
Discovery of Known and Novel Viruses in Wild and Cultivated Blueberry in Florida through Viral Metagenomic Approaches

 ,
, 
 ,
, 
 and
and
Abstract
:1. Introduction
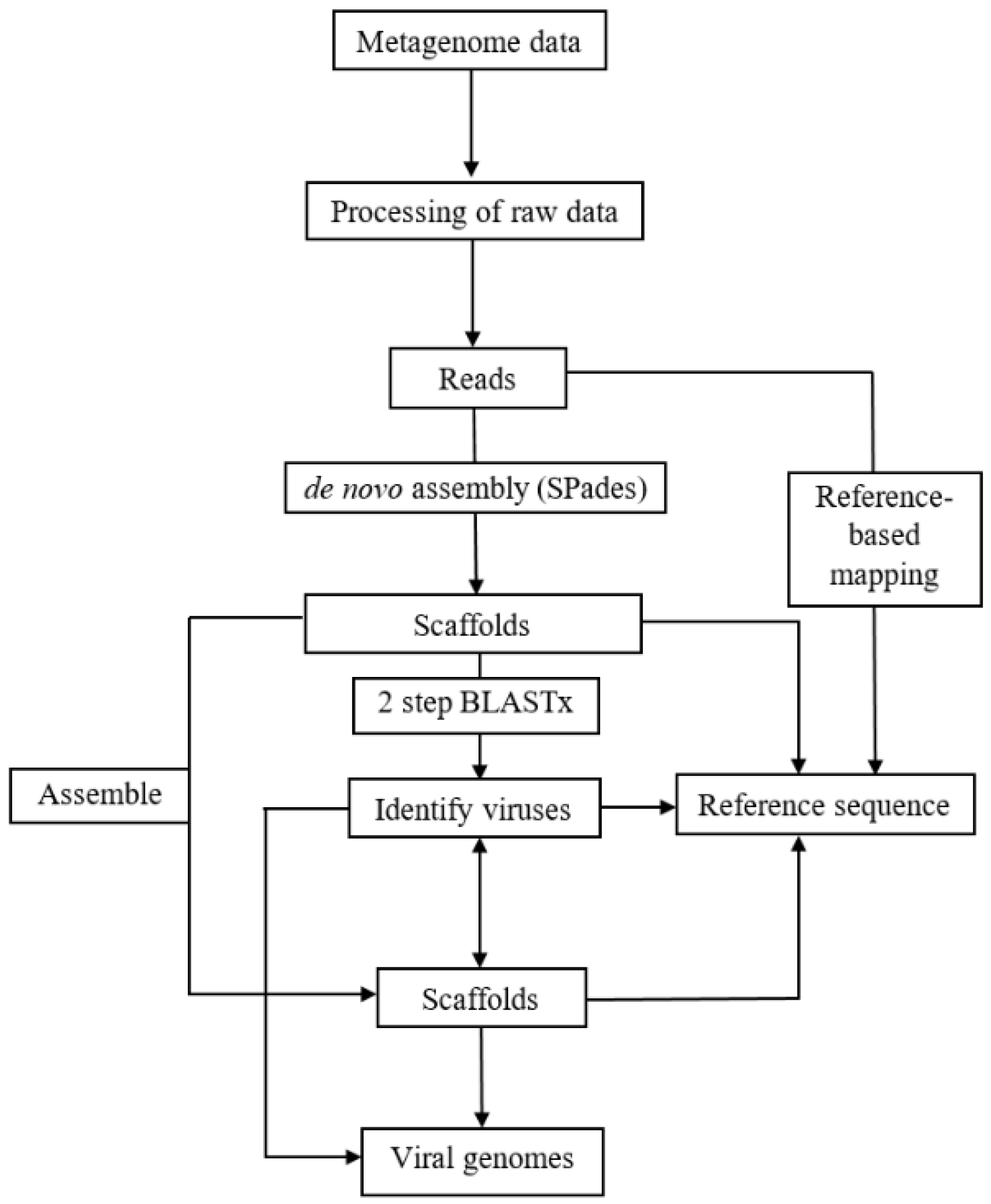
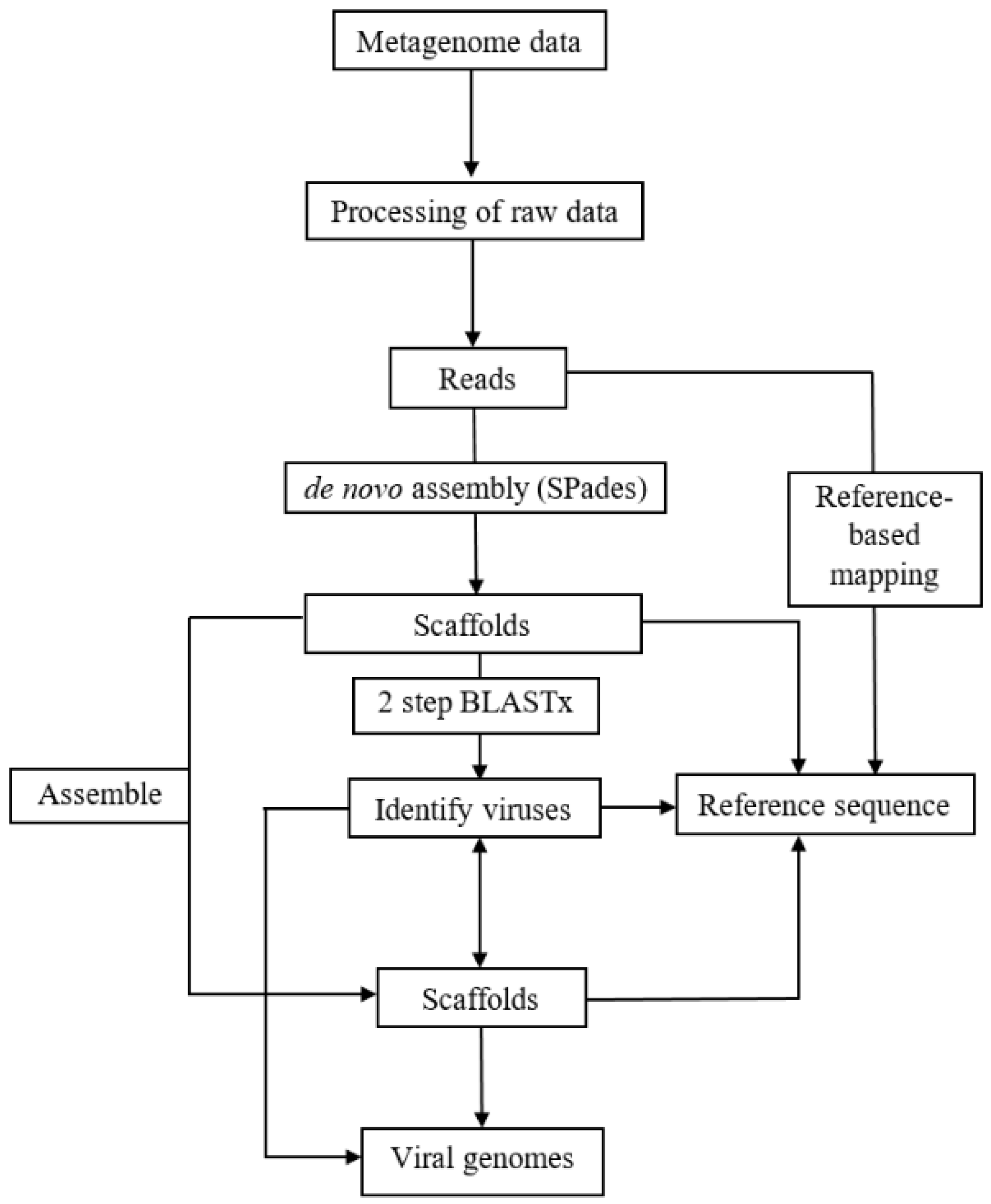
2. Materials and Methods
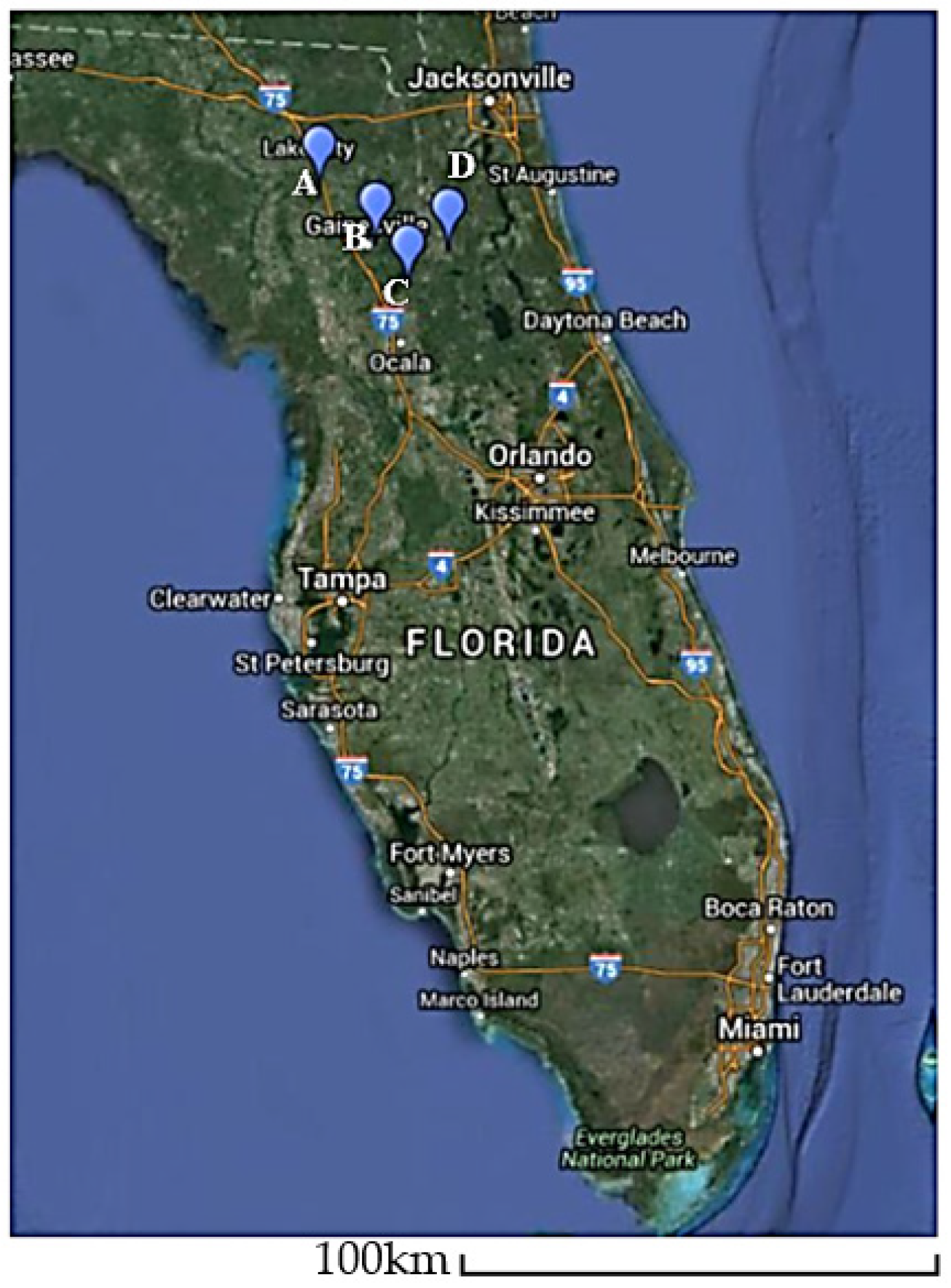
2.1. Plant Materials, Sample Preparation and Generation of V. corymbosum RNA Plant Viromes
2.2. Analyses of RNA Plant Viromes
2.3. Sequence and Phylogenetic Analyses
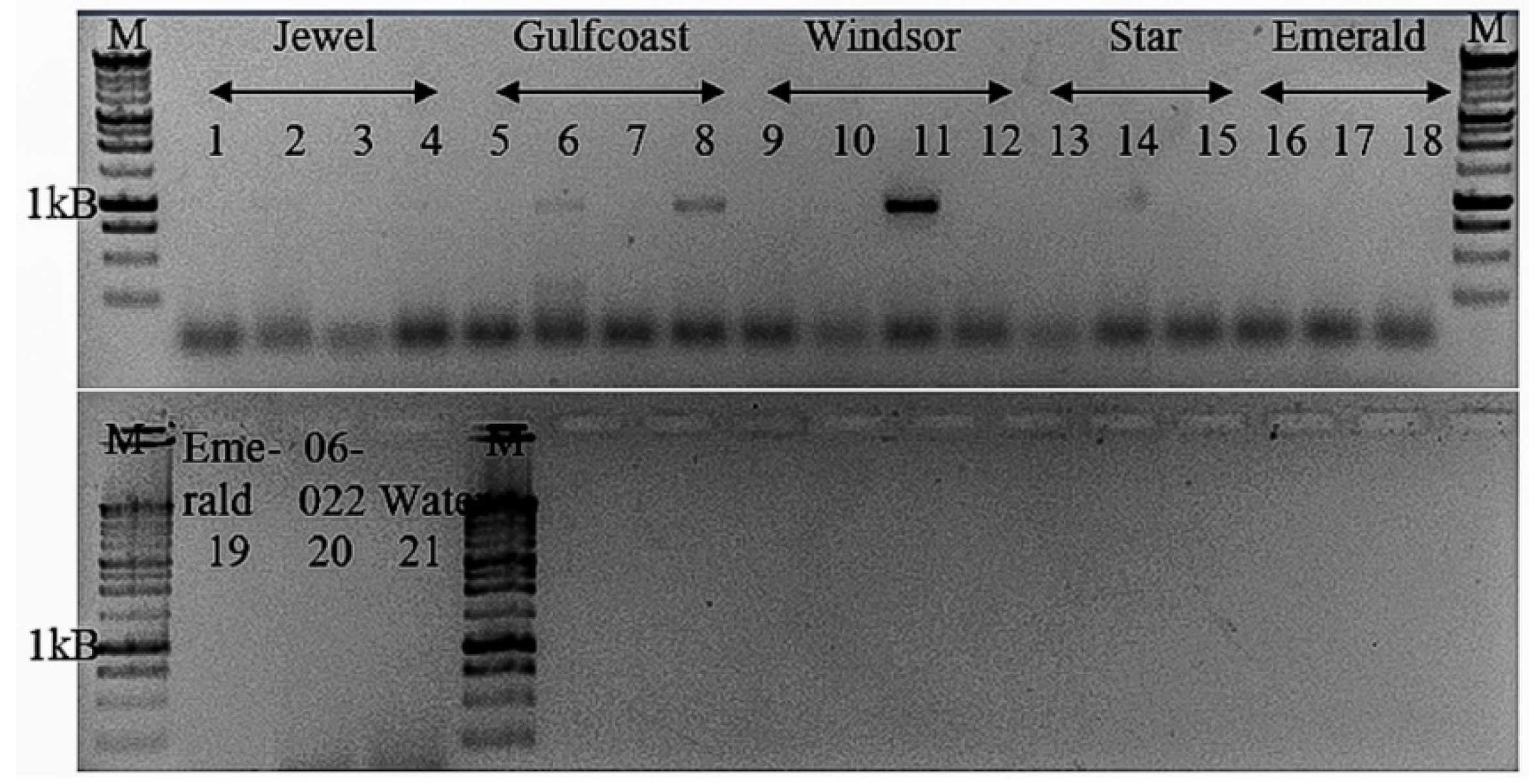
2.4. Validation and Detection In Vitro of Novel Tepovirus
3. Results
3.1. General Analyses of the RNA Plant Viromes
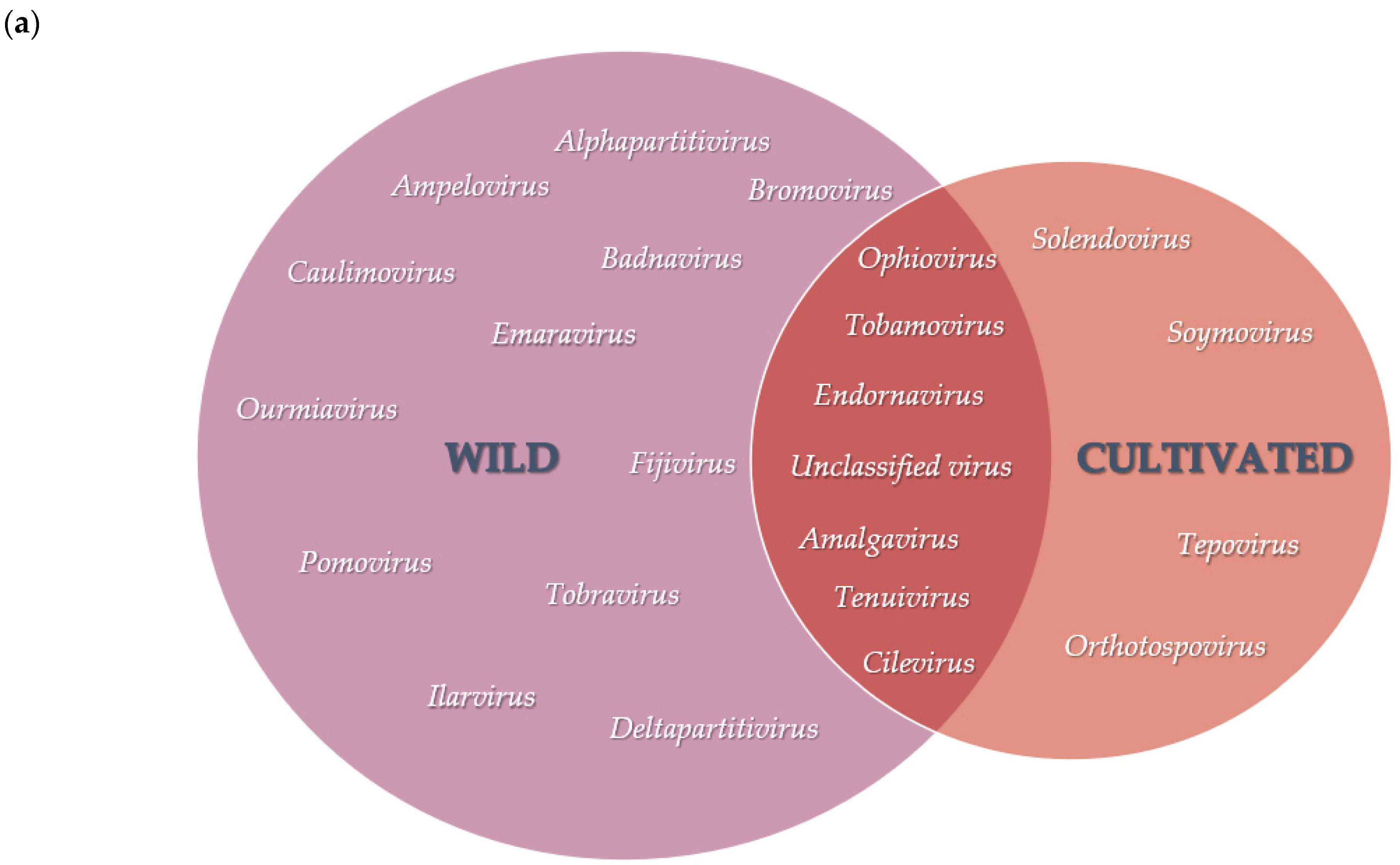
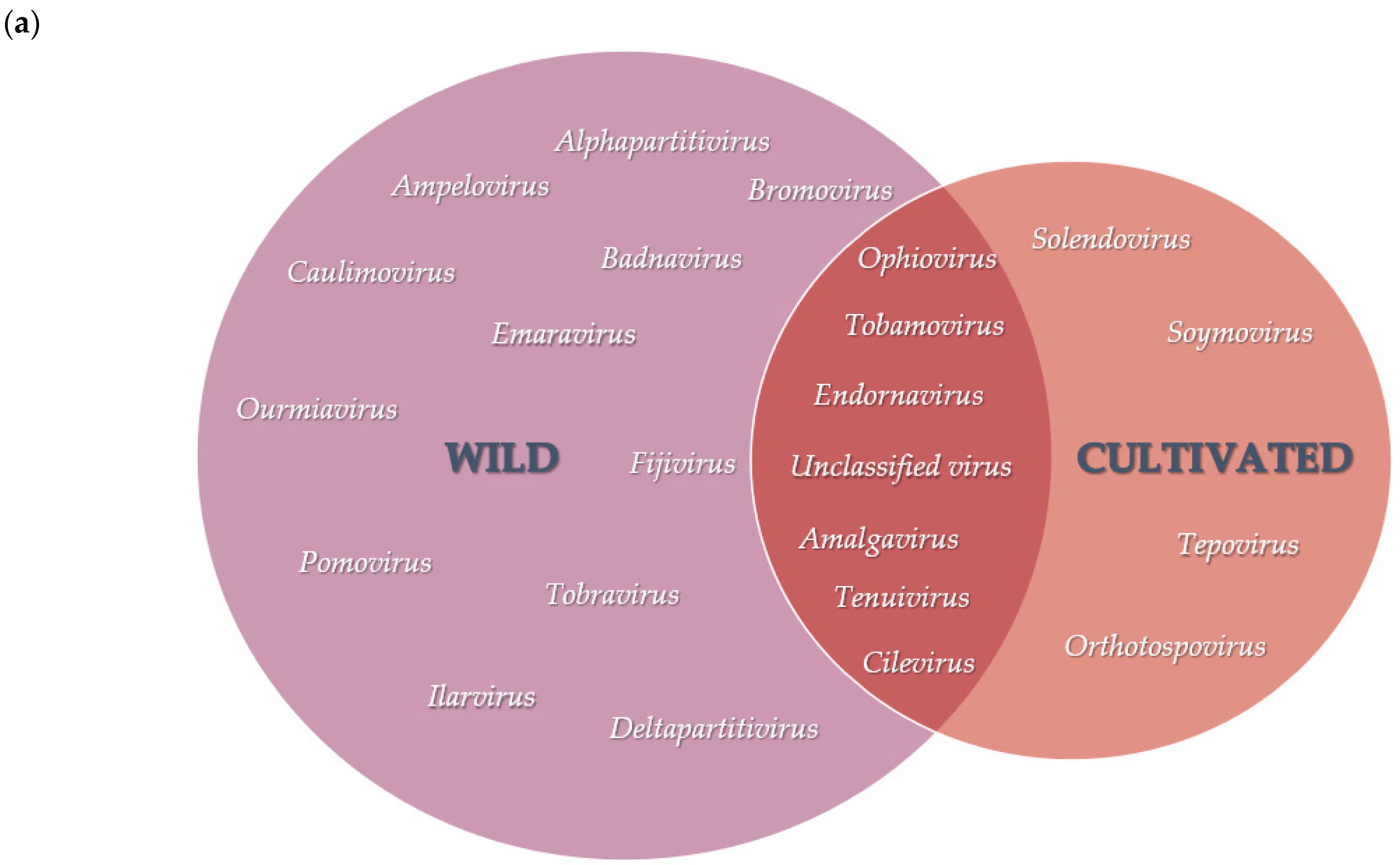
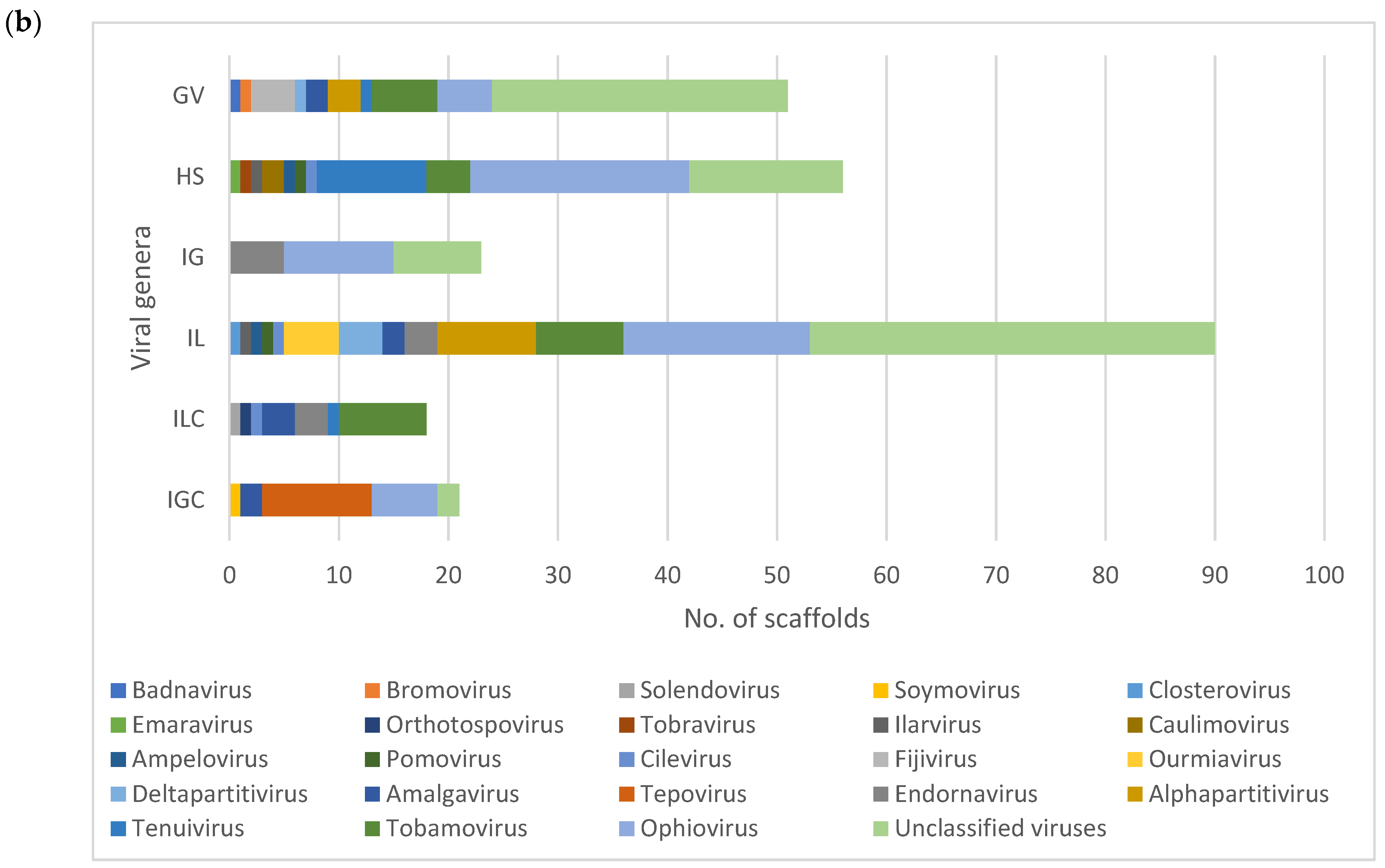
3.2. Diversity and Comparison of Viral Populations among the Viromes
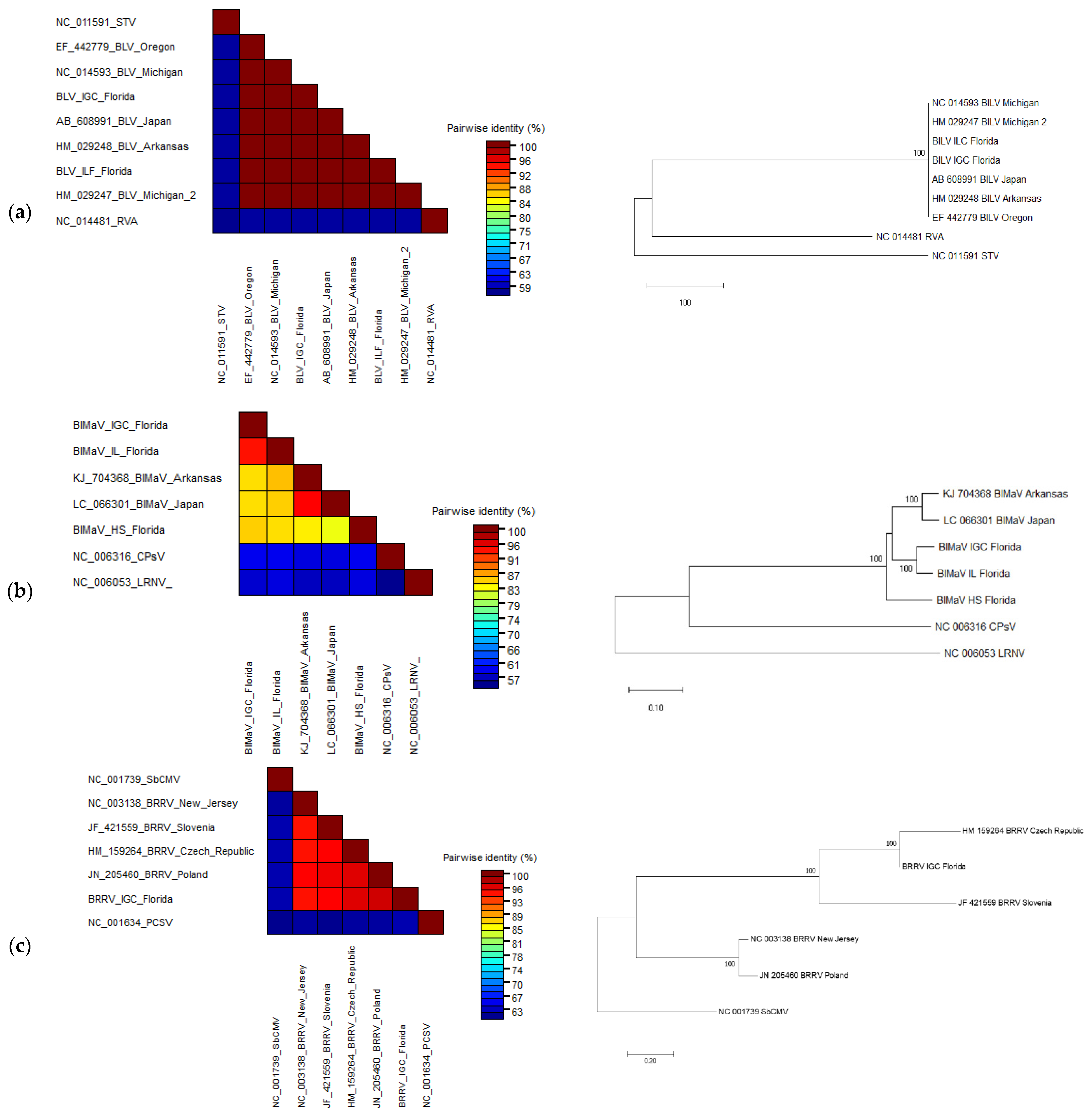
3.3. Sequence Comparison and Phylogenetic Analyses of the Complete Viral Genomes
3.3.1. Blueberry Latent Virus
3.3.2. Blueberry Mosaic Associated Ophiovirus
3.3.3. Blueberry Red Ringspot Virus
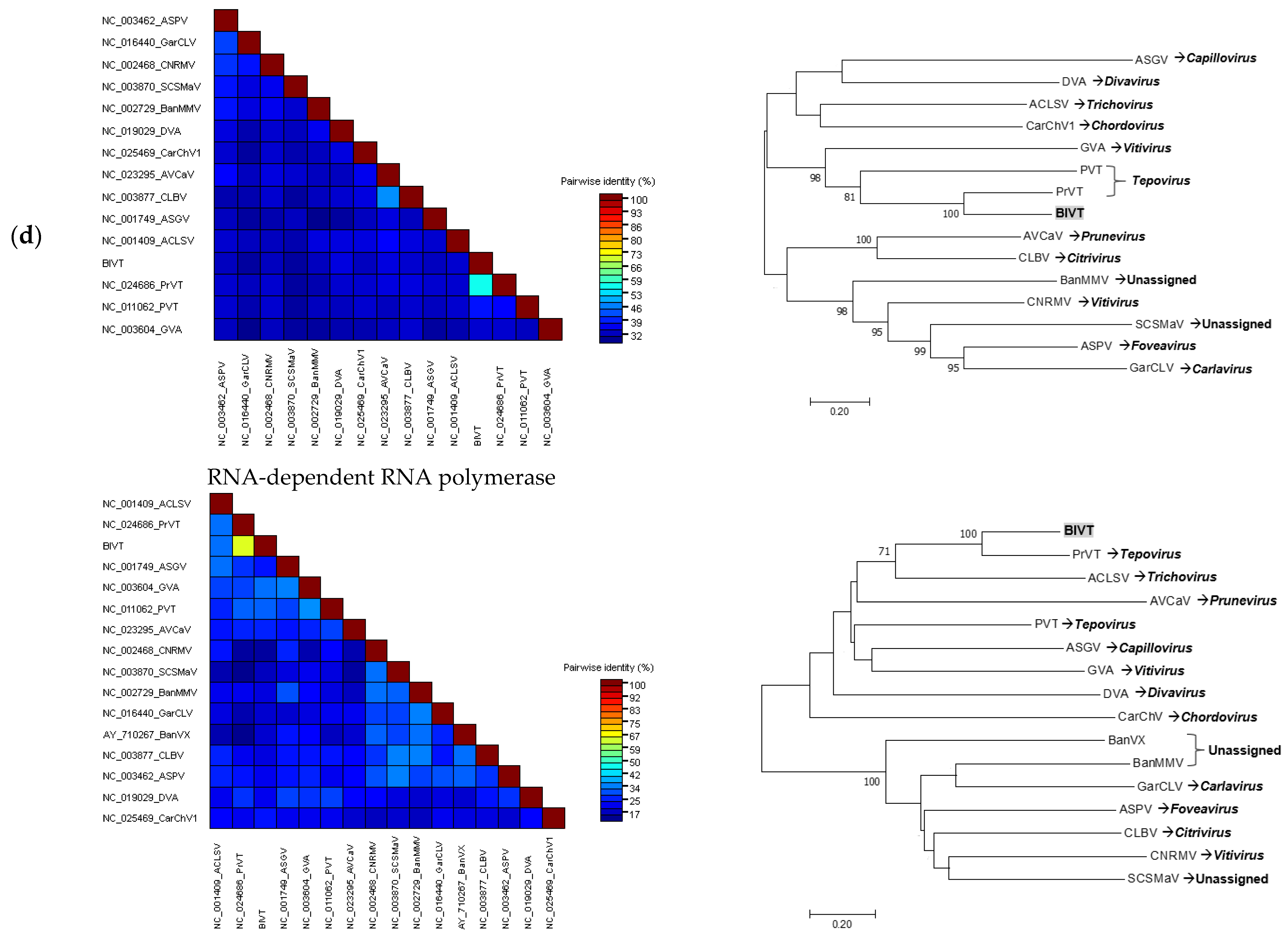
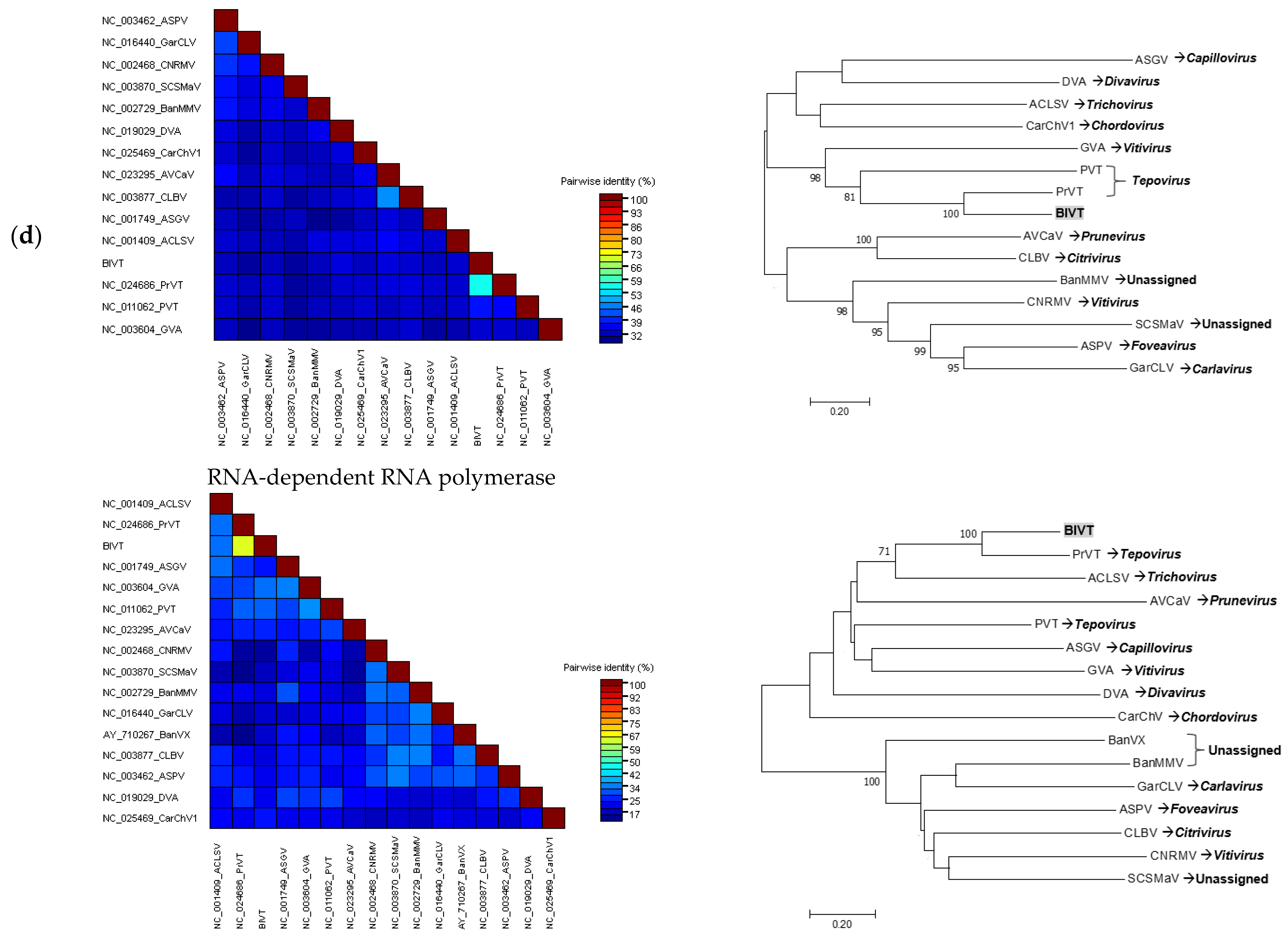
3.3.4. Tentative New Species in the Genus Tepovirus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barba, M.; Czosnek, H.; Hadidi, A. Historical perspective, development and applications of next-generation sequencing in plant virology. Viruses 2014, 6, 106–136. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant Virus Metagenomics: Advances in Virus Discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.A.C. Disease Pandemics and Major Epidemics Arising from New Encounters between Indigenous Viruses and Introduced Crops. Viruses 2020, 12, 1388. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J.; García-Arenal, F. Ecosystem simplification, biodiversity loss and plant virus emergence. Curr. Opin. Virol. 2015, 10, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Saad, N. Discovery of Known and Novel Viruses in Wild and Cultivated Blueberry through Transcriptomic and Viral Metagenomics Approaches. Ph.D. Thesis, University of Florida, Gainesville, FL, USA, 16 December 2017. University of Florida Digital Collections. Available online: https://ufdc.ufl.edu/UFE00 51785/00001 (accessed on 29 March 2021).
- Cantu-Iris, M.; Harmon, P.F.; Londono, A.; Polston, J.E. A variant of blueberry necrotic ring blotch virus associated with red lesions in blueberry. Arch. Virol. 2013, 158, 2197–2200. [Google Scholar] [CrossRef] [PubMed]
- Saad, N.; Alcalá-Briseño, R.; Polston, J.; Olmstead, J.; Varsani, A.; Harmon, P. Blueberry red ringspot virus genomes from Florida inferred through analysis of blueberry root transcriptomes. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Ho, T.; Martin, R.R.; Tzanetakis, I.E. Next-generation sequencing of elite berry germplasm and data analysis using a bioinformatics pipeline for virus detection and discovery. Methods Mol. Biol. 2015, 1302, 301–313. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.-S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.R.; Zhou, J.; Tzanetakis, I.E. Blueberry latent virus: An amalgam of the Partitiviridae and Totiviridae. Virus Res. 2011, 155, 175–180. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2016, 45, D200–D203. [Google Scholar] [CrossRef] [Green Version]
- Thekke-Veetil, T.; Ho, T.; Keller, K.E.; Martin, R.R.; Tzanetakis, I.E. A new ophiovirus is associated with blueberry mosaic disease. Virus Res. 2014, 189, 92–96. [Google Scholar] [CrossRef]
- Marais, A.; Faure, C.; Mustafayev, E.; Barone, M.; Alioto, D.; Candresse, T. Characterization by Deep Sequencing of Prunus virus T, a Novel Tepovirus Infecting Prunus Species. Phytopathology 2015, 105, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Muthukumar, V.; Melcher, U.; Pierce, M.; Wiley, G.B.; Roe, B.A.; Palmer, M.W.; Thapa, V.; Ali, A.; Ding, T. Non-cultivated plants of the Tallgrass Prairie Preserve of northeastern Oklahoma frequently contain virus-like sequences in particulate fractions. Virus Res. 2009, 141, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J.; Saha, P.; Wiley, G.B.; Quan, J.; White, J.D.; Lai, H.; Chavarria, F.; Shen, G.; Roe, B.A. Ecogenomics: Using massively parallel pyrosequencing to understand virus ecology. Mol. Ecol. 2010, 19 (Suppl. 1), 81–88. [Google Scholar] [CrossRef]
- Wylie, S.; Nouri, S.; Coutts, B.; Jones, M. Narcissus late season yellows virus and Vallota speciosa virus found infecting domestic and wild populations of Narcissus species in Australia. Arch. Virol. 2010, 155, 1171–1174. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.; Phillips, R.; Dixon, K.; Jones, M.G.; Wylie, S. Characterization of the first two viruses described from wild populations of hammer orchids (Drakaea spp.) in Australia. Plant Pathol. 2016, 65, 163–172. [Google Scholar] [CrossRef]
- Koh, S.H.; Ong, J.W.; Admiraal, R.; Sivasithamparam, K.; Jones, M.G.; Wylie, S.J. A novel member of the Tombusviridae from a wild legume, Gompholobium preissii. Arch. Virol. 2016, 161, 2893–2898. [Google Scholar] [CrossRef]
- Roossinck, M.J. Plant virus metagenomics: Biodiversity and ecology. Annu. Rev. Genet. 2012, 46, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Isogai, M.; Nakamura, T.; Ishii, K.; Watanabe, M.; Yamagishi, N.; Yoshikawa, N. Histochemical detection of Blueberry latent virus in highbush blueberry plant. J. Gen. Plant Pathol. 2011, 77, 304–306. [Google Scholar] [CrossRef]
- Martin, R.R.; Polashock, J.J.; Tzanetakis, I.E. New and emerging viruses of blueberry and cranberry. Viruses 2012, 4, 2831–2852. [Google Scholar] [CrossRef] [Green Version]
- Thekke-Veetil, T.; Polashock, J.J.; Marn, M.V.; Plesko, I.M.; Schilder, A.C.; Keller, K.E.; Martin, R.R.; Tzanetakis, I.E. Population structure of blueberry mosaic associated virus: Evidence of reassortment in geographically distinct isolates. Virus Res. 2015, 201, 79–84. [Google Scholar] [CrossRef]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Arch. Virol. 2016, 161, 2921–2949. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Libraries | No. of Reads | No. of Scaffolds ≥500 | No. of Putative Plant Virus Scaffolds | % of Putative Plant Virus Scaffolds | |
|---|---|---|---|---|---|
| Raw | Processed | ||||
| GV | 52,166,912 | 48,198,079 | 4256 | 52 | 1.22 |
| HS | 50,757,234 | 46,322,605 | 3485 | 60 | 1.72 |
| IL | 44,181,866 | 41,424,158 | 4890 | 91 | 1.86 |
| IG | 50,931,730 | 47,527,410 | 3899 | 24 | 0.62 |
| ILC | 44,632,114 | 41,527,161 | 3361 | 20 | 0.60 |
| IGC | 30,729,018 | 27,504,778 | 2790 | 23 | 0.82 |
| Virus | Genus | Vector | Transmission | GV | HS | IL | IG | ILC | IGC |
|---|---|---|---|---|---|---|---|---|---|
| BlLV | Amalgavirus | No | Transmitted through seed | ✓ | ✓ | ||||
| BlMaV | Ophiovirus | UK | UK | ✓ | ✓ | ||||
| BRRV | Soymovirus | UK | Vegetative propagation | ✓ | |||||
| BlVT | Tepovirus | UK | UK | ✓ | |||||
| TMV | Tobamovirus | No | Mechanical | ✓ | ✓ | ✓ | ✓ |
| RNA Segments | % of Mapped Reads (Average Coverage) | % Pairwise nt Identity to Ref. Seq. | ||||
|---|---|---|---|---|---|---|
| IGC | HS | IL | IGC | HS | IL | |
| 1 | 0.01 (47×) | 0.013 (102×) | - | 81.2 | 79.9 | - |
| 2 | 0.004 (83×) | 0.0005 (16×) | - | 79.5 | 79.7 | - |
| 3 | 0.003 (61×) | 0.007 (252×) | 0.01 (368x) | 83.4 | 82.7 | 84.6 |
| RNA Segments | ORFs | Length of RNA (nt) | Length of ORFs (nt) | 5′/3′ UTRs | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RS | IGC | HS | IL | RS | IGC | HS | IL | RS | IGC | HS | IL | ||
| 1 | RdRp/23 kDa | 7963 | 7747 | 7946 | - | 7014/585 | 7014/585 | 7014/576 | - | 238/19 | -/25 | 190/43 | - |
| 2 | MP | 1934 | 1981 | 1939 | - | 1548 | 1545 | 1545 | - | 317/69 | 334/60 | 337/99 | - |
| 3 | NP | 1570 | 1543 | 1602 | 1591 | 1368 | 1368 | 1368 | 1368 | 128/77 | 111/64 | 165/127 | 163/61 |
| Isolates | ORFs | Total Length | |||||||
|---|---|---|---|---|---|---|---|---|---|
| I (MP) | A | B | C | IV (CP) | V (RT) | VI (TA) | VII | ||
| CZ | 1101 | 312 | 561 | 600 | 1488 | 2004 | 1284 | 462 | 8302 |
| IGC-FL | 1197 | 369 | 561 | 597 | 1488 | 2007 | 1284 | 522 | 8392 |
| NJ | 939 | 369 | 561 | 600 | 1461 | 1974 | 1287 | 429 | 8303 |
| PL | 939 | 369 | 561 | 594 | 1455 | 1974 | 1284 | 462 | 8265 |
| SL | 1110 | 369 | 561 | 588 | 1476 | 2043 | 1284 | 462 | 8299 |
| Type of Region | Region | Length (nt) | % Pairwise nt Identity | |
|---|---|---|---|---|
| PrVT | BlVT | |||
| ORFs | RdRp | 5337 | 5457 | 61 |
| MP | 1152 | 1146 | 70 | |
| CP | 666 | 663 | 65 | |
| UTRs | 5′ | 46 | 109 | 68 |
| 3′ | 79 | 264 | 59 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saad, N.; Olmstead, J.W.; Varsani, A.; Polston, J.E.; Jones, J.B.; Folimonova, S.Y.; Harmon, P.F. Discovery of Known and Novel Viruses in Wild and Cultivated Blueberry in Florida through Viral Metagenomic Approaches. Viruses 2021, 13, 1165. https://doi.org/10.3390/v13061165
Saad N, Olmstead JW, Varsani A, Polston JE, Jones JB, Folimonova SY, Harmon PF. Discovery of Known and Novel Viruses in Wild and Cultivated Blueberry in Florida through Viral Metagenomic Approaches. Viruses. 2021; 13(6):1165. https://doi.org/10.3390/v13061165
Chicago/Turabian StyleSaad, Norsazilawati, James W. Olmstead, Arvind Varsani, Jane E. Polston, Jeffrey B. Jones, Svetlana Y. Folimonova, and Philip F. Harmon. 2021. "Discovery of Known and Novel Viruses in Wild and Cultivated Blueberry in Florida through Viral Metagenomic Approaches" Viruses 13, no. 6: 1165. https://doi.org/10.3390/v13061165
APA StyleSaad, N., Olmstead, J. W., Varsani, A., Polston, J. E., Jones, J. B., Folimonova, S. Y., & Harmon, P. F. (2021). Discovery of Known and Novel Viruses in Wild and Cultivated Blueberry in Florida through Viral Metagenomic Approaches. Viruses, 13(6), 1165. https://doi.org/10.3390/v13061165