
Herpes Simplex Virus Type 2 Mucin-Like Glycoprotein mgG Promotes Virus Release from the Surface of Infected Cells


 , and
, and {kind=link}
{kind=link}
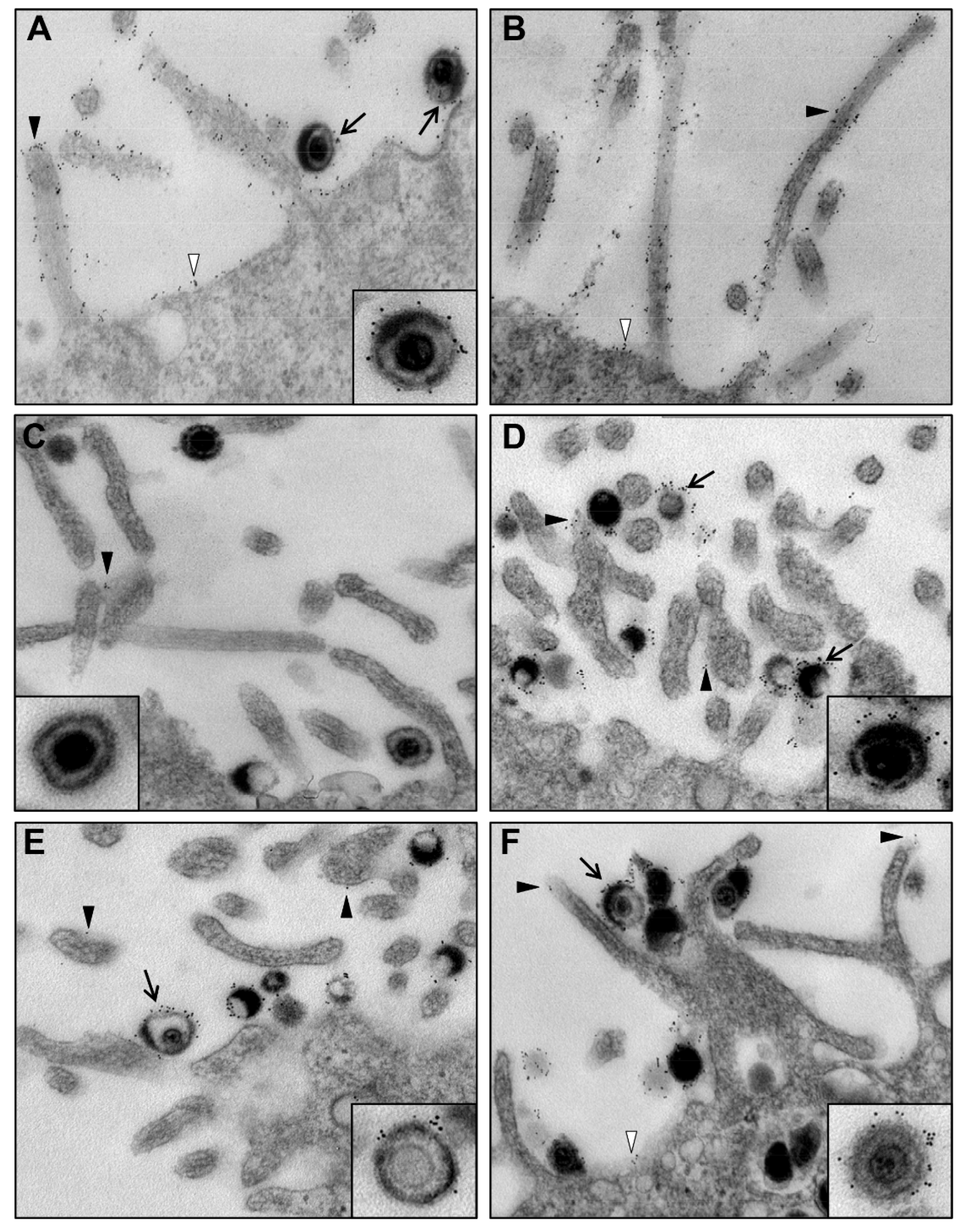{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cells and Viruses
2.3. Purification of HSV-2 Virions, Glycoprotein gG, and Quantification of Viral DNA
2.4. Virus Replication Kinetics and Forcible Virus Liberation Assays
2.5. Total Internal Reflection Fluorescence Microscopy
2.6. Binding of Virus to Infected Cells
2.7. Immunogold Electron Microscopy
2.8. Assay to Detect a GAG Lyase Activity
2.9. Preparation of Cellular Extracts, Electrophoresis, and Immunoblot Assay
2.10. Statistical Analysis
3. Results
3.1. Biological Activities of the mgG Deficient Mutant of HSV-2
3.2. Release of the mgG-def Virions from the Surface of Infected Cells
3.3. Interaction of mgG-def Virions with Surface-Bound CS Chains
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hirst, G.K. Adsorption of Influenza Hemagglutinins and Virus by Red Blood Cells. J. Exp. Med. 1942, 76, 195–209. [Google Scholar] [CrossRef]
- Gottschalk, A. Neuraminidase: The specific enzyme of influenza virus and Vibrio cholerae. Biochim. Biophys. Acta. 1957, 23, 645–646. [Google Scholar] [CrossRef]
- Palese, P.; Compans, R.W. Inhibition of influenza virus replication in tissue culture by 2-deoxy-2,3-dehydro-N-trifluoroacetylneuraminic acid (FANA): Mechanism of action. J. Gen. Virol. 1976, 33, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Palese, P.; Tobita, K.; Ueda, M.; Compans, R.W. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 1974, 61, 397–410. [Google Scholar] [CrossRef]
- von Itzstein, M.; Wu, W.Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Van Phan, T.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef] [PubMed]
- WuDunn, D.; Spear, P.G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989, 63, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Feyzi, E.; Trybala, E.; Bergstrom, T.; Lindahl, U.; Spillmann, D. Structural requirement of heparan sulfate for interaction with herpes simplex virus type 1 virions and isolated glycoprotein C. J. Biol. Chem. 1997, 272, 24850–24857. [Google Scholar] [CrossRef]
- Bergefall, K.; Trybala, E.; Johansson, M.; Uyama, T.; Naito, S.; Yamada, S.; Kitagawa, H.; Sugahara, K.; Bergstrom, T. Chondroitin sulfate characterized by the E-disaccharide unit is a potent inhibitor of herpes simplex virus infectivity and provides the virus binding sites on gro2C cells. J. Biol. Chem. 2005, 280, 32193–32199. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Spear, P.G. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J. Clin. Investig. 2001, 108, 503–510. [Google Scholar] [CrossRef]
- Blondeau, C.; Pelchen-Matthews, A.; Mlcochova, P.; Marsh, M.; Milne, R.S.; Towers, G.J. Tetherin restricts herpes simplex virus 1 and is antagonized by glycoprotein M. J. Virol. 2013, 87, 13124–13133. [Google Scholar] [CrossRef]
- Zenner, H.L.; Mauricio, R.; Banting, G.; Crump, C.M. Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J. Virol. 2013, 87, 13115–13123. [Google Scholar] [CrossRef]
- Bacsa, S.; Karasneh, G.; Dosa, S.; Liu, J.; Valyi-Nagy, T.; Shukla, D. Syndecan-1 and syndecan-2 play key roles in herpes simplex virus type-1 infection. J. Gen. Virol. 2011, 92, 733–743. [Google Scholar] [CrossRef] [PubMed]
- de Lima, B.D.; May, J.S.; Stevenson, P.G. Murine gammaherpesvirus 68 lacking gp150 shows defective virion release but establishes normal latency in vivo. J. Virol. 2004, 78, 5103–5112. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hadigal, S.R.; Agelidis, A.M.; Karasneh, G.A.; Antoine, T.E.; Yakoub, A.M.; Ramani, V.C.; Djalilian, A.R.; Sanderson, R.D.; Shukla, D. Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nat. Commun. 2015, 6, 6985. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, M.; Adamiak, B.; Bergefall, K.; Nenonen, H.; Roth, A.; Bergstrom, T.; Ferro, V.; Trybala, E. Molecular basis for resistance of herpes simplex virus type 1 mutants to the sulfated oligosaccharide inhibitor PI-88. Virology 2007, 367, 244–252. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Adamiak, B.; Ekblad, M.; Bergstrom, T.; Ferro, V.; Trybala, E. Herpes simplex virus type 2 glycoprotein G is targeted by the sulfated oligo- and polysaccharide inhibitors of virus attachment to cells. J. Virol. 2007, 81, 13424–13434. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, N.; Hutt-Fletcher, L.M. Synthesis and processing of glycoprotein gG of herpes simplex virus type 2. J. Virol. 1985, 54, 825–832. [Google Scholar] [CrossRef]
- Marsden, H.S.; Buckmaster, A.; Palfreyman, J.W.; Hope, R.G.; Minson, A.C. Characterization of the 92,000-dalton glycoprotein induced by herpes simplex virus type 2. J. Virol. 1984, 50, 547–554. [Google Scholar] [CrossRef]
- Su, H.K.; Eberle, R.; Courtney, R.J. Processing of the herpes simplex virus type 2 glycoprotein gG-2 results in secretion of a 34,000-Mr cleavage product. J. Virol. 1987, 61, 1735–1737. [Google Scholar] [CrossRef]
- Su, H.K.; Fetherston, J.D.; Smith, M.E.; Courtney, R.J. Orientation of the cleavage site of the herpes simplex virus glycoprotein G-2. J. Virol. 1993, 67, 2954–2959. [Google Scholar] [CrossRef]
- Viejo-Borbolla, A.; Martinez-Martin, N.; Nel, H.J.; Rueda, P.; Martin, R.; Blanco, S.; Arenzana-Seisdedos, F.; Thelen, M.; Fallon, P.G.; Alcami, A. Enhancement of chemokine function as an immunomodulatory strategy employed by human herpesviruses. PLoS Pathog. 2012, 8, e1002497. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martin, N.; Viejo-Borbolla, A.; Martin, R.; Blanco, S.; Benovic, J.L.; Thelen, M.; Alcami, A. Herpes simplex virus enhances chemokine function through modulation of receptor trafficking and oligomerization. Nat. Commun. 2015, 6, 6163. [Google Scholar] [CrossRef]
- Liljeqvist, J.A.; Trybala, E.; Hoebeke, J.; Svennerholm, B.; Bergstrom, T. Monoclonal antibodies and human sera directed to the secreted glycoprotein G of herpes simplex virus type 2 recognize type-specific antigenic determinants. J. Gen. Virol. 2002, 83, 157–165. [Google Scholar] [CrossRef]
- Liljeqvist, J.A.; Trybala, E.; Svennerholm, B.; Jeansson, S.; Sjogren-Jansson, E.; Bergstrom, T. Localization of type-specific epitopes of herpes simplex virus type 2 glycoprotein G recognized by human and mouse antibodies. J. Gen. Virol. 1998, 79 Pt 5, 1215–1224. [Google Scholar] [CrossRef]
- Bergstrom, T.; Sjogren-Jansson, E.; Jeansson, S.; Lycke, E. Mapping neuroinvasiveness of the herpes simplex virus type 1 encephalitis-inducing strain 2762 by the use of monoclonal antibodies. Mol. Cell Probes 1992, 6, 41–49. [Google Scholar] [CrossRef]
- Sjogren-Jansson, E.; Jeansson, S. Large-scale production of monoclonal antibodies in dialysis tubing. J. Immunol. Methods 1985, 84, 359–364. [Google Scholar] [CrossRef]
- Finnen, R.L.; Roy, B.B.; Zhang, H.; Banfield, B.W. Analysis of filamentous process induction and nuclear localization properties of the HSV-2 serine/threonine kinase Us3. Virology 2010, 397, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Gunalp, A. Growth and Cytopathic Effect of Rubella Virus in a Line of Green Monkey Kidney Cells. Proc. Soc. Exp. Biol. Med. 1964, 118. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell. Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Banfield, B.W.; Leduc, Y.; Esford, L.; Schubert, K.; Tufaro, F. Sequential isolation of proteoglycan synthesis mutants by using herpes simplex virus as a selective agent: Evidence for a proteoglycan-independent virus entry pathway. J. Virol. 1995, 69, 3290–3298. [Google Scholar] [CrossRef]
- Duff, R.; Rapp, F. Oncogenic transformation of hamster cells after exposure to herpes simplex virus type 2. Nat. New Biol. 1971, 233, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Trybala, E.; Liljeqvist, J.A.; Svennerholm, B.; Bergstrom, T. Herpes simplex virus types 1 and 2 differ in their interaction with heparan sulfate. J. Virol. 2000, 74, 9106–9114. [Google Scholar] [CrossRef] [PubMed]
- Gorander, S.; Harandi, A.M.; Lindqvist, M.; Bergstrom, T.; Liljeqvist, J.A. Glycoprotein G of herpes simplex virus 2 as a novel vaccine antigen for immunity to genital and neurological disease. J. Virol. 2012, 86, 7544–7553. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, K.; Ekblad, M.; Bergstrom, T.; Freeman, C.; Parish, C.R.; Ferro, V.; Trybala, E. The low molecular weight heparan sulfate-mimetic, PI-88, inhibits cell-to-cell spread of herpes simplex virus. Antiviral Res. 2004, 63, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, M.; Bergstrom, T.; Banwell, M.G.; Bonnet, M.; Renner, J.; Ferro, V.; Trybala, E. Anti-herpes simplex virus activities of two novel disulphated cyclitols. Antivir. Chem. Chemother. 2006, 17, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Bally, M.; Graule, M.; Parra, F.; Larson, G.; Hook, F. A virus biosensor with single virus-particle sensitivity based on fluorescent vesicle labels and equilibrium fluctuation analysis. Biointerphases 2013, 8, 4. [Google Scholar] [CrossRef]
- Gunnarsson, A.; Jonsson, P.; Marie, R.; Tegenfeldt, J.O.; Hook, F. Single-molecule detection and mismatch discrimination of unlabeled DNA targets. Nano Lett. 2008, 8, 183–188. [Google Scholar] [CrossRef]
- Peerboom, N.; Block, S.; Altgarde, N.; Wahlsten, O.; Moller, S.; Schnabelrauch, M.; Trybala, E.; Bergstrom, T.; Bally, M. Binding Kinetics and Lateral Mobility of HSV-1 on End-Grafted Sulfated Glycosaminoglycans. Biophys. J. 2017, 113, 1223–1234. [Google Scholar] [CrossRef]
- Trybala, E.; Bergstrom, T.; Svennerholm, B.; Jeansson, S.; Glorioso, J.C.; Olofsson, S. Localization of a functional site on herpes simplex virus type 1 glycoprotein C involved in binding to cell surface heparan sulphate. J. Gen. Virol. 1994, 75 Pt 4, 743–752. [Google Scholar] [CrossRef]
- Widehn, S.; Kindblom, L.G. Agarose embedding: A new method for the ultrastructural examination of the in-situ morphology of cell cultures. Ultrastruct. Pathol. 1990, 14, 81–85. [Google Scholar] [CrossRef]
- Lyon, M.; Deakin, J.A.; Mizuno, K.; Nakamura, T.; Gallagher, J.T. Interaction of hepatocyte growth factor with heparan sulfate. Elucidation of the major heparan sulfate structural determinants. J. Biol. Chem. 1994, 269, 11216–11223. [Google Scholar] [CrossRef]
- Demmin, G.L.; Clase, A.C.; Randall, J.A.; Enquist, L.W.; Banfield, B.W. Insertions in the gG gene of pseudorabies virus reduce expression of the upstream Us3 protein and inhibit cell-to-cell spread of virus infection. J. Virol. 2001, 75, 10856–10869. [Google Scholar] [CrossRef] [PubMed]
- Uyama, T.; Ishida, M.; Izumikawa, T.; Trybala, E.; Tufaro, F.; Bergstrom, T.; Sugahara, K.; Kitagawa, H. Chondroitin 4-O-sulfotransferase-1 regulates E disaccharide expression of chondroitin sulfate required for herpes simplex virus infectivity. J. Biol. Chem 2006, 281, 38668–38674. [Google Scholar] [CrossRef] [PubMed]
- Smiley, J.R. Herpes simplex virus virion host shutoff protein: Immune evasion mediated by a viral RNase? J. Virol. 2004, 78, 1063–1068. [Google Scholar] [CrossRef]
- Asano, S.; Honda, T.; Goshima, F.; Watanabe, D.; Miyake, Y.; Sugiura, Y.; Nishiyama, Y. US3 protein kinase of herpes simplex virus type 2 plays a role in protecting corneal epithelial cells from apoptosis in infected mice. J. Gen. Virol. 1999, 80 Pt 1, 51–56. [Google Scholar] [CrossRef]
- Parish, C.R.; Freeman, C.; Brown, K.J.; Francis, D.J.; Cowden, W.B. Identification of sulfated oligosaccharide-based inhibitors of tumor growth and metastasis using novel in vitro assays for angiogenesis and heparanase activity. Cancer Res. 1999, 59, 3433–3441. [Google Scholar]
- Vaheri, A. Heparin and Related Polyionic Substances as Virus Inhibitors. Acta Pathol. Microbiol. Scand. 1964, 171, 171–198. [Google Scholar]
- Ekblad, M.; Adamiak, B.; Bergstrom, T.; Johnstone, K.D.; Karoli, T.; Liu, L.; Ferro, V.; Trybala, E. A highly lipophilic sulfated tetrasaccharide glycoside related to muparfostat (PI-88) exhibits virucidal activity against herpes simplex virus. Antivir. Res. 2010, 86, 196–203. [Google Scholar] [CrossRef]
- Machiels, B.; Lete, C.; Guillaume, A.; Mast, J.; Stevenson, P.G.; Vanderplasschen, A.; Gillet, L. Antibody evasion by a gammaherpesvirus O-glycan shield. PLoS Pathog. 2011, 7, e1002387. [Google Scholar] [CrossRef]
- Cheshenko, N.; Herold, B.C. Glycoprotein B plays a predominant role in mediating herpes simplex virus type 2 attachment and is required for entry and cell-to-cell spread. J. Gen. Virol. 2002, 83, 2247–2255. [Google Scholar] [CrossRef]
- Ostberg, J.R.; Barth, R.K.; Frelinger, J.G. The Roman god Janus: A paradigm for the function of CD43. Immunol. Today 1998, 19, 546–550. [Google Scholar] [CrossRef]
- Zarbock, A.; Ley, K.; McEver, R.P.; Hidalgo, A. Leukocyte ligands for endothelial selectins: Specialized glycoconjugates that mediate rolling and signaling under flow. Blood 2011, 118, 6743–6751. [Google Scholar] [CrossRef]
- Crabb, B.S.; Allen, G.P.; Studdert, M.J. Characterization of the major glycoproteins of equine herpesviruses 4 and 1 and asinine herpesvirus 3 using monoclonal antibodies. J. Gen. Virol. 1991, 72 Pt 9, 2075–2082. [Google Scholar] [CrossRef]
- Gillet, L.; May, J.S.; Colaco, S.; Stevenson, P.G. The murine gammaherpesvirus-68 gp150 acts as an immunogenic decoy to limit virion neutralization. PLoS ONE 2007, 2, e705. [Google Scholar] [CrossRef]
- Jensen, H.L.; Norrild, B. The morphogenesis of herpes simplex virus type 1 in infected parental mouse L fibroblasts and mutant gro29 cells. APMIS 2003, 111, 1037–1052. [Google Scholar] [CrossRef] [PubMed]
- Campadelli-Fiume, G.; Arsenakis, M.; Farabegoli, F.; Roizman, B. Entry of herpes simplex virus 1 in BJ cells that constitutively express viral glycoprotein D is by endocytosis and results in degradation of the virus. J. Virol. 1988, 62, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.M.; Spear, P.G. Herpes simplex virus glycoprotein D mediates interference with herpes simplex virus infection. J. Virol. 1989, 63, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Lange, K. Fundamental role of microvilli in the main functions of differentiated cells: Outline of an universal regulating and signaling system at the cell periphery. J. Cell Physiol. 2011, 226, 896–927. [Google Scholar] [CrossRef] [PubMed]
- Sagar, M. Origin of the transmitted virus in HIV infection: Infected cells versus cell-free virus. J. Infect. Dis. 2014, 210 (Suppl. 3), S667–S673. [Google Scholar] [CrossRef] [PubMed]
- Altgarde, N.; Eriksson, C.; Peerboom, N.; Phan-Xuan, T.; Moeller, S.; Schnabelrauch, M.; Svedhem, S.; Trybala, E.; Bergstrom, T.; Bally, M. Mucin-like Region of Herpes Simplex Virus Type 1 Attachment Protein Glycoprotein C (gC) Modulates the Virus-Glycosaminoglycan Interaction. J. Biol. Chem. 2015, 290, 21473–21485. [Google Scholar] [CrossRef]
- Sun, Y.; MacLean, A.R.; Aitken, J.D.; Brown, S.M. The role of the gene 71 product in the life cycle of equine herpesvirus 1. J. Gen. Virol. 1996, 77 Pt 3, 493–500. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trybala, E.; Peerboom, N.; Adamiak, B.; Krzyzowska, M.; Liljeqvist, J.-Å.; Bally, M.; Bergström, T. Herpes Simplex Virus Type 2 Mucin-Like Glycoprotein mgG Promotes Virus Release from the Surface of Infected Cells. Viruses 2021, 13, 887. https://doi.org/10.3390/v13050887
Trybala E, Peerboom N, Adamiak B, Krzyzowska M, Liljeqvist J-Å, Bally M, Bergström T. Herpes Simplex Virus Type 2 Mucin-Like Glycoprotein mgG Promotes Virus Release from the Surface of Infected Cells. Viruses. 2021; 13(5):887. https://doi.org/10.3390/v13050887
Chicago/Turabian StyleTrybala, Edward, Nadia Peerboom, Beata Adamiak, Malgorzata Krzyzowska, Jan-Åke Liljeqvist, Marta Bally, and Tomas Bergström. 2021. "Herpes Simplex Virus Type 2 Mucin-Like Glycoprotein mgG Promotes Virus Release from the Surface of Infected Cells" Viruses 13, no. 5: 887. https://doi.org/10.3390/v13050887
APA StyleTrybala, E., Peerboom, N., Adamiak, B., Krzyzowska, M., Liljeqvist, J.-Å., Bally, M., & Bergström, T. (2021). Herpes Simplex Virus Type 2 Mucin-Like Glycoprotein mgG Promotes Virus Release from the Surface of Infected Cells. Viruses, 13(5), 887. https://doi.org/10.3390/v13050887