Carrascosa, A.L. Plaque assay for African swine fever virus on swine macrophages. Arch. Virol. 2002, 147, 1453–1459. [Google Scholar] [CrossRef]
- Tombácz, D.; Sharon, D.; Szűcs, A.; Moldován, N.; Snyder, M.; Boldogkői, Z. Transcriptome-wide survey of pseudorabies virus using next- and third-generation sequencing platforms. Sci. Data 2018, 5, 180119. [Google Scholar] [CrossRef]
- Tombácz, D.; Prazsák, I.; Szucs, A.; Dénes, B.; Snyder, M.; Boldogkoi, Z. Dynamic transcriptome profiling dataset of vaccinia virus obtained from long-read sequencing techniques. Gigascience 2018, 7, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Boldogkői, Z.; Szűcs, A.; Balázs, Z.; Sharon, D.; Snyder, M.; Tombácz, D. Transcriptomic study of herpes simplex virus type-1 using full-length sequencing techniques. Sci. Data 2018, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Wang, Z.; Jia, L.; Li, J.; Liu, H.; Liu, D. Pan-Genomic Analysis of African Swine Fever Virus. Virol. Sin. 2019, 35, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Galindo, I.; Alonso, C. African swine fever virus: A review. Viruses 2017, 9, 103. [Google Scholar] [CrossRef]
- Olasz, F.; Tombácz, D.; Torma, G.; Csabai, Z.; Moldován, N.; Dörmő, Á.; Prazsák, I.; Mészáros, I.; Magyar, T.; Tamás, V.; et al. Short and Long-Read Sequencing Survey of the Dynamic Transcriptomes of African Swine Fever Virus and the Host Cells. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Sessegolo, C.; Cruaud, C.; Da Silva, C.; Cologne, A.; Dubarry, M.; Derrien, T.; Lacroix, V.; Aury, J.M. Transcriptome profiling of mouse samples using nanopore sequencing of cDNA and RNA molecules. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Lee, S.; Liu, B.; Lee, S.; Huang, S.X.; Shen, B.; Qian, S.B. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 2012, 109, E2424–E2432. [Google Scholar] [CrossRef]
- Boldogkői, Z.; Balázs, Z.; Moldován, N.; Prazsák, I.; Tombácz, D. Novel classes of replication-associated transcripts discovered in viruses. RNA Biol. 2019, 16, 166–175. [Google Scholar] [CrossRef]
- Prazsák, I.; Moldován, N.; Balázs, Z.; Tombácz, D.; Megyeri, K.; Szűcs, A.; Csabai, Z.; Boldogkői, Z. Long-read sequencing uncovers a complex transcriptome topology in varicella zoster virus. BMC Genom. 2018, 19, 873. [Google Scholar] [CrossRef] [PubMed]
- Psaila, A.M.; Vohralik, E.J.; Quinlan, K.G.R. Shades of White: New insights into tissue-resident leukocyte heterogeneity. FEBS J. 2021, 1–11. [Google Scholar] [CrossRef]
- Franzoni, G.; Graham, S.P.; Giudici, S.D.; Bonelli, P.; Pilo, G.; Anfossi, A.G.; Pittau, M.; Nicolussi, P.S.; Laddomada, A.; Oggiano, A. Characterization of the interaction of African swine fever virus with monocytes and derived macrophage subsets. Vet. Microbiol. 2017, 198, 88–98. [Google Scholar] [CrossRef]
- Tombácz, D.; Prazsák, I.; Csabai, Z.; Moldován, N.; Dénes, B.; Snyder, M.; Boldogkői, Z. Long-read assays shed new light on the transcriptome complexity of a viral pathogen. Sci. Rep. 2020, 10, 13822. [Google Scholar] [CrossRef]
- Stacey, S.N.; Jordan, D.; Williamson, A.J.K.; Brown, M.; Coote, J.H.; Arrand, J.R. Leaky Scanning Is the Predominant Mechanism for Translation of Human Papillomavirus Type 16 E7 Oncoprotein from E6/E7 Bicistronic mRNA. J. Virol. 2000, 74, 7284–7297. [Google Scholar] [CrossRef]
- Kronstad, L.M.; Brulois, K.F.; Jung, J.U.; Glaunsinger, B.A. Dual short upstream open reading frames control translation of a herpesviral polycistronic mRNA. PLoS Pathog. 2013, 9, e1003156. [Google Scholar] [CrossRef]
- Vilela, C.; McCarthy, J.E.G. Regulation of fungal gene expression via short open reading frames in the mRNA 5′untranslated region. Mol. Microbiol. 2003, 49, 859–867. [Google Scholar] [CrossRef]
- Geballe, A.P.; Mocarski, E.S. Translational control of cytomegalovirus gene expression is mediated by upstream AUG codons. J. Virol. 1988, 62, 3334–3340. [Google Scholar] [CrossRef]
- Calvo, S.E.; Pagliarini, D.J.; Mootha, V.K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA 2009, 106, 7507–7512. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.; Romão, L. Translation of the human erythropoietin transcript is regulated by an upstream open reading frame in response to hypoxia. Rna 2014, 20, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Tombácz, D.; Balázs, Z.; Csabai, Z.; Snyder, M.; Boldogkői, Z. Long-Read Sequencing Revealed an Extensive Transcript Complexity in Herpesviruses. Front. Genet. 2018, 9, 259. [Google Scholar] [CrossRef]
- Dellino, G.I.; Cittaro, D.; Piccioni, R.; Luzi, L.; Banfi, S.; Segalla, S.; Cesaroni, M.; Mendoza-Maldonado, R.; Giacca, M.; Pelicci, P.G. Genome-wide mapping of human DNA-replication origins: Levels of transcription at ORC1 sites regulate origin selection and replication timing. Genome Res. 2013, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tikhanovich, I.; Liang, B.; Seoighe, C.; Folk, W.R.; Nasheuer, H.P. Inhibition of Human BK Polyomavirus Replication by Small Noncoding RNAs. J. Virol. 2011, 85, 6930–6940. [Google Scholar] [CrossRef]
- Boldogköi, Z. Transcriptional interference networks coordinate the expression of functionally related genes clustered in the same genomic loci. Front. Genet. 2012, 3, 122. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ORF | ATG (+)/Stop (−) | ATG (−)/Stop (+) | Strand |
|---|---|---|---|
| DP60R | 403 | 582 | − |
| 285L | 11,042 | 11,326 | − |
| MGF 110-8L | 11,455 | 11,838 | − |
| MGF-100-1R | 12,056 | 12,430 | + |
| MGF 110-9L | 12,589 | 13,461 | − |
| MGF 110-10L/MGF 110-14L | 13,752 | 14,570 | − |
| MGF 110-12L | 14,760 | 15,119 | − |
| MGF 110-13Lb | 15,205 | 16,029 | − |
| MGF 360-4L | 16,210 | 17,373 | − |
| MGF 360-6L | 18,188 | 19,315 | − |
| MGF 360-10L | 26,367 | 27,404 | − |
| MGF 360-11L | 27,432 | 28,493 | − |
| MGF 505-1R | 28,701 | 30,296 | + |
| MGF 360-12L | 30,349 | 31,401 | − |
| MGF 360-13L | 31,562 | 32,623 | − |
| MGF 360-14L | 32,808 | 33,881 | − |
| I7L | 181,681 | 181,989 | − |
| I8L | 182,203 | 182,514 | − |
| I9R | 182,709 | 182,999 | + |
| I10L | 183,075 | 183,587 | − |
| I11L | 183,826 | 184,107 | − |
| DP60R | 190,010 | 190,189 | + |
| Name | TSS (+)/TES (−) | TSS (−)/TES (+) | Strand |
|---|---|---|---|
| MGF 110-3L.3-AT-L | 8183 | 8458 | − |
| MGF 110-3L.3 | 8210 | 8458 | − |
| MGF 110-4L.3-AT-L | 8785 | 9146 | − |
| MGF 110-4L.3 | 8902 | 9146 | − |
| MGF 110-5L-6L.2-AT-L2 | 9468 | 9961 | − |
| MGF 110-5L-6L.1 | 9468 | 10,064 | − |
| MGF 110-5L-6L.6 | 9468 | 9585 | − |
| MGF 110-7L.1-AT-L | 10,220 | 10,563 | − |
| MGF 110-7L.1 | 10,270 | 10,563 | − |
| MGF 505-4R.13 | 37,956 | 38,295 | + |
| A224L.5 | 47,236 | 47,621 | − |
| A151R.3 | 49,961 | 50,173 | + |
| A137R.4 | 55,895 | 56,146 | + |
| F334L.2 | 56,946 | 57,937 | − |
| EP402R.2 | 74,801 | 75,785 | + |
| CP204L.2 | 125,685 | 126,344 | − |
| CP312R.6 | 128,878 | 129,347 | + |
| MGF 100-1L.2 | 180,386 | 180,891 | − |
| I8L.2 | 182,120 | 182,425 | − |
| Name | TSS (+)/TES (−) | TSS (−)/TES (+) | Strand |
|---|---|---|---|
| nc-MGF 110-3L | 8455 | 8680 | − |
| nc-MGF 110-3L-AT-S | 8526 | 8680 | − |
| nc-MGF 110-4L | 9146 | 9368 | − |
| nc-MGF 110-5L-6L | 9705 | 9961 | − |
| nc-MGF 110-5L-6L-L | 9705 | 10,162 | − |
| nc-MGF 110-7L-AT-L | 10,481 | 10,814 | − |
| nc-MGF 110-7L | 10,619 | 10,814 | − |
| nc-MGF 300-4L | 23,203 | 23,976 | − |
| nc-MGF 360-9L | 25,832 | 26,183 | − |
| nc-A151R | 49,646 | 49,784 | + |
| nc-EP152R-EP153R | 73,637 | 74,032 | + |
| nc-c275L | 86,209 | 86,487 | − |
| nc-CP204L | 126,247 | 126,344 | − |
| nc-NP419L | 135,550 | 135,755 | − |
| nc-H359L | 151,958 | 152,195 | − |
| nc-E184L | 162,670 | 163,182 | − |
| nc-DP238L | 176,989 | 177,547 | − |
| nc-MGF 100-1L | 180,386 | 180,613 | − |
| nc-MGF 100-3L-MGF 100-1L-AT-L2 | 180,671 | 181,084 | − |
| nc-MGF 100-3L-MGF 100-1L-AT-L3 | 180,743 | 181,084 | − |
| nc-MGF 100-3L | 181,362 | 181,585 | − |
| nc-MGF 100-3L-AT-S | 181,408 | 181,585 | − |
| Name | TSS (+)/TES (−) | TSS (−)/TES (+) | Strand | RNA Type |
|---|---|---|---|---|
| MGF 360-1La-KP93L | 2625 | 1367 | − | Bicistronic |
| MGF 360-1La-MGF 360-1Lb | 2914 | 1752 | − | Bicistronic |
| L60L-L83L | 5405 | 4819 | − | Bicistronic |
| MGF 110-3L-MGF 110-2L | 8680 | 7800 | − | Bicistronic |
| MGF 110-5L-6L-MGF 110-4L | 10,162 | 8785 | − | Bicistronic |
| MGF 110-5L-6L-MGF 110-4L-AT-S | 10,162 | 8902 | − | Bicistronic |
| MGF 110-7L-MGF 110-5L-6L | 10,814 | 9468 | − | Bicistronic |
| 285L-MGF 110-7L | 11,372 | 10,270 | − | Bicistronic |
| MGF 110-8L-ASFV G ACD 00160-285L | 11,854 | 10,991 | − | Bicistronic |
| ASFV G ACD 00290-ASFV G ACD 00350-AT-L | 17,530 | 17,963 | + | Bicistronic |
| QGV56997.1-ASFV G ACD 00320 | 19,383 | 19,699 | + | Bicistronic |
| ASFV G ACD 00320-ASFV G ACD 00330 | 19,482 | 20,130 | + | Bicistronic |
| ASFV G ACD 00330-X69R | 19,712 | 20,421 | + | Bicistronic |
| ASFV G ACD 00350-X69R | 19,930 | 20,421 | + | Bicistronic |
| ASFV_G_ACD_00290-MGF 300.5-2R | 21,682 | 22,200 | + | Bicistronic |
| MGF 300.5-2R-MGF 300-2R | 21,935 | 22,888 | + | Bicistronic |
| MGF 360-10L-MGF 360-9L | 27,409 | 24,983 | − | Bicistronic |
| MGF 505-3R-MGF 505-4R | 35,730 | 38,296 | + | Bicistronic |
| ASFV G ACD 00600-A224L-AT-L2 | 48,230 | 45,900 | − | Bicistronic |
| ASFV G ACD 00600-A224L-AT-L | 48,240 | 46,868 | − | Bicistronic |
| ASFV G ACD 00600-A224L | 48,240 | 47,236 | − | Bicistronic |
| A151R-MGF 360-15R-L | 49,395 | 51,279 | + | Bicistronic |
| A151R-MGF 360-15R | 49,646 | 51,269 | + | Bicistronic |
| A151R-MGF 360-15R-AT-L | 49,646 | 51,387 | + | Bicistronic |
| EP152R-EP153R | 73,293 | 74,365 | + | Bicistronic |
| EP152R-EP153R-AT-L | 73,293 | 74,953 | + | Bicistronic |
| EP153R-EP402R | 73,637 | 75,785 | + | Bicistronic |
| M448R-C129R | 80,455 | 82,407 | + | Bicistronic |
| CP530R-CP80R | 126,388 | 129,348 | + | Bicistronic |
| CP80R-CP312R | 127,979 | 129,347 | + | Bicistronic |
| D339L-DP79L | 141,202 | 139,812 | − | Bicistronic |
| H108R-H233R | 154,764 | 156,604 | + | Bicistronic |
| EP296R-E111R-L | 168,803 | 170,408 | + | Bicistronic |
| I267L-E66L | 171,549 | 170,453 | − | Bicistronic |
| I177L-I215L | 176,031 | 174,820 | − | Bicistronic |
| I196L-I177L | 176,620 | 175,414 | − | Bicistronic |
| MGF 100-3L-MGF 100-1L | 181,585 | 180,386 | − | Bicistronic |
| I8L-I7L | 182,657 | 181,655 | − | Bicistronic |
| ASFV G ACD 01940-ASFV G ACD.5 01940 | 186,102 | 185,635 | − | Bicistronic |
| ASFV G ACD 01940-ASFV G ACD.5 01940-AT-L | 186,102 | 185,686 | − | Bicistronic |
| MGF 360-19Ra-MGF 360-19Rb | 186,225 | 187,363 | + | Bicistronic |
| MGF 110-2L-ASFV G ACD 00090-MGF 110-1L | 8153 | 6830 | − | Tricistronic |
| MGF 110-7L-MGF 110-5L-6L-MGF 110-4L | 10,814 | 8902 | − | Tricistronic |
| 285L-MGF 110-7L-MGF 110-5L-6L | 11,372 | 9468 | − | Tricistronic |
| QGV56997.1-ASFV G ACD 00320-ASFV G ACD 00330 | 19,383 | 20,130 | + | Tricistronic |
| ASFV G ACD 00290-MGF 300.5-2R-MGF 300-2R | 21,697 | 22,890 | + | Tricistronic |
| MGF 360-11L-MGF 360-10L-MGF 360-9L | 28,482 | 24,983 | − | Tricistronic |
| K78R-K196R-K145R | 64,869 | 66,167 | + | Tricistronic |
| I7L-MGF 100-3L-MGF 100-1L | 181,993 | 180,386 | − | Tricistronic |
| K205R-K78R-K196R-K145R | 64,142 | 66,167 | + | Tetracistronic |
| H171R-H124R-H339R-H108R | 153,246 | 155,658 | + | Tetracistronic |
| Name | TSS (+)/TES (−) | TSS (−)/TES (+) | Strand |
|---|---|---|---|
| L83L-KP177R | 4203 | 5129 | − |
| MGF 110-4L-MGF 110-3L | 8210 | 9368 | − |
| MGF 110-8L-MGF 100-1R-ASFV G ACD 00190 | 11,282 | 13,246 | + |
| MGF 110-12L-ASFV G ACD 00240 | 14,412 | 15,385 | − |
| ASFV G ACD 00290-ASFV G ACD 00350-ASFV G ACD 00300 | 17,530 | 18,383 | + |
| ASFV G ACD 00350-ASFV G ACD 00300 | 17,782 | 18,383 | + |
| A240L-A104R | 48,012 | 49,331 | − |
| A104R-A240L-A118R | 48,308 | 49,985 | + |
| C122R-C257L | 85,124 | 86,738 | + |
| C122R-C257L-C475L-C315R | 85,124 | 89,338 | + |
| C122R-C257L-AT-L | 85,144 | 87,363 | + |
| B125R-B117L | 106,380 | 107,270 | + |
| B263R-B66L | 109,076 | 110,269 | + |
| D205R-D129L-L | 137,005 | 140,038 | + |
| D205R-D129L | 138,451 | 140,032 | + |
| I267L-E66L-E111R | 169,555 | 171,535 | − |
| I9R-I10L-L11L | 182,703 | 184,322 | + |
| DP71L-MGF 360-18R | 184,247 | 185,256 | − |
| MGF 360-18R-DP71L-DP96R | 184329 | 185647 | + |
| MGF 360-18R-DP71L-DP96R-ASFV G ACD.5 01940 | 184,329 | 185,895 | + |
| MGF 360-18R-DP71L-DP96R-AT-L | 184,329 | 185,733 | + |
| DP96R-ASFV G ACD.5 01940 | 185,335 | 185,895 | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torma, G.; Tombácz, D.; Csabai, Z.; Moldován, N.; Mészáros, I.; Zádori, Z.; Boldogkői, Z. Combined Short and Long-Read Sequencing Reveals a Complex Transcriptomic Architecture of African Swine Fever Virus. Viruses 2021, 13, 579. https://doi.org/10.3390/v13040579
Torma G, Tombácz D, Csabai Z, Moldován N, Mészáros I, Zádori Z, Boldogkői Z. Combined Short and Long-Read Sequencing Reveals a Complex Transcriptomic Architecture of African Swine Fever Virus. Viruses. 2021; 13(4):579. https://doi.org/10.3390/v13040579
Chicago/Turabian StyleTorma, Gábor, Dóra Tombácz, Zsolt Csabai, Norbert Moldován, István Mészáros, Zoltán Zádori, and Zsolt Boldogkői. 2021. "Combined Short and Long-Read Sequencing Reveals a Complex Transcriptomic Architecture of African Swine Fever Virus" Viruses 13, no. 4: 579. https://doi.org/10.3390/v13040579
APA StyleTorma, G., Tombácz, D., Csabai, Z., Moldován, N., Mészáros, I., Zádori, Z., & Boldogkői, Z. (2021). Combined Short and Long-Read Sequencing Reveals a Complex Transcriptomic Architecture of African Swine Fever Virus. Viruses, 13(4), 579. https://doi.org/10.3390/v13040579