
Top-Down and Bottom-Up Proteomics Methods to Study RNA Virus Biology

 , , and
, , and 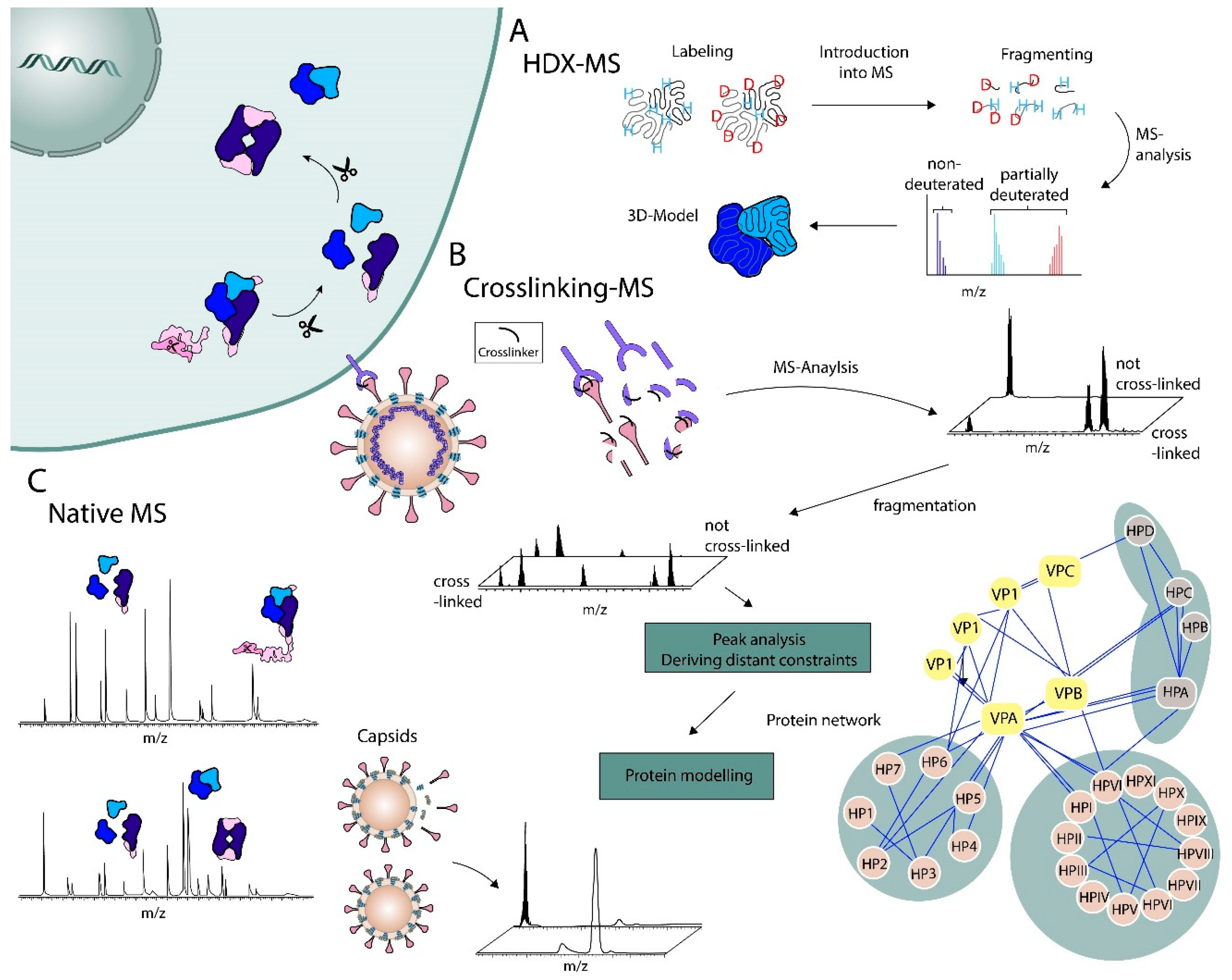{kind=link}
{kind=link}
Abstract
:1. Introduction
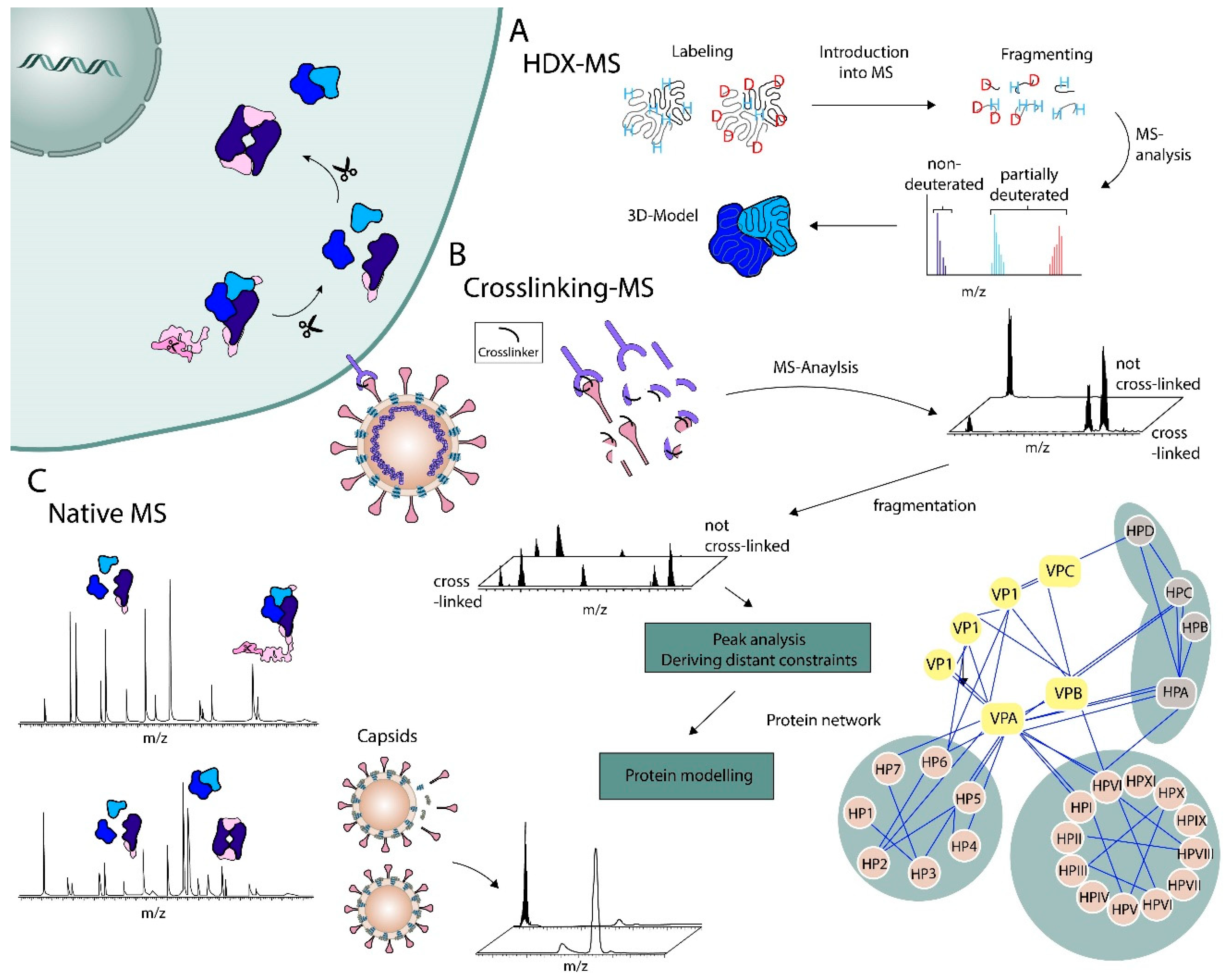
2. Top-Down Methods
2.1. RNA and Protein Complexes
2.2. Chemical Labeling Mass Spectrometry Methods
2.3. Structural Work on Viral Particles
2.4. Drug-Target Identification
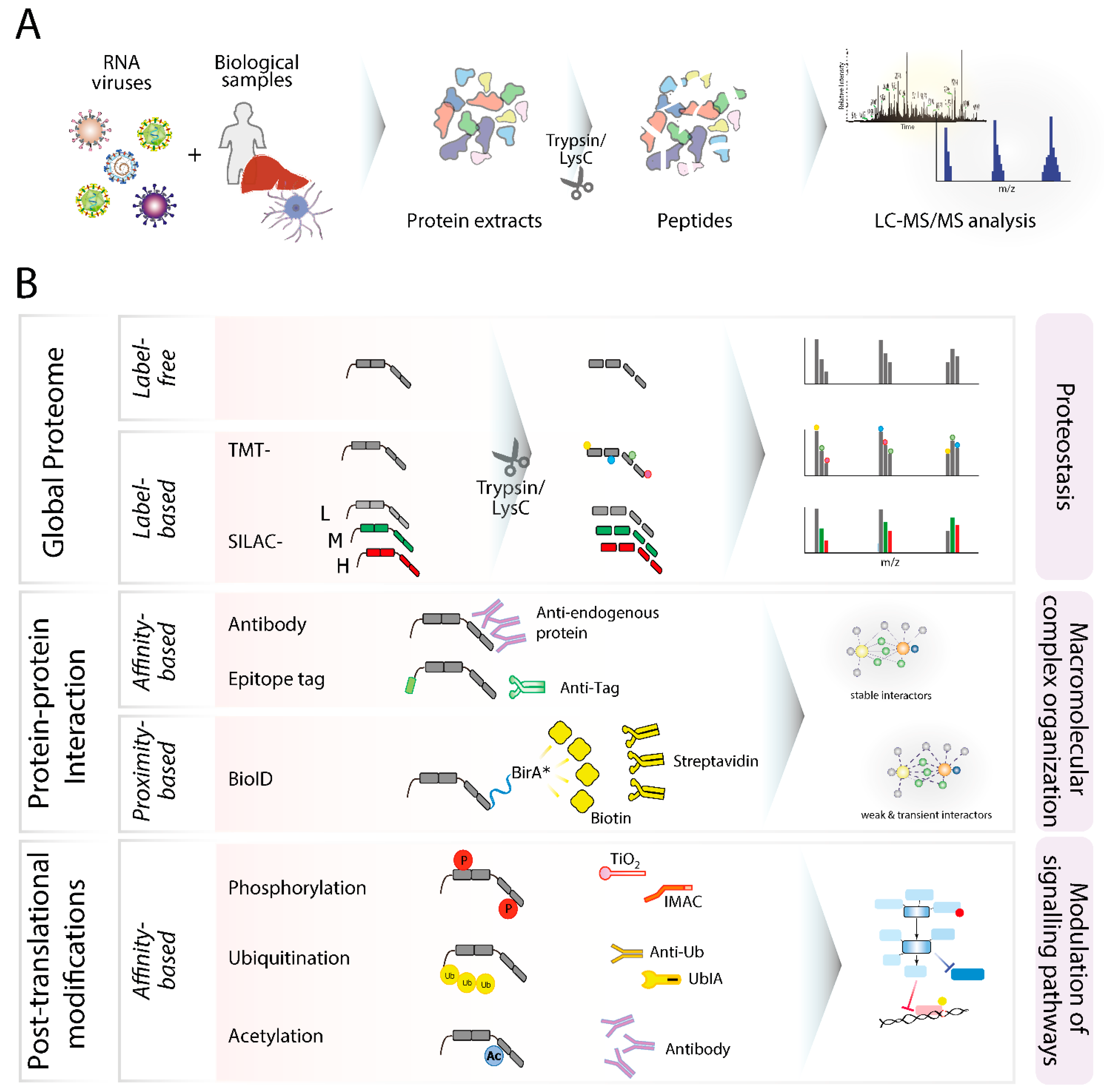
3. Bottom-Up Methods
3.1. Global Proteome Profiling
3.2. Protein–Protein Interactions
3.3. Post-Translational Modifications
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Chavez, J.D.; Cilia, M.; Weisbrod, C.R.; Ju, H.-J.; Eng, J.K.; Gray, S.M.; Bruce, J.E. Cross-Linking Measurements of the Potato Leafroll Virus Reveal Protein Interaction Topologies Required for Virion Stability, Aphid Transmission, and Virus-Plant Interactions. J. Proteome Res. 2012, 11, 2968–2981. [Google Scholar] [CrossRef] [Green Version]
- Mallagaray, A.; Creutznacher, R.; Dülfer, J.; Mayer, P.H.O.; Grimm, L.L.; Orduña, J.M.; Trabjerg, E.; Stehle, T.; Rand, K.D.; Blaum, B.S.; et al. A Post-Translational Modification of Human Norovirus Capsid Protein Attenuates Glycan Binding. Nat. Commun. 2019, 10, 1320. [Google Scholar] [CrossRef] [Green Version]
- Mann, M.; Kulak, N.A.; Nagaraj, N.; Cox, J. The Coming Age of Complete, Accurate, and Ubiquitous Proteomes. Mol. Cell 2013, 49, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siuti, N.; Kelleher, N.L. Decoding Protein Modifications Using Top-down Mass Spectrometry. Nat. Methods 2007, 4, 817–821. [Google Scholar] [CrossRef]
- Bond, K.M.; Lyktey, N.A.; Tsvetkova, I.B.; Dragnea, B.; Jarrold, M.F. Disassembly Intermediates of the Brome Mosaic Virus Identified by Charge Detection Mass Spectrometry. J. Phys. Chem. B 2020, 124, 2124–2131. [Google Scholar] [CrossRef] [PubMed]
- Pogan, R.; Dülfer, J.; Uetrecht, C. Norovirus Assembly and Stability. Curr. Opin. Virol. 2018, 31, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Olinares, P.D.B.; Kang, J.Y.; Llewellyn, E.; Chiu, C.; Chen, J.; Malone, B.; Saecker, R.M.; Campbell, E.A.; Darst, S.A.; Chait, B.T. Native Mass Spectrometry-Based Screening for Optimal Sample Preparation in Single-Particle Cryo-EM. Structure 2021, 29, 186–195.e6. [Google Scholar] [CrossRef] [PubMed]
- Greisch, J.-F.; Tamara, S.; Scheltema, R.A.; Maxwell, H.W.R.; Fagerlund, R.D.; Fineran, P.C.; Tetter, S.; Hilvert, D.; Heck, A.J.R. Expanding the Mass Range for UVPD-Based Native Top-down Mass Spectrometry. Chem. Sci. 2019, 10, 7163–7171. [Google Scholar] [CrossRef]
- Batra, J.; Hultquist, J.F.; Liu, D.; Shtanko, O.; Von Dollen, J.; Satkamp, L.; Jang, G.M.; Luthra, P.; Schwarz, T.M.; Small, G.I.; et al. Protein Interaction Mapping Identifies RBBP6 as a Negative Regulator of Ebola Virus Replication. Cell 2018, 175, 1917–1930.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichlmair, A.; Kandasamy, K.; Alvisi, G.; Mulhern, O.; Sacco, R.; Habjan, M.; Binder, M.; Stefanovic, A.; Eberle, C.-A.; Goncalves, A.; et al. Viral Immune Modulators Perturb the Human Molecular Network by Common and Unique Strategies. Nature 2012, 487, 486–490. [Google Scholar] [CrossRef]
- Scaturro, P.; Stukalov, A.; Haas, D.A.; Cortese, M.; Draganova, K.; Płaszczyca, A.; Bartenschlager, R.; Götz, M.; Pichlmair, A. An Orthogonal Proteomic Survey Uncovers Novel Zika Virus Host Factors. Nature 2018, 561, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Lantz, C.; Brown, K.A.; Ge, Y.; Paša-Tolić, L.; Loo, J.A.; Lermyte, F. Higher-Order Structural Characterisation of Native Proteins and Complexes by Top-down Mass Spectrometry. Chem. Sci. 2020, 11, 12918–12936. [Google Scholar] [CrossRef]
- Bacher, G.; Szymanski, W.W.; Kaufman, S.L.; Zöllner, P.; Blaas, D.; Allmaier, G. Charge-Reduced Nano Electrospray Ionization Combined with Differential Mobility Analysis of Peptides, Proteins, Glycoproteins, Noncovalent Protein Complexes and Viruses: ESI GEMMA of Proteins and Noncovalent Complexes. J. Mass Spectrom. 2001, 36, 1038–1052. [Google Scholar] [CrossRef]
- Petroff, J.T.; Tong, A.; Chen, L.J.; Dekoster, G.T.; Khan, F.; Abramson, J.; Frieden, C.; Cheng, W.W.L. Charge Reduction of Membrane Proteins in Native Mass Spectrometry Using Alkali Metal Acetate Salts. Anal. Chem. 2020, 92, 6622–6630. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Gavriilidou, A.F.M.; Zenobi, R. Influence of Alkylammonium Acetate Buffers on Protein–Ligand Noncovalent Interactions Using Native Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 341–346. [Google Scholar] [CrossRef]
- Dülfer, J.; Kadek, A.; Kopicki, J.-D.; Krichel, B.; Uetrecht, C. Structural mass spectrometry goes viral. Adv. Virus Res. 2019, 105, 189–238. [Google Scholar] [PubMed]
- Schmidt, C.; Kramer, K.; Urlaub, H. Investigation of Protein–RNA Interactions by Mass Spectrometry—Techniques and Applications. J. Proteom. 2012, 75, 3478–3494. [Google Scholar] [CrossRef]
- Hagan, N.; Fabris, D. Direct Mass Spectrometric Determination of the Stoichiometry and Binding Affinity of the Complexes between Nucleocapsid Protein and RNA Stem−Loop Hairpins of the HIV-1 Ψ-Recognition Element. Biochemistry 2003, 42, 10736–10745. [Google Scholar] [CrossRef] [PubMed]
- Weiss, V.U.; Bereszcazk, J.Z.; Havlik, M.; Kallinger, P.; Gösler, I.; Kumar, M.; Blaas, D.; Marchetti-Deschmann, M.; Heck, A.J.R.; Szymanski, W.W.; et al. Analysis of a Common Cold Virus and Its Subviral Particles by Gas-Phase Electrophoretic Mobility Molecular Analysis and Native Mass Spectrometry. Anal. Chem. 2015, 87, 8709–8717. [Google Scholar] [CrossRef] [PubMed]
- Elliott, A.G.; Harper, C.C.; Lin, H.-W.; Williams, E.R. Mass, Mobility and MS n Measurements of Single Ions Using Charge Detection Mass Spectrometry. Analyst 2017, 142, 2760–2769. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Burlingame, A.L. Mass Spectrometry-Based Detection and Assignment of Protein Posttranslational Modifications. ACS Chem. Biol. 2015, 10, 63–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagan, N.A.; Fabris, D. Dissecting the Protein–RNA and RNA–RNA Interactions in the Nucleocapsid-Mediated Dimerization and Isomerization of HIV-1 Stemloop 1. J. Mol. Biol. 2007, 365, 396–410. [Google Scholar] [CrossRef] [Green Version]
- Schneeberger, E.; Breuker, K. Native Top-Down Mass Spectrometry of TAR RNA in Complexes with a Wild-Type Tat Peptide for Binding Site Mapping. Angew. Chem. 2017, 129, 1274–1278. [Google Scholar] [CrossRef]
- Krichel, B.; Falke, S.; Hilgenfeld, R.; Redecke, L.; Uetrecht, C. Processing of the SARS-CoV Pp1a/Ab Nsp7–10 Region. Biochem. J. 2020, 477, 1009–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krichel, B.; Bylapudi, G.; Schmidt, C.; Blanchet, C.; Schubert, R.; Brings, L.; Koehler, M.; Zenobi, R.; Svergun, D.; Lorenzen, K.; et al. Hallmarks of Alpha- and Betacoronavirus Non-Structural Protein 7+8 Complexes. Sci. Adv. 2021, 7, eabf1004. [Google Scholar] [CrossRef]
- Chen, J.; Malone, B.; Llewellyn, E.; Grasso, M.; Shelton, P.M.M.; Olinares, P.D.B.; Maruthi, K.; Eng, E.T.; Vatandaslar, H.; Chait, B.T.; et al. Structural Basis for Helicase-Polymerase Coupling in the SARS-CoV-2 Replication-Transcription Complex. Cell 2020, 182, 1560–1573.e13. [Google Scholar] [CrossRef] [PubMed]
- Baslé, E.; Joubert, N.; Pucheault, M. Protein Chemical Modification on Endogenous Amino Acids. Chem. Biol. 2010, 17, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, V.L.; Vachet, R.W. Probing Protein Structure by Amino Acid-Specific Covalent Labeling and Mass Spectrometry. Mass Spectrom. Rev. 2009, 28, 785–815. [Google Scholar] [CrossRef] [Green Version]
- Iacob, R.E.; Keck, Z.; Olson, O.; Foung, S.K.H.; Tomer, K.B. Structural Elucidation of Critical Residues Involved in Binding of Human Monoclonal Antibodies to Hepatitis C Virus E2 Envelope Glycoprotein. Biochim. Biophys. Acta 2008, 1784, 530–542. [Google Scholar] [CrossRef] [Green Version]
- Limpikirati, P.; Liu, T.; Vachet, R.W. Covalent Labeling-Mass Spectrometry with Non-Specific Reagents for Studying Protein Structure and Interactions. Methods 2018, 144, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Merkley, E.D.; Rysavy, S.; Kahraman, A.; Hafen, R.P.; Daggett, V.; Adkins, J.N. Distance Restraints from Crosslinking Mass Spectrometry: Mining a Molecular Dynamics Simulation Database to Evaluate Lysine-Lysine Distances: Evaluating Lysine-Lysine Distances by MD for XL-MS. Protein Sci. 2014, 23, 747–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaim, C.L.; Smith, J.B.; Smith, D.L. Unexpected Products from the Reaction of the Synthetic Cross-Linker 3,3′-Dithiobis(Sulfosuccinimidyl Propionate), DTSSP with Peptides. J. Am. Soc. Mass Spectrom. 2004, 15, 736–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leavell, M.D.; Novak, P.; Behrens, C.R.; Schoeniger, J.S.; Kruppa, G.H. Strategy for Selective Chemical Cross-Linking of Tyrosine and Lysine Residues. J. Am. Soc. Mass Spectrom. 2004, 15, 1604–1611. [Google Scholar] [CrossRef] [Green Version]
- Sinz, A. Chemical Cross-Linking and Mass Spectrometry to Map Three-Dimensional Protein Structures and Protein–Protein Interactions. Mass Spectrom. Rev. 2006, 25, 663–682. [Google Scholar] [CrossRef]
- Sinz, A. The Advancement of Chemical Cross-Linking and Mass Spectrometry for Structural Proteomics: From Single Proteins to Protein Interaction Networks. Expert. Rev. Proteom. 2014, 11, 733–743. [Google Scholar] [CrossRef]
- Rappsilber, J. The Beginning of a Beautiful Friendship: Cross-Linking/Mass Spectrometry and Modelling of Proteins and Multi-Protein Complexes. J. Struct. Biol. 2011, 173, 530–540. [Google Scholar] [CrossRef] [Green Version]
- Kosinski, J.; Mosalaganti, S.; von Appen, A.; Teimer, R.; DiGuilio, A.L.; Wan, W.; Bui, K.H.; Hagen, W.J.H.; Briggs, J.A.G.; Glavy, J.S.; et al. Molecular Architecture of the Inner Ring Scaffold of the Human Nuclear Pore Complex. Science 2016, 352, 363–365. [Google Scholar] [CrossRef] [Green Version]
- Sinz, A.; Arlt, C.; Chorev, D.; Sharon, M. Chemical Cross-Linking and Native Mass Spectrometry: A Fruitful Combination for Structural Biology: Chemical Crosslinking and Native MS. Protein Sci. 2015, 24, 1193–1209. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Urlaub, H. Combining Cryo-Electron Microscopy (Cryo-EM) and Cross-Linking Mass Spectrometry (CX-MS) for Structural Elucidation of Large Protein Assemblies. Curr. Opin. Struct. Biol. 2017, 46, 157–168. [Google Scholar] [CrossRef]
- Bullock, J.M.A.; Sen, N.; Thalassinos, K.; Topf, M. Modeling Protein Complexes Using Restraints from Crosslinking Mass Spectrometry. Structure 2018, 26, 1015–1024.e2. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.M.; Mohr, J.P.; DeBlasio, S.L.; Chavez, J.D.; Ziegler-Graff, V.; Brault, V.; Bruce, J.E.; Heck, M. (Cilia) Insights in Luteovirid Structural Biology Guided by Chemical Cross-Linking and High Resolution Mass Spectrometry. Virus Res. 2017, 241, 42–52. [Google Scholar] [CrossRef]
- DeBlasio, S.L.; Chavez, J.D.; Alexander, M.M.; Ramsey, J.; Eng, J.K.; Mahoney, J.; Gray, S.M.; Bruce, J.E.; Cilia, M. Visualization of Host-Polerovirus Interaction Topologies Using Protein Interaction Reporter Technology. J. Virol. 2016, 90, 1973–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Gülbakan, B.; Weidmann, S.; Fagerer, S.R.; Ibáñez, A.J.; Zenobi, R. Applying Mass Spectrometry to Study Non-Covalent Biomolecule Complexes: Applying Mass Spectrometry to Study Biomolecule Complexes. Mass Spec. Rev. 2016, 35, 48–70. [Google Scholar] [CrossRef] [PubMed]
- Ooi, Y.S.; Majzoub, K.; Flynn, R.A.; Mata, M.A.; Diep, J.; Li, J.K.; van Buuren, N.; Rumachik, N.; Johnson, A.G.; Puschnik, A.S.; et al. An RNA-Centric Dissection of Host Complexes Controlling Flavivirus Infection. Nat. Microbiol. 2019, 4, 2369–2382. [Google Scholar] [CrossRef]
- Phillips, S.L.; Soderblom, E.J.; Bradrick, S.S.; Garcia-Blanco, M.A. Identification of Proteins Bound to Dengue Viral RNA In Vivo Reveals New Host Proteins Important for Virus Replication. mBio 2016, 7, e01865-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, R.A.; Belk, J.A.; Qi, Y.; Yasumoto, Y.; Schmitz, C.O.; Mumbach, M.R.; Limaye, A.; Wei, J.; Alfajaro, M.M.; Parker, K.R.; et al. Systematic Discovery and Functional Interrogation of SARS-CoV-2 Viral RNA-Host Protein Interactions during Infection. bioRxiv. Available online: https://www.biorxiv.org/content/10.1101/2020.10.06.327445v1 (accessed on 1 April 2021).
- Kalocsay, M.; Maliga, Z.; Nirmal, A.J.; Eisert, R.J.; Bradshaw, G.A.; Solomon, I.H.; Chen, Y.-A.; Pelletier, R.J.; Jacobson, C.A.; Mintseris, J.; et al. Multiplexed Proteomics and Imaging of Resolving and Lethal SARS-CoV-2 Infection in the Lung. Pathology 2020. in revision. [Google Scholar]
- Schmidt, N.; Lareau, C.A.; Keshishian, H.; Melanson, R.; Zimmer, M.; Kirschner, L.; Ade, J.; Werner, S.; Caliskan, N.; Lander, E.S.; et al. A Direct RNA-Protein Interaction Atlas of the SARS-CoV-2 RNA in Infected Human Cells. Nat. Microbiol. 2021, 6, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Jack, A.; Ferro, L.S.; Trnka, M.J.; Wehri, E.; Nadgir, A.; Nguyenla, X.; Costa, K.; Stanley, S.; Schaletzky, J.; Yildiz, A. SARS-CoV-2 Nucleocapsid Protein Forms Condensates with Viral Genomic RNA. bioRxiv. Available online: https://www.biorxiv.org/content/10.1101/2020.09.14.295824v3 (accessed on 1 April 2021).
- Chanthamontri, C.K.; Jordan, D.S.; Wang, W.; Wu, C.; Lin, Y.; Brett, T.J.; Gross, M.L.; Leung, D.W. The Ebola Viral Protein 35 N-Terminus Is a Parallel Tetramer. Biochemistry 2019, 58, 657–664. [Google Scholar] [CrossRef]
- Puchades, C.; Kűkrer, B.; Diefenbach, O.; Sneekes-Vriese, E.; Juraszek, J.; Koudstaal, W.; Apetri, A. Epitope Mapping of Diverse Influenza Hemagglutinin Drug Candidates Using HDX-MS. Sci. Rep. 2019, 9, 4735. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.K.; Westfield, G.H.; Xiao, K.; Reis, R.I.; Huang, L.-Y.; Tripathi-Shukla, P.; Qian, J.; Li, S.; Blanc, A.; Oleskie, A.N.; et al. Visualization of Arrestin Recruitment by a G-Protein-Coupled Receptor. Nature 2014, 512, 218–222. [Google Scholar] [CrossRef]
- Wales, T.E.; Engen, J.R. Hydrogen Exchange Mass Spectrometry for the Analysis of Protein Dynamics. Mass Spectrom. Rev. 2006, 25, 158–170. [Google Scholar] [CrossRef]
- Zhang, Q.; Willison, L.N.; Tripathi, P.; Sathe, S.K.; Roux, K.H.; Emmett, M.R.; Blakney, G.T.; Zhang, H.-M.; Marshall, A.G. Epitope Mapping of a 95 KDa Antigen in Complex with Antibody by Solution-Phase Amide Backbone Hydrogen/Deuterium Exchange Monitored by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 2011, 83, 7129–7136. [Google Scholar] [CrossRef] [Green Version]
- Renaud, J.-P.; Chung, C.-W.; Danielson, U.H.; Egner, U.; Hennig, M.; Hubbard, R.E.; Nar, H. Biophysics in Drug Discovery: Impact, Challenges and Opportunities. Nat. Rev. Drug Discov. 2016, 15, 679–698. [Google Scholar] [CrossRef] [Green Version]
- Silva, L.P.; Vanzile, M.; Bavari, S.; Aman, J.M.J.; Schriemer, D.C. Assembly of Ebola Virus Matrix Protein VP40 Is Regulated by Latch-Like Properties of N and C Terminal Tails. PLoS ONE 2012, 7, e39978. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Smith, D.L. Capsid Structure and Dynamics of a Human Rhinovirus Probed by Hydrogen Exchange Mass Spectrometry. Protein Sci. 2009, 14, 1661–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Lane, L.C.; Smith, D.L. Detecting Structural Changes in Viral Capsids by Hydrogen Exchange and Mass Spectrometry. Protein Sci. 2001, 10, 1234–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bale, S.; Liu, T.; Li, S.; Wang, Y.; Abelson, D.; Fusco, M.; Woods, V.L.; Ollmann Saphire, E. Ebola Virus Glycoprotein Needs an Additional Trigger, beyond Proteolytic Priming for Membrane Fusion. PLoS Negl. Trop. Dis. 2011, 5, e1395. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.K.; Guttman, M.; Ebner, J.L.; Lee, K.K. Dynamic Changes during Acid-Induced Activation of Influenza Hemagglutinin. Structure 2015, 23, 665–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.G.; Whidby, J.; Miller, M.T.; Scarborough, H.; Zatorski, A.V.; Cygan, A.; Price, A.A.; Yost, S.A.; Bohannon, C.D.; Jacob, J.; et al. Structure of the Core Ectodomain of the Hepatitis C Virus Envelope Glycoprotein 2. Nature 2014, 509, 381–384. [Google Scholar] [CrossRef] [Green Version]
- Benhaim, M.A.; Mangala Prasad, V.; Garcia, N.K.; Guttman, M.; Lee, K.K. Structural Monitoring of a Transient Intermediate in the Hemagglutinin Fusion Machinery on Influenza Virions. Sci. Adv. 2020, 6, eaaz8822. [Google Scholar] [CrossRef]
- Abzalimov, R.R.; Kaplan, D.A.; Easterling, M.L.; Kaltashov, I.A. Protein Conformations Can Be Probed in Top-down HDX MS Experiments Utilizing Electron Transfer Dissociation of Protein Ions without Hydrogen Scrambling. J. Am. Soc. Mass Spectrom. 2009, 20, 1514–1517. [Google Scholar] [CrossRef] [Green Version]
- Landgraf, R.R.; Chalmers, M.J.; Griffin, P.R. Automated Hydrogen/Deuterium Exchange Electron Transfer Dissociation High Resolution Mass Spectrometry Measured at Single-Amide Resolution. J. Am. Soc. Mass Spectrom. 2012, 23, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Percy, A.J.; Rey, M.; Burns, K.M.; Schriemer, D.C. Probing Protein Interactions with Hydrogen/Deuterium Exchange and Mass Spectrometry-a Review. Anal. Chim. Acta 2012, 721, 7–21. [Google Scholar] [CrossRef]
- Morton, V.L.; Stockley, P.G.; Stonehouse, N.J.; Ashcroft, A.E. Insights into Virus Capsid Assembly from Non-Covalent Mass Spectrometry: VIRUS CAPSID ASSEMBLY STUDIED BY MASS SPECTROMETRY. Mass Spectrom. Rev. 2008, 27, 575–595. [Google Scholar] [CrossRef] [PubMed]
- Fuerstenau, S.D.; Benner, W.H.; Thomas, J.J.; Brugidou, C.; Bothner, B.; Siuzdak, G. Mass Spectrometry of an Intact Virus. Angew. Chem. Int. Ed. Engl. 2001, 40, 541–544. [Google Scholar] [CrossRef]
- Broo, K.; Wei, J.; Marshall, D.; Brown, F.; Smith, T.J.; Johnson, J.E.; Schneemann, A.; Siuzdak, G. Viral Capsid Mobility: A Dynamic Conduit for Inactivation. Proc. Natl. Acad. Sci. USA 2001, 98, 2274–2277. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, A.E. Mass Spectrometry-Based Studies of Virus Assembly. Curr. Opin. Virol. 2019, 36, 17–24. [Google Scholar] [CrossRef]
- Bakhtiar, R.; Thomas, J.J.; Siuzdak, G. Mass Spectrometry in Viral Proteomics. Acc. Chem. Res. 2000, 33, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Uetrecht, C.; Heck, A.J.R. Modern Biomolecular Mass Spectrometry and Its Role in Studying Virus Structure, Dynamics, and Assembly. Angew. Chem. Int. Ed. 2011, 50, 8248–8262. [Google Scholar] [CrossRef] [PubMed]
- Wörner, T.P.; Shamorkina, T.M.; Snijder, J.; Heck, A.J.R. Mass Spectrometry-Based Structural Virology. Anal. Chem. 2021, 93, 620–640. [Google Scholar] [CrossRef]
- Snijder, J.; Uetrecht, C.; Rose, R.J.; Sanchez-Eugenia, R.; Marti, G.A.; Agirre, J.; Guérin, D.M.A.; Wuite, G.J.L.; Heck, A.J.R.; Roos, W.H. Probing the Biophysical Interplay between a Viral Genome and Its Capsid. Nat. Chem 2013, 5, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Tri, P.; Do, T.-O.; Nguyen, T.A.; Le, V.T.; Assadi, A.A. Nanocontainer: An introduction. In Smart Nanocontainers; Elsevier: Amsterdam, The Netherlands, 2020; pp. 3–6. ISBN 978-0-12-816770-0. [Google Scholar]
- Dunbar, C.A.; Rayaprolu, V.; Wang, J.C.-Y.; Brown, C.J.; Leishman, E.; Jones-Burrage, S.; Trinidad, J.C.; Bradshaw, H.B.; Clemmer, D.E.; Mukhopadhyay, S.; et al. Dissecting the Components of Sindbis Virus from Arthropod and Vertebrate Hosts: Implications for Infectivity Differences. ACS Infect. Dis. 2019, 5, 892–902. [Google Scholar] [CrossRef]
- van de Waterbeemd, M.; Snijder, J.; Tsvetkova, I.B.; Dragnea, B.G.; Cornelissen, J.J.; Heck, A.J.R. Examining the Heterogeneous Genome Content of Multipartite Viruses BMV and CCMV by Native Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 1000–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoemaker, G.K.; van Duijn, E.; Crawford, S.E.; Uetrecht, C.; Baclayon, M.; Roos, W.H.; Wuite, G.J.L.; Estes, M.K.; Prasad, B.V.V.; Heck, A.J.R. Norwalk Virus Assembly and Stability Monitored by Mass Spectrometry. Mol. Cell. Proteom. 2010, 9, 1742–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pogan, R.; Schneider, C.; Reimer, R.; Hansman, G.; Uetrecht, C. Norovirus-like VP1 Particles Exhibit Isolate Dependent Stability Profiles. J. Phys. Condens. Matter 2018, 30, 064006. [Google Scholar] [CrossRef] [Green Version]
- Pogan, R.; Weiss, V.U.; Bond, K.; Dülfer, J.; Krisp, C.; Lyktey, N.; Müller-Guhl, J.; Zoratto, S.; Allmaier, G.; Jarrold, M.F.; et al. N-Terminal VP1 Truncations Favor T = 1 Norovirus-Like Particles. Vaccines 2020, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Bereszczak, J.Z.; Barbu, I.M.; Tan, M.; Xia, M.; Jiang, X.; van Duijn, E.; Heck, A.J.R. Structure, Stability and Dynamics of Norovirus P Domain Derived Protein Complexes Studied by Native Mass Spectrometry. J. Struct. Biol. 2012, 177, 273–282. [Google Scholar] [CrossRef]
- Dülfer, J.; Yan, H.; Brodmerkel, M.N.; Creutznacher, R.; Mallagaray, A.; Peters, T.; Caleman, C.; Marklund, E.G.; Uetrecht, C. Glycan-Induced Protein Dynamics in Human Norovirus P Dimers Depend on Virus Strain and Deamidation Status. Molecules 2021, 26, 2125. [Google Scholar] [CrossRef]
- Bücher, K.S.; Yan, H.; Creutznacher, R.; Ruoff, K.; Mallagaray, A.; Grafmüller, A.; Dirks, J.S.; Kilic, T.; Weickert, S.; Rubailo, A.; et al. Fucose-Functionalized Precision Glycomacromolecules Targeting Human Norovirus Capsid Protein. Biomacromolecules 2018, 19, 3714–3724. [Google Scholar] [CrossRef]
- Xia, M.; Huang, P.; Sun, C.; Han, L.; Vago, F.S.; Li, K.; Zhong, W.; Jiang, W.; Klassen, J.S.; Jiang, X.; et al. Bioengineered Norovirus S60 Nanoparticles as a Multifunctional Vaccine Platform. ACS Nano 2018, 12, 10665–10682. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of Early Drug Discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Sobott, F. Chapter 4. Structural Studies using Electron-based Fragmentation Methods and Chemical Labelling of Proteins. In New Developments in Mass Spectrometry; Lermyte, F., Ed.; Royal Society of Chemistry: Cambridge, UK, 2020; pp. 72–101. ISBN 978-1-83916-104-9. [Google Scholar]
- Roy, A. Early Probe and Drug Discovery in Academia: A Minireview. High-Throughput 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uetrecht, C.; Rose, R.J.; van Duijn, E.; Lorenzen, K.; Heck, A.J.R. Ion Mobility Mass Spectrometry of Proteins and Proteinassemblies. Chem. Soc. Rev. 2010, 39, 1633–1655. [Google Scholar] [CrossRef] [PubMed]
- Vivat Hannah, V.; Atmanene, C.; Zeyer, D.; Van Dorsselaer, A.; Sanglier-Cianférani, S. Native MS: An ’ESI‚ Way to Support Structure- and Fragment-Based Drug Discovery. Future Med. Chem. 2010, 2, 35–50. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and Calpain Inhibitors II, XII Inhibit SARS-CoV-2 Viral Replication by Targeting the Viral Main Protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Sacco, M.D.; Ma, C.; Lagarias, P.; Gao, A.; Townsend, J.A.; Meng, X.; Dube, P.; Zhang, X.; Hu, Y.; Kitamura, N.; et al. Structure and Inhibition of the SARS-CoV-2 Main Protease Reveal Strategy for Developing Dual Inhibitors against M pro and Cathepsin L. Sci. Adv. 2020, 6, eabe0751. [Google Scholar] [CrossRef]
- Günther, S.; Reinke, P.Y.A.; Fernández-García, Y.; Lieske, J.; Lane, T.J.; Ginn, H.M.; Koua, F.H.M.; Ehrt, C.; Ewert, W.; Ober-thuer, D.; et al. Inhibition of SARS-CoV-2 Main Protease by Allosteric Drug-Binding. bioRxiv. Available online: https://www.biorxiv.org/content/10.1101/2020.11.12.378422v2.full (accessed on 1 April 2021).
- Mehaffey, M.R.; Lee, J.; Jung, J.; Lanzillotti, M.B.; Escobar, E.E.; Morgenstern, K.R.; Georgiou, G.; Brodbelt, J.S. Mapping a Conformational Epitope of Hemagglutinin A Using Native Mass Spectrometry and Ultraviolet Photodissociation. Anal. Chem. 2020, 92, 11869–11878. [Google Scholar] [CrossRef]
- Holm, T.; Kopicki, J.-D.; Busch, C.; Olschewski, S.; Rosenthal, M.; Uetrecht, C.; Günther, S.; Reindl, S. Biochemical and Structural Studies Reveal Differences and Commonalities among Cap-Snatching Endonucleases from Segmented Negative-Strand RNA Viruses. J. Biol. Chem. 2018, 293, 19686–19698. [Google Scholar] [CrossRef] [Green Version]
- Mirza, U.A.; Chen, G.; Liu, Y.-H.; Doll, R.J.; Girijavallabhan, V.M.; Ganguly, A.K.; Pramanik, B.N. Mass Spectrometric Studies of Potent Inhibitors of Farnesyl Protein Transferase-Detection of Pentameric Noncovalent Complexes. J. Mass Spectrom. 2008, 43, 1393–1401. [Google Scholar] [CrossRef]
- Rogniaux, H.; Dorsselaer, A.; Barth, P.; Biellmann, J.F.; Barbanton, J.; Zandt, M.; Chevrier, B.; Howard, E.; Mitschler, A.; Potier, N.; et al. Binding of Aldose Reductase Inhibitors: Correlation of Crystallographic and Mass Spectrometric Studies. J Am. Soc. Mass Spectrom. 1999, 10, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Meng, B.; Lan, K.; Xie, J.; Lerner, R.A.; Wilson, I.A.; Yang, B. Inhibitory Antibodies Identify Unique Sites of Therapeutic Vulnerability in Rhinovirus and Other Enteroviruses. Proc. Natl. Acad. Sci. USA 2020, 117, 13499–13508. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Lee, D.E.; Kadam, R.U.; Liu, T.; Giang, E.; Nieusma, T.; Garces, F.; Tzarum, N.; Woods, V.L.; Ward, A.B.; et al. Structural Flexibility at a Major Conserved Antibody Target on Hepatitis C Virus E2 Antigen. Proc. Natl. Acad. Sci. USA 2016, 113, 12768–12773. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Soh, T.S.; Zheng, J.; Chan, K.W.K.; Phoo, W.W.; Lee, C.C.; Tay, M.Y.F.; Swaminathan, K.; Cornvik, T.C.; Lim, S.P.; et al. A Crystal Structure of the Dengue Virus NS5 Protein Reveals a Novel Inter-Domain Interface Essential for Protein Flexibility and Virus Replication. PLoS Pathog. 2015, 11, e1004682. [Google Scholar] [CrossRef] [PubMed]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of This Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef]
- Kim, B.; Araujo, R.; Howard, M.; Magni, R.; Liotta, L.A.; Luchini, A. Affinity Enrichment for Mass Spectrometry: Improving the Yield of Low Abundance Biomarkers. Expert Rev. Proteom. 2018, 15, 353–366. [Google Scholar] [CrossRef]
- Gatto, L.; Vizcaíno, J.A.; Hermjakob, H.; Huber, W.; Lilley, K.S. Organelle Proteomics Experimental Designs and Analysis. Proteomics 2010, 10, 3957–3969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bache, N.; Geyer, P.E.; Bekker-Jensen, D.B.; Hoerning, O.; Falkenby, L.; Treit, P.V.; Doll, S.; Paron, I.; Müller, J.B.; Meier, F.; et al. A Novel LC System Embeds Analytes in Pre-Formed Gradients for Rapid, Ultra-Robust Proteomics. Mol. Cell. Proteom. 2018, 17, 2284–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossegesse, M.; Hartkopf, F.; Nitsche, A.; Schaade, L.; Doellinger, J.; Muth, T. Perspective on Proteomics for Virus Detection in Clinical Samples. J. Proteome Res. 2020, 19, 4380–4388. [Google Scholar] [CrossRef]
- Krieger, J.R.; Wybenga-Groot, L.E.; Tong, J.; Bache, N.; Tsao, M.S.; Moran, M.F. Evosep One Enables Robust Deep Proteome Coverage Using Tandem Mass Tags While Significantly Reducing Instrument Time. J. Proteome Res. 2019, 18, 2346–2353. [Google Scholar] [CrossRef]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent Acquisition-based SWATH - MS for Quantitative Proteomics: A Tutorial. Mol. Syst. Biol. 2018, 14. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.-D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation–Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef] [Green Version]
- Hale, O.J.; Illes-Toth, E.; Mize, T.H.; Cooper, H.J. High-Field Asymmetric Waveform Ion Mobility Spectrometry and Native Mass Spectrometry: Analysis of Intact Protein Assemblies and Protein Complexes. Anal. Chem. 2020, 92, 6811–6816. [Google Scholar] [CrossRef]
- Ogata, K.; Ishihama, Y. Extending the Separation Space with Trapped Ion Mobility Spectrometry Improves the Accuracy of Isobaric Tag-Based Quantitation in Proteomic LC/MS/MS. Anal. Chem. 2020, 92, 8037–8040. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Bernhardt, O.M.; Hogrebe, A.; Martinez-Val, A.; Verbeke, L.; Gandhi, T.; Kelstrup, C.D.; Reiter, L.; Olsen, J.V. Rapid and Site-Specific Deep Phosphoproteome Profiling by Data-Independent Acquisition without the Need for Spectral Libraries. Nat. Commun. 2020, 11, 787. [Google Scholar] [CrossRef] [Green Version]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Prianichnikov, N.; Koch, H.; Koch, S.; Lubeck, M.; Heilig, R.; Brehmer, S.; Fischer, R.; Cox, J. MaxQuant Software for Ion Mobility Enhanced Shotgun Proteomics. Mol. Cell. Proteom. 2020, 19, 1058–1069. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wei, S.; Ji, Y.; Guo, X.; Yang, F. Quantitative Proteomics Using SILAC: Principles, Applications, and Developments. Proteomics 2015, 15, 3175–3192. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Wölmer, N.; Koncarevic, S.; Selzer, S.; Böhm, G.; Legner, H.; Schmid, P.; Kienle, S.; Penning, P.; Höhle, C.; et al. TMTpro: Design, Synthesis, and Initial Evaluation of a Proline-Based Isobaric 16-Plex Tandem Mass Tag Reagent Set. Anal. Chem. 2019, 91, 15941–15950. [Google Scholar] [CrossRef] [PubMed]
- Savitski, M.M.; Mathieson, T.; Zinn, N.; Sweetman, G.; Doce, C.; Becher, I.; Pachl, F.; Kuster, B.; Bantscheff, M. Measuring and Managing Ratio Compression for Accurate ITRAQ/TMT Quantification. J. Proteome Res. 2013, 12, 3586–3598. [Google Scholar] [CrossRef] [PubMed]
- Dejarnac, O.; Hafirassou, M.L.; Chazal, M.; Versapuech, M.; Gaillard, J.; Perera-Lecoin, M.; Umana-Diaz, C.; Bonnet-Madin, L.; Carnec, X.; Tinevez, J.-Y.; et al. TIM-1 Ubiquitination Mediates Dengue Virus Entry. Cell Rep. 2018, 23, 1779–1793. [Google Scholar] [CrossRef] [PubMed]
- Gerold, G.; Meissner, F.; Bruening, J.; Welsch, K.; Perin, P.M.; Baumert, T.F.; Vondran, F.W.; Kaderali, L.; Marcotrigiano, J.; Khan, A.G.; et al. Quantitative Proteomics Identifies Serum Response Factor Binding Protein 1 as a Host Factor for Hepatitis C Virus Entry. Cell Rep. 2015, 12, 864–878. [Google Scholar] [CrossRef] [Green Version]
- Matheson, N.J.; Sumner, J.; Wals, K.; Rapiteanu, R.; Weekes, M.P.; Vigan, R.; Weinelt, J.; Schindler, M.; Antrobus, R.; Costa, A.S.H.; et al. Cell Surface Proteomic Map of HIV Infection Reveals Antagonism of Amino Acid Metabolism by Vpu and Nef. Cell Host Microbe 2015, 18, 409–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Münch, C. Proteomics of SARS-CoV-2-Infected Host Cells Reveals Therapy Targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]
- Bogdanow, B.; Wang, X.; Eichelbaum, K.; Sadewasser, A.; Husic, I.; Paki, K.; Budt, M.; Hergeselle, M.; Vetter, B.; Hou, J.; et al. The Dynamic Proteome of Influenza A Virus Infection Identifies M Segment Splicing as a Host Range Determinant. Nat. Commun. 2019, 10, 5518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naamati, A.; Williamson, J.C.; Greenwood, E.J.; Marelli, S.; Lehner, P.J.; Matheson, N.J. Functional Proteomic Atlas of HIV Infection in Primary Human CD4+ T Cells. eLife 2019, 8, e41431. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59–72.e15. [Google Scholar] [CrossRef] [PubMed]
- Messner, C.B.; Demichev, V.; Wendisch, D.; Michalick, L.; White, M.; Freiwald, A.; Textoris-Taube, K.; Vernardis, S.I.; Egger, A.-S.; Kreidl, M.; et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst. 2020, 11, 11–24.e4. [Google Scholar] [CrossRef]
- Korba, B.E.; Montero, A.B.; Farrar, K.; Gaye, K.; Mukerjee, S.; Ayers, M.S.; Rossignol, J.-F. Nitazoxanide, Tizoxanide and Other Thiazolides Are Potent Inhibitors of Hepatitis B Virus and Hepatitis C Virus Replication. Antivir. Res. 2008, 77, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.-F. Nitazoxanide, a New Drug Candidate for the Treatment of Middle East Respiratory Syndrome Coronavirus. J. Infect. Public Health 2016, 9, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and Chloroquine Effectively Inhibit the Recently Emerged Novel Coronavirus (2019-NCoV) in Vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Yamamoto, K.A.; Blackburn, K.; Migowski, E.; Goshe, M.B.; Brown, D.T.; Ferreira, D.F.; Soares, M.R. Quantitative Proteomic Analysis of the Tizoxanide Effect in Vero Cells. Sci. Rep. 2020, 10, 14733. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; LaCount, D.J.; Kuhn, R.J.; Randall, G. Dengue Virus Nonstructural Protein 3 Redistributes Fatty Acid Synthase to Sites of Viral Replication and Increases Cellular Fatty Acid Synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17345–17350. [Google Scholar] [CrossRef] [Green Version]
- Ohol, Y.M.; Wang, Z.; Kemble, G.; Duke, G. Direct Inhibition of Cellular Fatty Acid Synthase Impairs Replication of Respiratory Syncytial Virus and Other Respiratory Viruses. PLoS ONE 2015, 10, e0144648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, R.; Sur, S.; Cheng, Q.; Steele, R.; Ray, R.B. Repression of MicroRNA-30e by Hepatitis C Virus Enhances Fatty Acid Synthesis. Hepatol. Commun. 2019, 3, 943–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poenisch, M.; Metz, P.; Blankenburg, H.; Ruggieri, A.; Lee, J.-Y.; Rupp, D.; Rebhan, I.; Diederich, K.; Kaderali, L.; Domingues, F.S.; et al. Identification of HNRNPK as Regulator of Hepatitis C Virus Particle Production. PLoS Pathog. 2015, 11, e1004573. [Google Scholar] [CrossRef] [PubMed]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Salokas, K.; Weldatsadik, R.G.; Gawriyski, L.; Varjosalo, M. Combined Proximity Labeling and Affinity Purification−mass Spectrometry Workflow for Mapping and Visualizing Protein Interaction Networks. Nat. Protoc. 2020, 15, 3182–3211. [Google Scholar] [CrossRef]
- Yang, B.; Tang, S.; Ma, C.; Li, S.-T.; Shao, G.-C.; Dang, B.; DeGrado, W.F.; Dong, M.-Q.; Wang, P.G.; Ding, S.; et al. Spontaneous and Specific Chemical Cross-Linking in Live Cells to Capture and Identify Protein Interactions. Nat. Commun. 2017, 8, 2240. [Google Scholar] [CrossRef]
- Sperk, M.; van Domselaar, R.; Rodriguez, J.E.; Mikaeloff, F.; Sá Vinhas, B.; Saccon, E.; Sönnerborg, A.; Singh, K.; Gupta, S.; Végvári, Á.; et al. Utility of Proteomics in Emerging and Re-Emerging Infectious Diseases Caused by RNA Viruses. J. Proteome Res. 2020, 19, 4259–4274. [Google Scholar] [CrossRef]
- Shah, P.S.; Link, N.; Jang, G.M.; Sharp, P.P.; Zhu, T.; Swaney, D.L.; Johnson, J.R.; Von Dollen, J.; Ramage, H.R.; Satkamp, L.; et al. Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis. Cell 2018, 175, 1931–1945.e18. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Dong, S.; Luo, Z.; Xie, X.; Fu, B.; Li, P.; Liu, C.; Yang, X.; Chen, Y.; Wang, X.; et al. The Zika Virus Capsid Disrupts Corticogenesis by Suppressing Dicer Activity and MiRNA Biogenesis. Cell Stem Cell 2020, 27, 618–632.e9. [Google Scholar] [CrossRef]
- Link, N.; Chung, H.; Jolly, A.; Withers, M.; Tepe, B.; Arenkiel, B.R.; Shah, P.S.; Krogan, N.J.; Aydin, H.; Geckinli, B.B.; et al. Mutations in ANKLE2, a ZIKA Virus Target, Disrupt an Asymmetric Cell Division Pathway in Drosophila Neuroblasts to Cause Microcephaly. Dev. Cell 2019, 51, 713–729.e6. [Google Scholar] [CrossRef]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.-S.; Lee, S.-A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-MTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Hafirassou, M.L.; Meertens, L.; Umaña-Diaz, C.; Labeau, A.; Dejarnac, O.; Bonnet-Madin, L.; Kümmerer, B.M.; Delaugerre, C.; Roingeard, P.; Vidalain, P.-O.; et al. A Global Interactome Map of the Dengue Virus NS1 Identifies Virus Restriction and Dependency Host Factors. Cell Rep. 2017, 21, 3900–3913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uckeley, Z.M.; Moeller, R.; Kühn, L.I.; Nilsson, E.; Robens, C.; Lasswitz, L.; Lindqvist, R.; Lenman, A.; Passos, V.; Voss, Y.; et al. Quantitative Proteomics of Uukuniemi Virus-Host Cell Interactions Reveals GBF1 as Proviral Host Factor for Phleboviruses. Mol. Cell. Proteom. 2019, 18, 2401–2417. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J.; et al. Comparative Host-Coronavirus Protein Interaction Networks Reveal Pan-Viral Disease Mechanisms. Science 2020, 370. [Google Scholar] [CrossRef] [PubMed]
- Sage, V.; Cinti, A.; Mouland, A.J. Proximity-Dependent Biotinylation for Identification of Interacting Proteins. Curr. Protoc. Cell Biol. 2016, 73. [Google Scholar] [CrossRef]
- Coyaud, E.; Ranadheera, C.; Cheng, D.; Gonçalves, J.; Dyakov, B.J.A.; Laurent, E.M.N.; St-Germain, J.; Pelletier, L.; Gingras, A.-C.; Brumell, J.H.; et al. Global Interactomics Uncovers Extensive Organellar Targeting by Zika Virus. Mol. Cell. Proteom. 2018, 17, 2242–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-C.; Peterson, S.E.; Loring, J.F. Protein Post-Translational Modifications and Regulation of Pluripotency in Human Stem Cells. Cell Res. 2014, 24, 143–160. [Google Scholar] [CrossRef] [Green Version]
- Fíla, J.; Honys, D. Enrichment Techniques Employed in Phosphoproteomics. Amino Acids 2012, 43, 1025–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klann, K.; Bojkova, D.; Tascher, G.; Ciesek, S.; Münch, C.; Cinatl, J. Growth Factor Receptor Signaling Inhibition Prevents SARS-CoV-2 Replication. Mol. Cell 2020, 80, 164–174.e4. [Google Scholar] [CrossRef]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Correa Marrero, M.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712.e19. [Google Scholar] [CrossRef]
- Nayak, T.K.; Mamidi, P.; Sahoo, S.S.; Kumar, P.S.; Mahish, C.; Chatterjee, S.; Subudhi, B.B.; Chattopadhyay, S.; Chattopadhyay, S. P38 and JNK Mitogen-Activated Protein Kinases Interact With Chikungunya Virus Non-Structural Protein-2 and Regulate TNF Induction During Viral Infection in Macrophages. Front. Immunol. 2019, 10, 786. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-C.; Simanjuntak, Y.; Chu, L.-W.; Ping, Y.-H.; Lee, Y.-L.; Lin, Y.-L.; Li, W.-S. Benzenesulfonamide Derivatives as Calcium/Calmodulin-Dependent Protein Kinase Inhibitors and Antiviral Agents against Dengue and Zika Virus Infections. J. Med. Chem. 2020, 63, 1313–1327. [Google Scholar] [CrossRef]
- Söderholm, S.; Kainov, D.E.; Öhman, T.; Denisova, O.V.; Schepens, B.; Kulesskiy, E.; Imanishi, S.Y.; Corthals, G.; Hintsanen, P.; Aittokallio, T.; et al. Phosphoproteomics to Characterize Host Response During Influenza A Virus Infection of Human Macrophages. Mol. Cell. Proteom. 2016, 15, 3203–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, A.; Dam, S.; Saul, V.V.; Kuznetsova, I.; Müller, C.; Fritz-Wolf, K.; Becker, K.; Linne, U.; Gu, H.; Stokes, M.P.; et al. Phosphoproteome Analysis of Cells Infected with Adapted and Nonadapted Influenza A Virus Reveals Novel Pro- and Antiviral Signaling Networks. J. Virol. 2019, 93, e00528-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yángüez, E.; Hunziker, A.; Dobay, M.P.; Yildiz, S.; Schading, S.; Elshina, E.; Karakus, U.; Gehrig, P.; Grossmann, J.; Dijkman, R.; et al. Phosphoproteomic-Based Kinase Profiling Early in Influenza Virus Infection Identifies GRK2 as Antiviral Drug Target. Nat. Commun. 2018, 9, 3679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Sun, J.; Ye, J.; Ashraf, U.; Chen, Z.; Zhu, B.; He, W.; Xu, Q.; Wei, Y.; Chen, H.; et al. Quantitative Label-Free Phosphoproteomics Reveals Differentially Regulated Protein Phosphorylation Involved in West Nile Virus-Induced Host Inflammatory Response. J. Proteome Res. 2015, 14, 5157–5168. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, H.; He, W.; Zhu, B.; Zhou, D.; Chen, Z.; Ashraf, U.; Wei, Y.; Liu, Z.; Fu, Z.F.; et al. Quantitative Phosphoproteomic Analysis Identifies the Critical Role of JNK1 in Neuroinflammation Induced by Japanese Encephalitis Virus. Sci. Signal. 2016, 9, ra98. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, M.I.; Xia, H.; Aguilera-Aguirre, L.; Hage, A.; van Tol, S.; Shan, C.; Xie, X.; Sturdevant, G.L.; Robertson, S.J.; McNally, K.L.; et al. Envelope Protein Ubiquitination Drives Entry and Pathogenesis of Zika Virus. Nature 2020, 585, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Barouch-Bentov, R.; Xiao, F.; Schor, S.; Pu, S.; Biquand, E.; Lu, A.; Lindenbach, B.D.; Jacob, Y.; Demeret, C.; et al. MARCH8 Ubiquitinates the Hepatitis C Virus Nonstructural 2 Protein and Mediates Viral Envelopment. Cell Rep. 2019, 26, 1800–1814.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Smits, A.H.; van Tilburg, G.B.; Ovaa, H.; Huber, W.; Vermeulen, M. Proteome-Wide Identification of Ubiquitin Interactions Using UbIA-MS. Nat. Protoc. 2018, 13, 530–550. [Google Scholar] [CrossRef] [PubMed]
- Giese, S.; Ciminski, K.; Bolte, H.; Moreira, É.A.; Lakdawala, S.; Hu, Z.; David, Q.; Kolesnikova, L.; Götz, V.; Zhao, Y.; et al. Role of Influenza A Virus NP Acetylation on Viral Growth and Replication. Nat. Commun. 2017, 8, 1259. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Wu, R.; Xu, G.; Cheng, Y.; Wang, Z.; Wang, H.; Yan, Y.; Li, J.; Sun, J. Acetylation at K108 of the NS1 Protein Is Important for the Replication and Virulence of Influenza Virus. Vet. Res. 2020, 51, 20. [Google Scholar] [CrossRef] [Green Version]
- Murray, L.A.; Combs, A.N.; Rekapalli, P.; Cristea, I.M. Methods for characterizing protein acetylation during viral infection. Methods Enzymol. 2019, 626, 587–620. [Google Scholar]
- Bagdonaite, I.; Wandall, H.H. Global Aspects of Viral Glycosylation. Glycobiology 2018, 28, 443–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Suttapitugsakul, S.; Sun, F.; Wu, R. Mass Spectrometry-Based Chemical and Enzymatic Methods for Global Analysis of Protein Glycosylation. Acc. Chem. Res. 2018, 51, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. A Dream of Single-Cell Proteomics. Nat. Methods 2019, 16, 809–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simanjuntak, Y.; Schamoni-Kast, K.; Grün, A.; Uetrecht, C.; Scaturro, P. Top-Down and Bottom-Up Proteomics Methods to Study RNA Virus Biology. Viruses 2021, 13, 668. https://doi.org/10.3390/v13040668
Simanjuntak Y, Schamoni-Kast K, Grün A, Uetrecht C, Scaturro P. Top-Down and Bottom-Up Proteomics Methods to Study RNA Virus Biology. Viruses. 2021; 13(4):668. https://doi.org/10.3390/v13040668
Chicago/Turabian StyleSimanjuntak, Yogy, Kira Schamoni-Kast, Alice Grün, Charlotte Uetrecht, and Pietro Scaturro. 2021. "Top-Down and Bottom-Up Proteomics Methods to Study RNA Virus Biology" Viruses 13, no. 4: 668. https://doi.org/10.3390/v13040668
APA StyleSimanjuntak, Y., Schamoni-Kast, K., Grün, A., Uetrecht, C., & Scaturro, P. (2021). Top-Down and Bottom-Up Proteomics Methods to Study RNA Virus Biology. Viruses, 13(4), 668. https://doi.org/10.3390/v13040668