
HIV-1 Natural Antisense Transcription and Its Role in Viral Persistence


Abstract
:1. Introduction
2. Non-Coding RNAs
2.1. Phylogenetic Distribution and Complexity of ncRNAs
2.2. Classification of ncRNAs
3. Natural Antisense Transcripts (NATs)
3.1. Phylogenetic Distribution and Conservation
3.2. Characteristics of NATs Expression
3.3. Mechanisms of Gene Regulation by NATs
3.4. Protein-Based vs. NAT-Based Gene Regulation
3.5. Examples of NATs in Eukaryotic Cell Systems
3.6. Examples of NATs in Viral Systems
4. Natural Antisense Transcription in the HIV-1 Proviral Genome
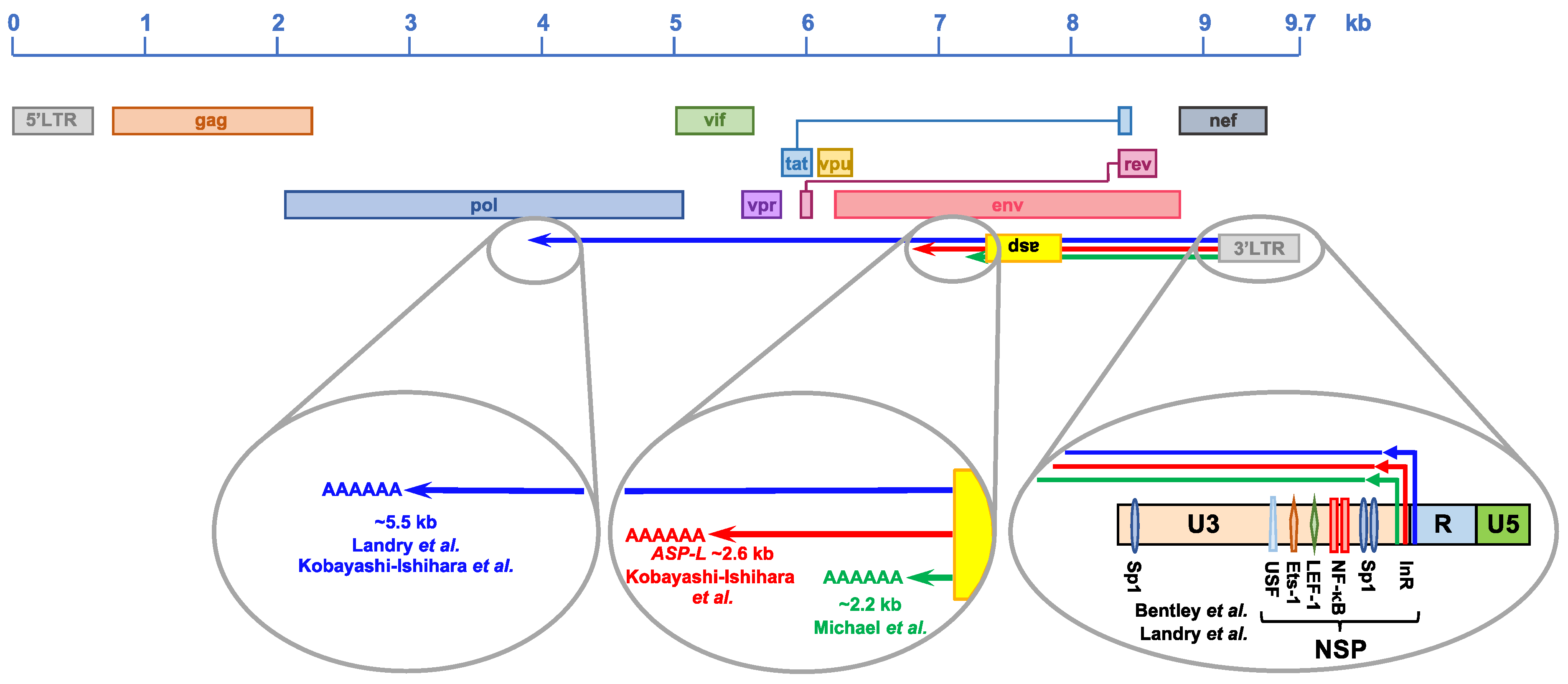
4.1. Discovery of HIV-1 Natural Antisense Transcription
4.2. Structure of the HIV-1 Natural Antisense Transcripts
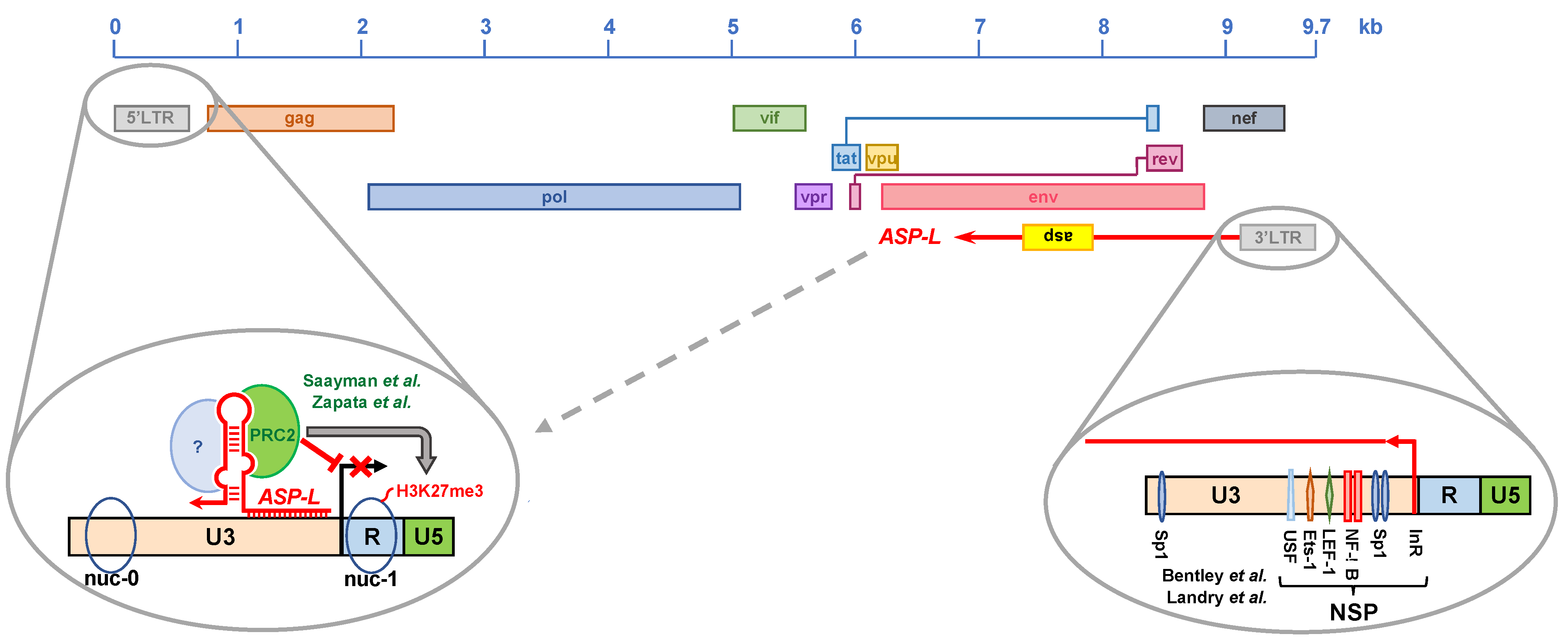
4.3. Regulation of HIV-1 Natural Antisense Transcription
4.4. The HIV-1 Natural Antisense Transcript as a Protein-Coding RNA
4.5. Role of HIV-1 Antisense Transcripts in Viral Expression
5. Open Questions and Future Research Avenues
5.1. One or Multiple HIV-1 Antisense Transcripts?
5.2. Does HIV-1 Express Antisense Transcripts In Vivo?
5.3. Does Ast Affect Host Gene Expression?
5.4. Is Ast Expression Regulated at the Epigenetic Level?
5.5. Why Does HIV-1 Need Antisense Transcription?
5.6. Can HIV-1 NATs Be Exploited in Cure Strategies?
5.7. What Would Be the Advantage of an Ast-Based Cure Strategy?
5.8. Are NAT-Induced Epigenetic Changes Inheritable by Daughter Cells after Cell Division?
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- The ENCODE Project Consortium. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivas, E.; Klein, R.J.; Jones, T.A.; Eddy, S.R. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr. Biol. 2001, 11, 1369–1373. [Google Scholar] [CrossRef] [Green Version]
- Vogel, J.; Bartels, V.; Tang, T.H.; Churakov, G.; Slagter-Jager, J.G.; Huttenhofer, A.; Wagner, E.G. RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res. 2003, 31, 6435–6443. [Google Scholar] [CrossRef] [Green Version]
- Kawano, M.; Reynolds, A.A.; Miranda-Rios, J.; Storz, G. Detection of 5’- and 3’-UTR-derived small RNAs and cis-encoded antisense RNAs in Escherichia coli. Nucleic Acids Res. 2005, 33, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Wilderman, P.J.; Sowa, N.A.; Fitzgerald, D.J.; Fitzgerald, P.C.; Gottesman, S.; Ochsner, U.A.; Vasil, M.L. Identification of tandem duplicate regulatory small RNAs in Pseudomonas aeruginosa involved in iron homeostasis. Proc. Natl. Acad. Sci. USA 2004, 101, 9792–9797. [Google Scholar] [CrossRef] [Green Version]
- Axmann, I.M.; Kensche, P.; Vogel, J.; Kohl, S.; Herzel, H.; Hess, W.R. Identification of cyanobacterial non-coding RNAs by comparative genome analysis. Genome Biol. 2005, 6, R73. [Google Scholar] [CrossRef] [Green Version]
- Dennis, P.P.; Omer, A. Small non-coding RNAs in Archaea. Curr. Opin. Microbiol. 2005, 8, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Simons, R.W. Naturally occurring antisense RNA control—A brief review. Gene 1988, 72, 35–44. [Google Scholar] [CrossRef]
- Mattick, J.S. RNA regulation: A new genetics? Nat. Rev. Genet. 2004, 5, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Makino, K.; Ohnishi, M.; Kurokawa, K.; Ishii, K.; Yokoyama, K.; Han, C.G.; Ohtsubo, E.; Nakayama, K.; Murata, T.; et al. Complete Genome Sequence of Enterohemorrhagic Eschelichia coli O157:H7 and Genomic Comparison with a Laboratory Strain K-12. DNA Res. 2001, 8, 11–22. [Google Scholar] [CrossRef] [Green Version]
- van Nimwegen, E. Scaling laws in the functional content of genomes. Trends Genet. 2003, 19, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Mattick, J.S.; Gagen, M.J. MATHEMATICS/COMPUTATION: Accelerating Networks. Science 2005, 307, 856–858. [Google Scholar] [CrossRef] [PubMed]
- Gagen, M.J.; Mattick, J.S. Inherent size constraints on prokaryote gene networks due to? accelerating? growth. Theory Biosci. 2005, 123, 381–411. [Google Scholar] [CrossRef] [Green Version]
- Timmons, J.A.; Good, L. Does everything now make (anti)sense? Biochem. Soc. Trans. 2006, 34, 1148–1150. [Google Scholar] [CrossRef] [PubMed]
- Frith, M.C.; Pheasant, M.; Mattick, J.S. Genomics: The amazing complexity of the human transcriptome. Eur. J. Hum. Genet. 2005, 13, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S. Non-coding RNAs: The architects of eukaryotic complexity. EMBO Rep. 2001, 2, 986–991. [Google Scholar] [CrossRef]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The Transcriptional Landscape of the Mammalian Genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Human Genome Sequencing Consortium (IHGS). Finishing the euchromatic sequence of the human genome. Nat. Cell Biol. 2004, 431, 931–945. [Google Scholar] [CrossRef]
- Mattick, J.S.; Gagen, M.J. The Evolution of Controlled Multitasked Gene Networks: The Role of Introns and Other Noncoding RNAs in the Development of Complex Organisms. Mol. Biol. Evol. 2001, 18, 1611–1630. [Google Scholar] [CrossRef] [Green Version]
- Mattick, J.S. Challenging the dogma: The hidden layer of non-protein-coding RNAs in complex organisms. BioEssays 2003, 25, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.-M. Fewer Genes, More Noncoding RNA. Science 2005, 309, 1529–1530. [Google Scholar] [CrossRef]
- Kaikkonen, M.U.; Lam, M.T.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Khorkova, O.; Myers, A.J.; Hsiao, J.; Wahlestedt, C. Natural antisense transcripts. Hum. Mol. Genet. 2014, 23, R54–R63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [Green Version]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L.; et al. RNA Maps Reveal New RNA Classes and a Possible Function for Pervasive Transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Kim, Y.C.; Lu, J.; Xuan, Z.; Chen, J.; Zheng, Y.; Zhou, T.; Zhang, M.Q.; Wu, C.-I.; Wang, S.M. Poly A- Transcripts Expressed in HeLa Cells. PLoS ONE 2008, 3, e2803. [Google Scholar] [CrossRef] [Green Version]
- Engström, P.G.; Suzuki, H.; Ninomiya, N.; Akalin, A.; Sessa, L.; Lavorgna, G.; Brozzi, A.; Luzi, L.; Tan, S.L.; Yang, L.; et al. Complex Loci in Human and Mouse Genomes. PLoS Genet. 2006, 2, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutter, C.; Watt, S.; Stefflova, K.; Wilson, M.D.; Goncalves, A.; Ponting, C.P.; Odom, D.T.; Marques, A.C. Rapid Turnover of Long Noncoding RNAs and the Evolution of Gene Expression. PLoS Genet. 2012, 8, e1002841. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.J.; Chin-Inmanu, K.; Jia, H.; Lipovich, L. Sense-antisense gene pairs: Sequence, transcription, and structure are not conserved between human and mouse. Front. Genet. 2013, 4, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnsson, P.; Ackley, A.; Vidarsdottir, L.; Lui, W.-O.; Corcoran, M.; Grandér, D.; Morris, K.V. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat. Struct. Mol. Biol. 2013, 20, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.F.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nat. Cell Biol. 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [Green Version]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and Functions of Long Noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torarinsson, E.; Sawera, M.; Havgaard, J.H.; Fredholm, M.; Gorodkin, J. Thousands of corresponding human and mouse genomic regions unalignable in primary sequence contain common RNA structure. Genome Res. 2006, 16, 885–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnsson, P.; Lipovich, L.; Grandér, D.; Morris, K.V. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, Evolution, and Mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacatena, R.M.; Cesareni, G. Base pairing of RNA I with its complementary sequence in the primer precursor inhibits ColE1 replication. Nat. Cell Biol. 1981, 294, 623–626. [Google Scholar] [CrossRef]
- Bøvre, K.; Szybalski, W. Patterns of convergent and overlapping transcription within the b2 region of coliphage λ. Virology 1969, 38, 614–626. [Google Scholar] [CrossRef]
- Inouye, M. Antisense RNA: Its functions and applications in gene regulation—A review. Gene 1988, 72, 25–34. [Google Scholar] [CrossRef]
- Henikoff, S.; Keene, M.A.; Fechtel, K.; Fristrom, J.W. Gene within a gene: Nested Drosophila genes encode unrelated proteins on opposite DNA strands. Cell 1986, 44, 33–42. [Google Scholar] [CrossRef]
- Spencer, C.A.; Gietz, R.D.; Hodgetts, R.B. Overlapping transcription units in the dopa decarboxylase region of Drosophila. Nat. Cell Biol. 1986, 322, 279–281. [Google Scholar] [CrossRef]
- Williams, T.; Fried, M. A mouse locus at which transcription from both DNA strands produces mRNAs complementary at their 3′ ends. Nat. Cell Biol. 1986, 322, 275–279. [Google Scholar] [CrossRef]
- Van Duin, M.; Tol, J.V.D.; Hoeijmakers, J.H.; Bootsma, D.; Rupp, I.P.; Reynolds, P.; Prakash, L.; Prakash, S. Conserved pattern of antisense overlapping transcription in the homologous human ERCC-1 and yeast RAD10 DNA repair gene regions. Mol. Cell. Biol. 1989, 9, 1794–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Chua, N.-H.; Wang, X.-J. Prediction of trans-antisense transcripts in Arabidopsis thaliana. Genome Biol. 2006, 7, R92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, L.; Huber, W.; Granovskaia, M.; Toedling, J.; Palm, C.J.; Bofkin, L.; Jones, T.; Davis, R.W.; Steinmetz, L.M. A high-resolution map of transcription in the yeast genome. Proc. Natl. Acad. Sci. USA 2006, 103, 5320–5325. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.; Rubinstein, W.S.; Jung, Y.-C.; Wu, Q. Genome-wide analysis of antisense transcription with Affymetrix exon array. BMC Genom. 2008, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Bao, J.; Zhou, G.; Shapiro, J.; Xu, J.; Shi, R.Z.; Lu, X.; Clark, T.; Johnson, D.; Kim, Y.C.; et al. Detecting novel low-abundant transcripts in Drosophila. RNA 2005, 11, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Katayama, S.; Tomaru, Y.; Kasukawa, T.; Waki, K.; Nakanishi, M.; Nakamura, M.; Nishida, H.; Yap, C.C.; Suzuki, M.; Kawai, J.; et al. Antisense Transcription in the Mammalian Transcriptome. Science 2005, 309, 1564–1566. [Google Scholar] [CrossRef]
- Chen, J.; Sun, M.; Kent, W.J.; Huang, X.; Xie, H.; Wang, W.; Zhou, G.; Shi, R.Z.; Rowley, J.D. Over 20% of human transcripts might form sense-antisense pairs. Nucleic Acids Res. 2004, 32, 4812–4820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapidot, M.; Pilpel, Y. Genome-wide natural antisense transcription: Coupling its regulation to its different regulatory mechanisms. EMBO Rep. 2006, 7, 1216–1222. [Google Scholar] [CrossRef] [Green Version]
- Conley, A.B.; Miller, W.J.; Jordan, I.K. Human cis natural antisense transcripts initiated by transposable elements. Trends Genet. 2008, 24, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Soumillon, M.; Necsulea, A.; Weier, M.; Brawand, D.; Zhang, X.; Gu, H.; Barthès, P.; Kokkinaki, M.; Nef, S.; Gnirke, A.; et al. Cellular Source and Mechanisms of High Transcriptome Complexity in the Mammalian Testis. Cell Rep. 2013, 3, 2179–2190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, X.S.; Liu, Q.-R.; Wei, L. Genome-wide in silico identification and analysis of cis natural antisense transcripts (cis-NATs) in ten species. Nucleic Acids Res. 2006, 34, 3465–3475. [Google Scholar] [CrossRef] [Green Version]
- Yassour, M.; Pfiffner, J.; Levin, J.Z.; Adiconis, X.; Gnirke, A.; Nusbaum, C.; Thompson, D.-A.; Friedman, N.; Regev, A. Strand-specific RNA sequencing reveals extensive regulated long antisense transcripts that are conserved across yeast species. Genome Biol. 2010, 11, R87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhind, N.; Chen, Z.; Yassour, M.; Thompson, D.A.; Haas, B.J.; Habib, N.; Wapinski, I.; Roy, S.; Lin, M.F.; Heiman, D.I.; et al. Comparative Functional Genomics of the Fission Yeasts. Science 2011, 332, 930–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, A.J.; Daugharthy, E.R.; Kim, J. Pervasive Antisense Transcription Is Evolutionarily Conserved in Budding Yeast. Mol. Biol. Evol. 2012, 30, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Raghavan, R.; Sloan, D.B.; Ochman, H. Antisense Transcription Is Pervasive but Rarely Conserved in Enteric Bacteria. mBio 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Pelechano, V.; Steinmetz, L.M. Gene regulation by antisense transcription. Nat. Rev. Genet. 2013, 14, 880–893. [Google Scholar] [CrossRef]
- He, Y.; Vogelstein, B.; Velculescu, V.E.; Papadopoulos, N.; Kinzler, K.W. The Antisense Transcriptomes of Human Cells. Science 2008, 322, 1855–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozsolak, F.; Kapranov, P.; Foissac, S.; Kim, S.W.; Fishilevich, E.; Monaghan, A.P.; John, B.; Milos, P.M. Comprehensive Polyadenylation Site Maps in Yeast and Human Reveal Pervasive Alternative Polyadenylation. Cell 2010, 143, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Struhl, K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat. Struct. Mol. Biol. 2007, 14, 103–105. [Google Scholar] [CrossRef]
- Rosikiewicz, W.; Makałowska, I. Biological Functions of Natural Antisense Transcripts. Acta Biochim. Pol. 2017, 63, 665–673. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Sun, M.; Hurst, L.D.; Carmichael, G.G.; Rowley, J.D. Genome-wide analysis of coordinate expression and evolution of human encoded sense-antisense transcripts. Trends Genet. 2005, 21, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Hurst, L.D.; Carmichael, G.G.; Chen, J. Evidence for a preferential targeting of 3’-UTRs by cis-encoded natural antisense transcripts. Nucleic Acids Res. 2005, 33, 5533–5543. [Google Scholar] [CrossRef] [Green Version]
- Su, W.-Y.; Li, J.-T.; Cui, Y.; Hong, J.; Du, W.; Wang, Y.-C.; Lin, Y.-W.; Xiong, H.; Wang, J.-L.; Kong, X.; et al. Bidirectional regulation between WDR83 and its natural antisense transcript DHPS in gastric cancer. Cell Res. 2012, 22, 1374–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkening, S.; Pelechano, V.; Järvelin, A.I.; Tekkedil, M.M.; Anders, S.; Benes, V.; Steinmetz, L.M. An efficient method for genome-wide polyadenylation site mapping and RNA quantification. Nucleic Acids Res. 2013, 41, e65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelechano, V.; Wei, W.; Steinmetz, L.M. Extensive transcriptional heterogeneity revealed by isoform profiling. Nat. Cell Biol. 2013, 497, 127–131. [Google Scholar] [CrossRef] [Green Version]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; Iii, G.S.L.; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of β-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef] [Green Version]
- Uchida, T.; Rossignol, F.; Matthay, M.A.; Mounier, R.; Couette, S.; Clottes, E.; Clerici, C. Prolonged Hypoxia Differentially Regulates Hypoxia-inducible Factor (HIF)-1α and HIF-2α Expression in Lung Epithelial Cells: Implication of natural antisense HIF-1alpha. J. Biol. Chem. 2004, 279, 14871–14878. [Google Scholar] [CrossRef] [Green Version]
- Wight, M.; Werner, A. The functions of natural antisense transcripts. Essays Biochem. 2013, 54, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Zinad, H.S.; Natasya, I.; Werner, A. Natural Antisense Transcripts at the Interface between Host Genome and Mobile Genetic Elements. Front. Microbiol. 2017, 8, 2292. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Wahlestedt, C. Regulatory roles of natural antisense transcripts. Nat. Rev. Mol. Cell Biol. 2009, 10, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Legewie, S.; Dienst, D.; Wilde, A.; Herzel, H.; Axmann, I.M. Small RNAs Establish Delays and Temporal Thresholds in Gene Expression. Biophys. J. 2008, 95, 3232–3238. [Google Scholar] [CrossRef] [Green Version]
- Dühring, U.; Axmann, I.M.; Hess, W.R.; Wilde, A. An internal antisense RNA regulates expression of the photosynthesis gene isiA. Proc. Natl. Acad. Sci. USA 2006, 103, 7054–7058. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Wei, W.; Gagneur, J.; Clauder-Münster, S.; Smolik, M.; Huber, W.; Steinmetz, L.M. Antisense expression increases gene expression variability and locus interdependency. Mol. Syst. Biol. 2011, 7, 468. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Mesnard, J.-M. HTLV-1 bZIP factor: The key viral gene for pathogenesis. Retrovirology 2020, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wanowska, E.; Kubiak, M.R.; Rosikiewicz, W.; Makałowska, I.; Szcześniak, M.W. Natural antisense transcripts in diseases: From modes of action to targeted therapies. Wiley Interdiscip. Rev. RNA 2018, 9, e1461. [Google Scholar] [CrossRef] [Green Version]
- Pontier, D.B.; Gribnau, J. Xist regulation and function eXplored. Qual. Life Res. 2011, 130, 223–236. [Google Scholar] [CrossRef] [Green Version]
- Patrat, C.; Ouimette, J.-F.; Rougeulle, C. X chromosome inactivation in human development. Development 2020, 147, dev183095. [Google Scholar] [CrossRef]
- Ohhata, T.; Matsumoto, M.; Leeb, M.; Shibata, S.; Sakai, S.; Kitagawa, K.; Niida, H.; Kitagawa, M.; Wutz, A. Histone H3 Lysine 36 Trimethylation Is Established over theXistPromoter by AntisenseTsixTranscription and Contributes to RepressingXistExpression. Mol. Cell. Biol. 2015, 35, 3909–3920. [Google Scholar] [CrossRef] [Green Version]
- Willard, H.F.; Carrel, L. Making sense (and antisense) of the X inactivation center. Proc. Natl. Acad. Sci. USA 2001, 98, 10025–10027. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Davidow, L.S.; Warshawsky, D. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat. Genet. 1999, 21, 400–404. [Google Scholar] [CrossRef]
- Congrains, A.; Kamide, K.; Ohishi, M.; Rakugi, H. ANRIL: Molecular Mechanisms and Implications in Human Health. Int. J. Mol. Sci. 2013, 14, 1278–1292. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Han, X.; Wittfeldt, A.; Sun, J.; Liu, C.; Wang, X.; Gan, L.-M.; Cao, H.; Liang, Z. Long non-coding RNA ANRIL regulates inflammatory responses as a novel component of NF-κB pathway. RNA Biol. 2016, 13, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.-M. Molecular Interplay of the Noncoding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Gius, D.; Onyango, P.; Muldoon-Jacobs, K.; Karp, J.E.; Feinberg, A.P.; Cui, H. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nat. Cell Biol. 2008, 451, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-DiNardo, D.; Kanduri, C. Kcnq1ot1 Antisense Noncoding RNA Mediates Lineage-Specific Transcriptional Silencing through Chromatin-Level Regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar] [CrossRef]
- Kacem, S.; Feil, R. Chromatin mechanisms in genomic imprinting. Mamm. Genome 2009, 20, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Mancini-DiNardo, D.; Steele, S.J.; Levorse, J.M.; Ingram, R.S.; Tilghman, S.M. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 2006, 20, 1268–1282. [Google Scholar] [CrossRef] [Green Version]
- Autuoro, J.M.; Pirnie, S.P.; Carmichael, G.G. Long Noncoding RNAs in Imprinting and X Chromosome Inactivation. Biomolecules 2014, 4, 76–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad, F.; Mondal, T.; Fau-Kanduri, C.; Kanduri, C. Epigenetics of imprinted long noncoding RNAs. Epigenetics 2009, 4, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelfand, B.; Mead, J.; Bruning, A.; Apostolopoulos, N.; Tadigotla, V.; Nagaraj, V.; Sengupta, A.M.; Vershon, A.K. Regulated Antisense Transcription Controls Expression of Cell-Type-Specific Genes in Yeast. Mol. Cell. Biol. 2011, 31, 1701–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hongay, C.F.; Grisafi, P.L.; Galitski, T.; Fink, G.R. Antisense Transcription Controls Cell Fate in Saccharomyces cerevisiae. Cell 2006, 127, 735–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modarresi, F.; Faghihi, M.A.; Patel, N.S.; Sahagan, B.G.; Wahlestedt, C.; Lopez-Toledano, M.A. Knockdown of BACE1-AS Nonprotein-Coding Transcript Modulates Beta-Amyloid-Related Hippocampal Neurogenesis. Int. J. Alzheimer’s Dis. 2011, 2011, 929042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassar, R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 89. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Li, J.; Ma, L.; Wen, L.; Wang, Q.; Yao, H.; Ruan, C.; Wu, D.; Zhang, X.; Chen, S. Overexpression of ZEB2-AS1 lncRNA is associated with poor clinical outcomes in acute myeloid leukemia. Oncol. Lett. 2019, 17, 4935–4947. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yan, T.; Wang, Z.; Wu, X.; Cao, G.; Zhang, C. LncRNA ZEB2-AS1 promotes bladder cancer cell proliferation and inhibits apoptosis by regulating miR-27b. Biomed. Pharmacother. 2017, 96, 299–304. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, X.; Chen, S.; Zhang, S. Natural antisense transcripts in the biological hallmarks of cancer: Powerful regulators hidden in the dark. J. Exp. Clin. Cancer Res. 2020, 39, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zeitz, M.J.; Wang, H.; Niu, B.; Ge, S.; Li, W.; Cui, J.; Wang, G.; Qian, G.; Higgins, M.J.; et al. Long noncoding RNA-mediated intrachromosomal interactions promote imprinting at the Kcnq1 locus. J. Cell Biol. 2014, 204, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, W.A.; Mann, M.R.W. Long noncoding RNA functionality in imprinted domain regulation. PLoS Genet. 2020, 16, e1008930. [Google Scholar] [CrossRef] [PubMed]
- Carter, K.L.; Ward, P.L.; Roizman, B. Characterization of the products of the U(L)43 gene of herpes simplex virus 1: Potential implications for regulation of gene expression by antisense transcription. J. Virol. 1996, 70, 7663–7668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bego, M.; Maciejewski, J.; Khaiboullina, S.; Pari, G.; Jeor, S.S. Characterization of an Antisense Transcript Spanning the UL81-82 Locus of Human Cytomegalovirus. J. Virol. 2005, 79, 11022–11034. [Google Scholar] [CrossRef] [Green Version]
- Cantello, J.L.; Anderson, A.S.; Morgan, R.W. Identification of latency-associated transcripts that map antisense to the ICP4 homolog gene of Marek’s disease virus. J. Virol. 1994, 68, 6280–6290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.E.; Menotti, L.; Filatov, F.; Campadelli-Fiume, G.; Roizman, B. UL27.5 Is a Novel γ2 Gene Antisense to the Herpes Simplex Virus 1 Gene Encoding Glycoprotein B. J. Virol. 1998, 72, 6056–6064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagunoff, M.; Roizman, B. Expression of a herpes simplex virus 1 open reading frame antisense to the gamma(1)34.5 gene and transcribed by an RNA 3’ coterminal with the unspliced latency-associated transcript. J. Virol. 1994, 68, 6021–6028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prang, N.; Wolf, H.; Schwarzmann, F. Epstein-Barr virus lytic replication is controlled by posttranscriptional negative regulation of BZLF1. J. Virol. 1995, 69, 2644–2648. [Google Scholar] [CrossRef] [Green Version]
- Prang, N.; Wolf, H.; Schwarzmann, F. Latency of Epstein-Barr virus is stabilized by antisense-mediated control of the viral immediate-early gene BZLF-1. J. Med. Virol. 1999, 59, 512–519. [Google Scholar] [CrossRef]
- Wirth, U.V.; Fraefel, C.; Vogt, B.; Vlcek, C.; Paces, V.; Schwyzer, M. Immediate-early RNA 2.9 and early RNA 2.6 of bovine herpesvirus 1 are 3’ coterminal and encode a putative zinc finger transactivator protein. J. Virol. 1992, 66, 2763–2772. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Kaplan, S.L.; Wakenell, P.; Schat, K.A. Transactivation of Latent Marek’s Disease Herpesvirus Genes in QT35, a Quail Fibroblast Cell Line, by Herpesvirus of Turkeys. J. Virol. 2000, 74, 10176–10186. [Google Scholar] [CrossRef] [Green Version]
- Priola, S.A.; Stevens, J.G. The 5′ and 3′ limits of transcription in the pseudorabies virus latency associated transcription unit. Virology 1991, 182, 852–856. [Google Scholar] [CrossRef]
- Bratanich, A.C.; Hanson, N.D.; Jones, C.J. The latency-related gene of bovine herpesvirus 1 inhibits the activity of immediate-early transcription unit 1. Virology 1992, 191, 988–991. [Google Scholar] [CrossRef]
- Stevens, J.G.; Wagner, E.K.; Devi-Rao, G.B.; Cook, M.L.; Feldman, L.T. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 1987, 235, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-Y.; Zhou, C.; Johnson, K.E.; Colgrove, R.C.; Coen, D.M.; Knipe, D.M. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 2005, 102, 16055–16059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordani, N.V.; Neumann, D.M.; Kwiatkowski, D.L.; Bhattacharjee, P.S.; McAnany, P.K.; Hill, J.M.; Bloom, D.C. During Herpes Simplex Virus Type 1 Infection of Rabbits, the Ability to Express the Latency-Associated Transcript Increases Latent-Phase Transcription of Lytic Genes. J. Virol. 2008, 82, 6056–6060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cliffe, A.R.; Garber, D.A.; Knipe, D.M. Transcription of the Herpes Simplex Virus Latency-Associated Transcript Promotes the Formation of Facultative Heterochromatin on Lytic Promoters. J. Virol. 2009, 83, 8182–8190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, D.A.; Schaffer, P.A.; Knipe, D.M. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J. Virol. 1997, 71, 5885–5893. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.H.; Kramer, M.F.; Schaffer, P.A.; Coen, D.M. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 1997, 71, 5878–5884. [Google Scholar] [CrossRef] [Green Version]
- Perng, G.-C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; et al. Virus-Induced Neuronal Apoptosis Blocked by the Herpes Simplex Virus Latency-Associated Transcript. Science 2000, 287, 1500–1503. [Google Scholar] [CrossRef] [Green Version]
- Henderson, G.; Peng, W.; Jin, L.; Perng, G.-C.; Nesburn, A.; Wechsler, S.; Jones, C. Regulation of Caspase 8- and Caspase 9-Induced Apoptosis by the Herpes Simplex Virus Type 1 Latency-Associated Transcript. J. Neuro Virol. 2002, 8 (Suppl. 2), 103–111. [Google Scholar] [CrossRef]
- Branco, F.J.; Fraser, N.W. Herpes Simplex Virus Type 1 Latency-Associated Transcript Expression Protects Trigeminal Ganglion Neurons from Apoptosis. J. Virol. 2005, 79, 9019–9025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Chentoufi, A.A.; Hsiang, C.; Carpenter, D.; Osorio, N.; Benmohamed, L.; Fraser, N.W.; Jones, C.; Wechsler, S.L. The Herpes Simplex Virus Type 1 Latency-Associated Transcript Can Protect Neuron-Derived C1300 and Neuro2A Cells from Granzyme B-Induced Apoptosis and CD8 T-Cell Killing. J. Virol. 2010, 85, 2325–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, D.; Hsiang, C.; Jiang, X.; Osorio, N.; Benmohamed, L.; Jones, C.; Wechsler, S.L. The herpes simplex virus type 1 (HSV-1) latency-associated transcript (LAT) protects cells against cold-shock-induced apoptosis by maintaining phosphorylation of protein kinase B (AKT). J. Neuro Virol. 2015, 21, 568–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depledge, D.P.; Ouwendijk, W.J.D.; Sadaoka, T.; Braspenning, S.E.; Mori, Y.; Cohrs, R.J.; Verjans, G.M.G.M.; Breuer, J. A spliced latency-associated VZV transcript maps antisense to the viral transactivator gene 61. Nat. Commun. 2018, 9, 1167. [Google Scholar] [CrossRef]
- Kulkarni, A.; Bangham, C.R.M. HTLV-1: Regulating the Balance Between Proviral Latency and Reactivation. Front. Microbiol. 2018, 9, 449. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, D.; Chao, L.A.; Seto, M.H.; Brunck, T.K. Human T-cell Leukemia Virus minus strand transcription in infected T-cells. Biochem. Biophys. Res. Commun. 1989, 163, 1006–1013. [Google Scholar] [CrossRef]
- Cavanagh, M.-H.; Landry, S.; Audet, B.; Arpin-André, C.; Hivin, P.; Paré, M.-E.; Thête, J.; Wattel, E.; Marriott, S.J.; Mesnard, J.-M.; et al. HTLV-I antisense transcripts initiating in the 3’LTR are alternatively spliced and polyadenylated. Retrovirology 2006, 3, 15. [Google Scholar] [CrossRef] [Green Version]
- Gazon, H.; Lemasson, I.; Polakowski, N.; Césaire, R.; Matsuoka, M.; Barbeau, B.; Mesnard, J.-M.; Peloponese, J.-M. Human T-Cell Leukemia Virus Type 1 (HTLV-1) bZIP Factor Requires Cellular Transcription Factor JunD To Upregulate HTLV-1 Antisense Transcription from the 3’ Long Terminal Repeat. J. Virol. 2012, 86, 9070–9078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arpin-André, C.; Laverdure, S.; Barbeau, B.; Gross, A.; Mesnard, J.-M. Construction of a reporter vector for analysis of bidirectional transcriptional activity of retrovirus LTR. Plasmid 2014, 74, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Yasunaga, J.-I.; Akari, H.; Matsuoka, M. TCF1 and LEF1 act as T-cell intrinsic HTLV-1 antagonists by targeting Tax. Proc. Natl. Acad. Sci. USA 2015, 112, 2216–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, M.F.; Trainor, C.D.; Mann, D.L.; Gallo, R.C.; Reitz, M.S. Methylation of human T-cell leukemia virus proviral DNA and viral RNA expression in short- and long-term cultures of infected cells. Virology 1984, 135, 97–104. [Google Scholar] [CrossRef]
- Saggioro, D.; Panozzo, M.; Chieco-Bianchi, L. Human T-lymphotropic virus type I transcriptional regulation by methylation. Cancer Res. 1990, 50, 4968–4973. [Google Scholar] [PubMed]
- Kitamura, T.; Takano, M.; Hoshino, H.; Shimotohno, K.; Shimoyama, M.; Miwa, M.; Takaku, F.; Sugimura, T. Methylation pattern of human T-cell leukemia virusin vivo andin vitro: pX and LTR regions are hypomethylatedin vivo. Int. J. Cancer 1985, 35, 629–635. [Google Scholar] [CrossRef]
- Koiwa, T.; Hamano-Usami, A.; Ishida, T.; Okayama, A.; Yamaguchi, K.; Kamihira, S.; Watanabe, T. 5′-Long Terminal Repeat-Selective CpG Methylation of Latent Human T-Cell Leukemia Virus Type 1 Provirus In Vitro and In Vivo. J. Virol. 2002, 76, 9389–9397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, M.; Shimizu, A.; Ikuta, K.; Okamoto, H.; Kashihara, M.; Uchiyama, T.; Honjo, T.; Yodoi, J. Origin of human T-lymphotrophic virus I-positive T cell lines in adult T cell leukemia. Analysis of T cell receptor gene rearrangement. J. Exp. Med. 1985, 162, 2169–2174. [Google Scholar] [CrossRef] [PubMed]
- Satou, Y.; Miyazato, P.; Ishihara, K.; Yaguchi, H.; Melamed, A.; Miura, M.; Fukuda, A.; Nosaka, K.; Watanabe, T.; Rowan, A.G.; et al. The retrovirus HTLV-1 inserts an ectopic CTCF-binding site into the human genome. Proc. Natl. Acad. Sci. USA 2016, 113, 3054–3059. [Google Scholar] [CrossRef] [Green Version]
- Murata, K.; Hayashibara, T.; Sugahara, K.; Uemura, A.; Yamaguchi, T.; Harasawa, H.; Hasegawa, H.; Tsuruda, K.; Okazaki, T.; Koji, T.; et al. A Novel Alternative Splicing Isoform of Human T-Cell Leukemia Virus Type 1 bZIP Factor (HBZ-SI) Targets Distinct Subnuclear Localization. J. Virol. 2006, 80, 2495–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usui, T.; Yanagihara, K.; Tsukasaki, K.; Murata, K.; Hasegawa, H.; Yamada, Y.; Kamihira, S. Characteristic expression of HTLV-1 basic zipper factor (HBZ) transcripts in HTLV-1 provirus-positive cells. Retrovirology 2008, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Satou, Y.; Yasunaga, J.-I.; Fujisawa, J.-I.; Matsuoka, M. Transcriptional Control of Spliced and Unspliced Human T-Cell Leukemia Virus Type 1 bZIP Factor (HBZ) Gene. J. Virol. 2008, 82, 9359–9368. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Yasunaga, J.-I.; Matsuoka, M. Multifaceted functions and roles of HBZ in HTLV-1 pathogenesis. Retrovirology 2016, 13, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, M.; Yasunaga, J.-I.; Taniguchi, Y.; Tamiya, S.; Nakahata, T.; Matsuoka, M. Preferential Selection of Human T-Cell Leukemia Virus Type 1 Provirus Lacking the 5′ Long Terminal Repeat during Oncogenesis. J. Virol. 2007, 81, 5714–5723. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, Y.; Nosaka, K.; Yasunaga, J.-I.; Maeda, M.; Mueller, N.; Okayama, A.; Matsuoka, M. Silencing of human T-cell leukemia virus type I gene transcription by epigenetic mechanisms. Retrovirology 2005, 2, 64. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Ma, G.; Nosaka, K.; Tanabe, J.; Satou, Y.; Koito, A.; Wain-Hobson, S.; Vartanian, J.-P.; Matsuoka, M. APOBEC3G Generates Nonsense Mutations in Human T-Cell Leukemia Virus Type 1 Proviral Genomes In Vivo. J. Virol. 2010, 84, 7278–7287. [Google Scholar] [CrossRef] [Green Version]
- Satou, Y.; Yasunaga, J.-I.; Yoshida, M.; Matsuoka, M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc. Natl. Acad. Sci. USA 2006, 103, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitobe, Y.; Yasunaga, J.-I.; Furuta, R.; Matsuoka, M. HTLV-1 bZIP Factor RNA and Protein Impart Distinct Functions on T-cell Proliferation and Survival. Cancer Res. 2015, 75, 4143–4152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clerc, I.; Polakowski, N.; André-Arpin, C.; Cook, P.; Barbeau, B.; Mesnard, J.-M.; Lemasson, I. An Interaction between the Human T Cell Leukemia Virus Type 1 Basic Leucine Zipper Factor (HBZ) and the KIX Domain of p300/CBP Contributes to the Down-regulation of Tax-dependent Viral Transcription by HBZ. J. Biol. Chem. 2008, 283, 23903–23913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schifano, J.M.; Corcoran, K.; Kelkar, H.; Dittmer, D.P. Expression of the Antisense-to-Latency Transcript Long Noncoding RNA in Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2016, 91, e01698-16. [Google Scholar] [CrossRef] [Green Version]
- Tormanen, K.; Allen, S.; Mott, K.R.; Ghiasi, H. The Latency-Associated Transcript Inhibits Apoptosis via Downregulation of Components of the Type I Interferon Pathway during Latent Herpes Simplex Virus 1 Ocular Infection. J. Virol. 2019, 93, 93. [Google Scholar] [CrossRef] [Green Version]
- Majerciak, V.; Yang, W.; Zheng, J.; Zhu, J.; Zheng, Z.-M. A Genome-Wide Epstein-Barr Virus Polyadenylation Map and Its Antisense RNA to EBNA. J. Virol. 2018, 93, 93. [Google Scholar] [CrossRef] [Green Version]
- Cantello, J.L.; Parcells, M.S.; Anderson, A.S.; Morgan, R.W. Marek’s disease virus latency-associated transcripts belong to a family of spliced RNAs that are antisense to the ICP4 homolog gene. J. Virol. 1997, 71, 1353–1361. [Google Scholar] [CrossRef] [Green Version]
- Chandriani, S.; Xu, Y.; Ganem, D. The Lytic Transcriptome of Kaposi’s Sarcoma-Associated Herpesvirus Reveals Extensive Transcription of Noncoding Regions, Including Regions Antisense to Important Genes. J. Virol. 2010, 84, 7934–7942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Ganem, D. Making Sense of Antisense: Seemingly Noncoding RNAs Antisense to the Master Regulator of Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication Do Not Regulate That Transcript but Serve as mRNAs Encoding Small Peptides. J. Virol. 2010, 84, 5465–5475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durkin, K.; Rosewick, N.; Artesi, M.M.; Hahaut, V.V.; Griebel, P.; Arsic, N.N.; Burny, A.; Georges, M.; Broeke, A.V.D. Characterization of novel Bovine Leukemia Virus (BLV) antisense transcripts by deep sequencing reveals constitutive expression in tumors and transcriptional interaction with viral microRNAs. Retrovirology 2016, 13, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, M.; Yasunaga, J.-I.; Tanabe, J.; Sugata, K.; Zhao, T.; Ma, G.; Miyazato, P.; Ohshima, K.; Kaneko, A.; Watanabe, A.; et al. Characterization of simian T-cell leukemia virus type 1 in naturally infected Japanese macaques as a model of HTLV-1 infection. Retrovirology 2013, 10, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, M.H.; Ballarín-González, B.; Liu, J.; Lassen, L.B.; FüchtbauerA., *!!! REPLACE !!!*; Fuchtbauer, E.-M.; Nielsen, A.L.; Pedersen, F.S. Antisense Transcription in Gammaretroviruses as a Mechanism of Insertional Activation of Host Genes. J. Virol. 2010, 84, 3780–3788. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zhao, X.; Shen, W.; Kong, X. Evidence for the antisense transcription in the proviral R29-127 strain of bovine immunodeficiency virus. Virol. Sin. 2015, 30, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Briquet, S.; Richardson, J.; Vanhée-Brossollet, C.; Vaquero, C. Natural antisense transcripts are detected in different cell lines and tissues of cats infected with feline immunodeficiency virus. Gene 2001, 267, 157–164. [Google Scholar] [CrossRef]
- Miller, R. Human immunodeficiency virus may encode a novel protein on the genomic DNA plus strand. Science 1988, 239, 1420–1422. [Google Scholar] [CrossRef]
- Casino, A.; Cipollaro, M.; Guerrini, A.; Mastrocinque, G.; Spena, A.; Scarlato, V. Coding capacity of complementary DNA strands. Nucleic Acids Res. 1981, 9, 1499–1518. [Google Scholar] [CrossRef] [Green Version]
- Bukrinsky, M.I.; Etkin, A.F. Plus Strand of the HIV Provirus DNA Is Expressed at Early Stages of Infection. AIDS Res. Hum. Retroviruses 1990, 6, 425–426. [Google Scholar] [CrossRef]
- Vanhée-Brossollet, C.; Thoreau, H.; Serpente, N.; D’Auriol, L.; Lévy, J.-P.; Vaquero, C. A natural antisense RNA derived from the HIV-1 env gene encodes a protein which is recognized by circulating antibodies of HIV+ individuals. Virology 1995, 206, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Michael, N.L.; Vahey, M.T.; D’Arcy, L.; Ehrenberg, P.K.; Mosca, J.D.; Rappaport, J.; Redfield, R.R. Negative-strand RNA transcripts are produced in human immunodeficiency virus type 1-infected cells and patients by a novel promoter downregulated by Tat. J. Virol. 1994, 68, 979–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landry, S.; Halin, M.; Lefort, S.; Audet, B.; Vaquero, C.; Mesnard, J.-M.; Barbeau, B. Detection, characterization and regulation of antisense transcripts in HIV-1. Retrovirology 2007, 4, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddad, F.; Qin, A.X.; Giger, J.M.; Guo, H.; Baldwin, K.M. Potential pitfalls in the accuracy of analysis of natural sense-antisense RNA pairs by reverse transcription-PCR. BMC Biotechnol. 2007, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi-Ishihara, M.; Yamagishi, M.; Hara, T.; Matsuda, Y.; Takahashi, R.; Miyake, A.; Nakano, K.; Yamochi, T.; Ishida, T.; Watanabe, T. HIV-1-encoded antisense RNA suppresses viral replication for a prolonged period. Retrovirology 2012, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Zapata, J.C.; Campilongo, F.; Barclay, R.A.; DeMarino, C.; Iglesias-Ussel, M.D.; Kashanchi, F.; Romerio, F. The Human Immunodeficiency Virus 1 ASP RNA promotes viral latency by recruiting the Polycomb Repressor Complex 2 and promoting nucleosome assembly. Virology 2017, 506, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, A.; Procopio, F.A.; Achsel, T.; De Crignis, E.; Foley, B.T.; Corradin, G.; Bagni, C.; Pantaleo, G.; Graziosi, C. Detection of antisense protein (ASP) RNA transcripts in individuals infected with human immunodeficiency virus type 1 (HIV-1). J. Gen. Virol. 2019, 100, 863–876. [Google Scholar] [CrossRef]
- Lefebvre, G.; Desfarges, S.; Uyttebroeck, F.; Muñoz, M.; Beerenwinkel, N.; Rougemont, J.; Telenti, A.; Ciuffi, A. Analysis of HIV-1 Expression Level and Sense of Transcription by High-Throughput Sequencing of the Infected Cell. J. Virol. 2011, 85, 6205–6211. [Google Scholar] [CrossRef] [Green Version]
- Champiat, S.; Raposo, R.A.S.; Maness, N.J.; Lehman, J.L.; Purtell, S.E.; Hasenkrug, A.M.; Miller, J.C.; Dean, H.; Koff, W.C.; Hong, M.A.; et al. Influence of HAART on Alternative Reading Frame Immune Responses over the Course of HIV-1 Infection. PLoS ONE 2012, 7, e39311. [Google Scholar] [CrossRef]
- Laverdure, S.; Gross, A.; Arpin-André, C.; Clerc, I.; Beaumelle, B.; Barbeau, B.; Mesnard, J.-M. HIV-1 Antisense Transcription Is Preferentially Activated in Primary Monocyte-Derived Cells. J. Virol. 2012, 86, 13785–13789. [Google Scholar] [CrossRef] [Green Version]
- Saayman, S.; Ackley, A.; Turner, A.-M.W.; Famiglietti, M.; Bosque, A.; Clemson, M.; Planelles, V.; Morris, K.V. An HIV-Encoded Antisense Long Noncoding RNA Epigenetically Regulates Viral Transcription. Mol. Ther. 2014, 22, 1164–1175. [Google Scholar] [CrossRef] [Green Version]
- Berger, C.T.; Llano, A.; Carlson, J.M.; Brumme, Z.L.; Brockman, M.A.; Cedeño, S.; Harrigan, P.R.; Kaufmann, D.E.; Heckerman, D.; Meyerhans, A.; et al. Immune Screening Identifies Novel T Cell Targets Encoded by Antisense Reading Frames of HIV-1. J. Virol. 2015, 89, 4015–4019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bet, A.; Maze, E.A.; Bansal, A.; Sterrett, S.; Gross, A.; Graff-Dubois, S.; Samri, A.; Guihot, A.; Katlama, C.; Theodorou, I.; et al. The HIV-1 Antisense Protein (ASP) induces CD8 T cell responses during chronic infection. Retrovirology 2015, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi-Ishihara, M.; Terahara, K.; Martinez, J.P.; Yamagishi, M.; Iwabuchi, R.; Brander, C.; Ato, M.; Watanabe, T.; Meyerhans, A.; Tsunetsugu-Yokota, Y. HIV LTR-Driven Antisense RNA by Itself Has Regulatory Function and May Curtail Virus Reactivation from Latency. Front. Microbiol. 2018, 9, 1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoret, J.; Chazal, N.; Moles, J.-P.; Tuaillon, E.; Boufassa, F.; Meyer, L.; Lecuroux, C.; Lambotte, O.; Van De Perre, P.; Mesnard, J.-M.; et al. A Pilot Study of the Humoral Response Against the AntiSense Protein (ASP) in HIV-1-Infected Patients. Front. Microbiol. 2020, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Bentley, K.; Deacon, N.; Sonza, S.; Zeichner, S.; Churchill, M. Mutational analysis of the HIV-1 LTR as a promoter of negative sense transcription. Arch. Virol. 2004, 149, 2277–2294. [Google Scholar] [CrossRef]
- Ludwig, L.B.; Ambrus, J.L., Jr.; Krawczyk, K.A.; Sharma, S.; Brooks, S.; Hsiao, C.-B.; Schwartz, S.A. Human Immunodeficiency Virus-Type 1 LTR DNA contains an intrinsic gene producing antisense RNA and protein products. Retrovirology 2006, 3, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, A.; Lambert, P.F.; Deacon, N.J. A fourth Sp1 site in the human immunodeficiency virus type 1 long terminal repeat is essential for negative-sense transcription. J. Virol. 1996, 70, 6665–6672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kugel, J.F.; Goodrich, J.A. Finding the start site: Redefining the human initiator element. Genes Dev. 2017, 31, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briquet, S.; Vaquero, C. Immunolocalization Studies of an Antisense Protein in HIV-1-Infected Cells and Viral Particles. Virology 2002, 292, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Bansal, A.; Carlson, J.; Yan, J.; Akinsiku, O.T.; Schaefer, M.; Sabbaj, S.; Bet, A.; Levy, D.N.; Heath, S.; Tang, J.; et al. CD8 T cell response and evolutionary pressure to HIV-1 cryptic epitopes derived from antisense transcription. J. Exp. Med. 2010, 207, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Clerc, I.; Laverdure, S.; Torresilla, C.; Landry, S.; Borel, S.; Vargas, A.; Arpin-André, C.; Gay, B.; Briant, L.; Gross, A.; et al. Polarized expression of the membrane ASP protein derived from HIV-1 antisense transcription in T cells. Retrovirology 2011, 8, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torresilla, C.; Larocque, É.; Landry, S.; Halin, M.; Coulombe, Y.; Masson, J.-Y.; Mesnard, J.-M.; Barbeau, B. Detection of the HIV-1 Minus-Strand-Encoded Antisense Protein and Its Association with Autophagy. J. Virol. 2013, 87, 5089–5105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassan, E.; Arigon-Chifolleau, A.-M.; Mesnard, J.-M.; Gross, A.; Gascuel, O. Concomitant emergence of the antisense protein gene of HIV-1 and of the pandemic. Proc. Natl. Acad. Sci. USA 2016, 113, 11537–11542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Torresilla, C.; Xiao, Y.; Nguyen, P.T.; Caté, C.; Barbosa, K.; Rassart, É.; Cen, S.; Bourgault, S. HIV-1 Antisense Protein of Different Clades Induces Autophagy and Associates with the Autophagy Factor p62. J. Virol. 2018, 93, e01757-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Affram, Y.; Zapata, J.C.; Gholizadeh, Z.; Tolbert, W.D.; Zhou, W.; Iglesias-Ussel, M.D.; Pazgier, M.; Ray, K.; Latinovic, O.S.; Romerio, F. The HIV-1 Antisense Protein ASP Is a Transmembrane Protein of the Cell Surface and an Integral Protein of the Viral Envelope. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.W.; Ardern, Z.; Wei, X. OLGenie: Estimating Natural Selection to Predict Functional Overlapping Genes. Mol. Biol. Evol. 2020, 37, 2440–2449. [Google Scholar] [CrossRef]
- Rancurel, C.; Khosravi, M.; Dunker, A.K.; Romero, P.R.; Karlin, D. Overlapping Genes Produce Proteins with Unusual Sequence Properties and Offer Insight into De Novo Protein Creation. J. Virol. 2009, 83, 10719–10736. [Google Scholar] [CrossRef] [Green Version]
- Sabath, N.; Wagner, A.; Karlin, D. Evolution of Viral Proteins Originated De Novo by Overprinting. Mol. Biol. Evol. 2012, 29, 3767–3780. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Ding, S.-W. Virus Counterdefense: Diverse Strategies for Evading the RNA-Silencing Immunity. Annu. Rev. Microbiol. 2006, 60, 503–531. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, A.; James, W. Inhibition of human immunodeficiency virus replication in cell culture by endogenously synthesized antisense RNA. J. Gen. Virol. 1990, 71 Pt 9, 1965–1974. [Google Scholar] [CrossRef]
- Rhodes, A.; James, W. Inhibition of heterologous strains of HIV by antisense RNA. AIDS 1991, 5, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Tagieva, N.E.; Vaquero, C. Expression of naturally occurring antisense RNA inhibits human immunodeficiency virus type 1 heterologous strain replication. J. Gen. Virol. 1997, 78 Pt 10, 2503–2511. [Google Scholar] [CrossRef] [Green Version]
- Berkhout, B.; Van Wamel, J.L. Inhibition of human immunodeficiency virus expression by sense transcripts encoding the retroviral leader RNA. Antivir. Res. 1995, 26, 101–115. [Google Scholar] [CrossRef]
- Lu, X.; Yu, Q.; Binder, G.K.; Chen, Z.; Slepushkina, T.; Rossi, J.; Dropulic, B. Antisense-Mediated Inhibition of Human Immunodeficiency Virus (HIV) Replication by Use of an HIV Type 1-Based Vector Results in Severely Attenuated Mutants Incapable of Developing Resistance. J. Virol. 2004, 78, 7079–7088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.; Cho, W.-K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic Silencing of HIV-1 by the Histone H3 Lysine 27 Methyltransferase Enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef] [Green Version]
- Klaver, B.; Berkhout, B. Comparison of 5’ and 3’ long terminal repeat promoter function in human immunodeficiency virus. J. Virol. 1994, 68, 3830–3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlestedt, C. Natural antisense and noncoding RNA transcripts as potential drug targets. Drug Discov. Today 2006, 11, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Barlow, D.P.; Bartolomei, M.S. Genomic Imprinting in Mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a018382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galupa, R.; Heard, E. X-Chromosome Inactivation: A Crossroads Between Chromosome Architecture and Gene Regulation. Annu. Rev. Genet. 2018, 52, 535–566. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.A.; Utrankar, A.; Reyes, J.A.; Simons, D.D.; Kosten, T.R. Epigenetics of drug abuse: Predisposition or response. Pharmacogenomics 2012, 13, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Sadri-Vakili, G. Cocaine triggers epigenetic alterations in the corticostriatal circuit. Brain Res. 2015, 1628, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Sani, E.; Herzyk, P.; Perrella, G.; Colot, V.; Amtmann, A. Hyperosmotic priming of Arabidopsis seedlings establishes a long-term somatic memory accompanied by specific changes of the epigenome. Genome Biol. 2013, 14, R59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, M.N.D.; Auler, P.A.; Rossatto, T.; Barros, P.M.; Oliveira, M.M.; Braga, E.J.B. Long-term somatic memory of salinity unveiled from physiological, biochemical and epigenetic responses in two contrasting rice genotypes. Physiol. Plant 2020, 170, 248–268. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Mechanism | NAT | Organism | Function/Effects | References |
|---|---|---|---|---|
| Epigenetic Silencing | Tsix | Mammals | Recruits DNA methyltransferases to induce Xist promoter methylation, which inhibits its expression and prevents gene inactivation | [79,80,81,82,83] |
| ANRIL | H. sapiens | Recruits PRC1 and PRC2 to induce histone methylation (H3K27) and mono-ubiquitination (H2A-K119) for silencing and repression of the INK4 locus | [84,85,86,87] | |
| Kcnq1ot1 | H. sapiens | Silences genes within Kcnq1 loci on the paternal allele by recruiting chromatin modifiers, which induce repressive histone modification and DNA methylation | [88,89,90,91,92] | |
| Transcriptional Interference | RME2 | S. cerevisiae | Blocks transcriptional elongation of IME4 transcript | [93,94] |
| SUT719 | S. cerevisiae | Acts as a regulatory hub linking the expression of divergent neighboring genes GAL80 and SUR7 and establishes a threshold-dependent on–off switch | [58,76] | |
| RNA Stability | BACE1-AS | H. sapiens | Masks the miR485-5p binding site and prevents miRNA-mediated degradation of BACE1 mRNA | [69,95,96] |
| RNA Masking | ZEB2-AS | H. sapiens | Prevents splicing of an IRES-containing intron, resulting in transcription of an alternate isoform of ZEB2 | [78,97,98,99] |
| Mechanism | NAT | Virus | Function/Effects | References |
|---|---|---|---|---|
| Epigenetic Silencing | LATs | Herpesvirus (HSV) | Regulates viral lytic gene expression by limiting transcripts and silencing their promoters via heterochromatinization during latency May promote latency reactivation by inhibiting apoptosis and promoting cell survival | [102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123] |
| VLT | Varicella zoster virus (VZV) | Suppresses the expression of ORF61 to regulate latency, similar to LATs in HSV | [124] | |
| Transcriptional Interference | Hbz | Human T cell leukemia virus 1 (HTLV-1) | Induces host genes involved in cell cycle progression and proliferation and anti-apoptosis factors, such as survivin May play a role in leukemogenesis with HBZ protein | [77,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146] |
| Unknown | ALT | Kaposi’s sarcoma-associated herpesvirus (KSHV) | May play a role in regulating the viral lifecycle | [147] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Sklutuis, R.; Groebner, J.L.; Romerio, F. HIV-1 Natural Antisense Transcription and Its Role in Viral Persistence. Viruses 2021, 13, 795. https://doi.org/10.3390/v13050795
Li R, Sklutuis R, Groebner JL, Romerio F. HIV-1 Natural Antisense Transcription and Its Role in Viral Persistence. Viruses. 2021; 13(5):795. https://doi.org/10.3390/v13050795
Chicago/Turabian StyleLi, Rui, Rachel Sklutuis, Jennifer L. Groebner, and Fabio Romerio. 2021. "HIV-1 Natural Antisense Transcription and Its Role in Viral Persistence" Viruses 13, no. 5: 795. https://doi.org/10.3390/v13050795