Abstract
Drug resistance studies on human γ-herpesviruses are hampered by the absence of an in vitro system that allows efficient lytic viral replication. Therefore, we employed murine γ-herpesvirus-68 (MHV-68) that efficiently replicates in vitro as a model to study the antiviral resistance of γ-herpesviruses. In this study, we investigated the mechanism of resistance to nucleoside (ganciclovir (GCV)), nucleotide (cidofovir (CDV), HPMP-5azaC, HPMPO-DAPy) and pyrophosphate (foscarnet (PFA)) analogues and the impact of these drug resistance mutations on viral fitness. Viral fitness was determined by dual infection competition assays, where MHV-68 drug-resistant viral clones competed with the wild-type virus in the absence and presence of antivirals. Using next-generation sequencing, the composition of the viral populations was determined at the time of infection and after 5 days of growth. Antiviral drug resistance selection resulted in clones harboring mutations in the viral DNA polymerase (DP), denoted Y383SGCV, Q827RHPMP-5azaC, G302WPFA, K442TPFA, G302W+K442TPFA, C297WHPMPO-DAPy and C981YCDV. Without antiviral pressure, viral clones Q827RHPMP-5azaC, G302WPFA, K442TPFA and G302W+K442TPFA grew equal to the wild-type virus. However, in the presence of antivirals, these mutants had a growth advantage over the wild-type virus that was moderately to very strongly correlated with antiviral resistance. The Y383SGCV mutant was more fit than the wild-type virus with and without antivirals, except in the presence of brivudin. The C297W and C981Y changes were associated with a mutator phenotype and had a severely impaired viral fitness in the absence and presence of antivirals. The mutator phenotype caused by C297W in MHV-68 DP was validated by using a CRISPR/Cas9 genome editing approach.
1. Introduction
The human γ-herpesviruses Epstein–Barr virus (EBV) and Kaposi’s sarcoma-associated virus (KSHV) distinguish themselves from the α-herpesviruses (herpes simplex virus 1 (HSV-1) and 2 (HSV-2), varicella-zoster virus (VZV)) and β-herpesviruses (human cytomegalovirus (HCMV), human herpesvirus 6A/6B/7 (HHV-6A/6B/7)) by their association with a large number of malignancies [1,2,3,4]. EBV is associated with the development of several lymphoproliferative disorders (Burkitt’s lymphoma, Hodgkin lymphoma, diffuse large B-cell lymphoma and lymphoproliferative disease in immunocompromised hosts) and carcinomas of epithelial origin (nasopharyngeal carcinoma and gastric carcinoma) [5]. KSHV is linked to endothelial sarcomas (Kaposi’s sarcoma) and malignancies of B-cell origin (multicentric Castleman’s disease and primary effusion lymphoma) [6].
Though EBV and KSHV oncogenesis is mostly associated with latent gene expression, recent investigations demonstrate a significant contribution of lytic replication to viral tumorigenesis [7,8]. Therefore, antiviral therapy intended at inhibiting the expression of lytic viral proteins should be beneficial for controlling the early stages of EBV-associated malignancies. The contribution of EBV lytic replication to lymphoproliferative disease has been highlighted by several studies [9,10]. The EBV immediately early (IE) proteins BZLF1 and BRLF1 promote interleukin-6 (IL-6) secretion in lytically infected cells, sustaining early lymphoproliferative disease [11] as IL-6 (a cytokine playing a key role in maintenance of immune functions, stimulation of hematopoietic cells differentiation and persistence of inflammation) is a decisive element in various epithelial and hematological malignancies. IL-6 promotes cell survival and induces signal transducer and activator of transcription 3 (STAT3), acting through paracrine and autocrine mechanisms [11]. EBV, a virus infecting both epithelial and lymphoid cells, induces IL-6 expression. Expression of cellular and viral interleukin 10 (IL-10) is induced in lytically infected EBV cells, resulting in a more efficient growth of B-cells [12]. The involvement of cellular IL10 and viral IL-6 in the growth, survival and spread of KSHV-associated PEL, MCD and KS was also established [13,14]. Vascular endothelial growth factor (VEGF), which contributes to angiogenesis in both B-cell and epithelial malignancies, is also increased following EBV infection [15]. The lytic cycle is important for KSHV-mediated disease development. For instance, KSHV viral load in peripheral blood mononuclear cells is correlated with KS progression [16].
Some antiviral drugs that inhibit viral lytic DNA replication, such as foscarnet and ganciclovir, can repress early KS development [17]. Tumor-forming spindle cells are latently infected in most KS lesions, though a small number of cells still undergo spontaneous lytic replication. EBV is a major risk factor for the development of post-transplant lymphoproliferative disorder (PTLD), a serious and often fatal complication. Persistence of elevated viral loads or a high level of EBV DNA in the blood of transplant recipients is associated with an increased risk of PTLD [18,19]. Although antiviral therapy is not useful in PTLD due to viral latency, antiviral drug prophylaxis is able to reduce its incidence [20,21], highlighting the role of antivirals in preventing PTLD in transplant recipients. Collectively, lytic replication not only plays an indispensable function during the life cycle of γ-herpesvirus, ensuring virus propagation, but also has a fundamental role in EBV- and KSHV-associated malignancies. Additionally, lytic EBV replication contributes to infectious mononucleosis, oral hairy leukoplakia and chronic active Epstein–Barr virus (CAEBV). Hence, considering that γ-herpesvirus reactivation is an important mechanism in the pathogenesis of EBV- and KSHV-associated diseases, antivirals targeting the lytic stage should be helpful for controlling the early stages of γ-associated malignancies. The anti-γ-herpesvirus activity of known antivirals is directed toward the impediment of viral replication; no antiviral is effective on the latent phase of infection, showing limited success clinically, and none of them have been licensed for treatment of EBV or KSHV infections [22,23,24].
In vitro, a number of nucleosides (acyclovir (ACV), ganciclovir (GCV), brivudin (BVDU)), nucleotides (cidofovir (CDV), HPMP-5azaC and HPMPO-DAPy) and pyrophosphate analogues (foscarnet (PFA)) inhibit γ-herpesvirus replication [25,26,27,28,29]. However, to date, no antiviral agent has been approved for the prevention or treatment of human γ-herpesvirus. The impact of long-term antiviral treatment on EBV and KSHV disease initiation and progression has been investigated in multiple clinical trials [17,20,30,31]. The goal was to evaluate the pre-emptive effect of antivirals on the development of lymphoproliferative malignancies, i.e., GCV prophylaxis on EBV-positive PTLD development [30,32], and GCV or PFA prophylaxis on AIDS-associated KS [17]. In these studies, the most used antivirals were ganciclovir and its oral prodrug valganciclovir, and no cases of drug resistance have been reported yet [17,22,23]. However, nearly all these studies evaluated antivirals in an immunocompromised setting, a condition known to favor the emergence of drug resistance.
Nucleoside, nucleotide and pyrophosphate analogues target the viral DNA polymerase (DP) but differ in their mode of activation. In γ-herpesviruses, purine and pyrimidine nucleosides require a first phosphorylation step by a viral protein kinase (PK) or thymidine kinase (TK), respectively, and are subsequently phosphorylated by cellular kinases. The active triphosphates can then serve as a substrate of the viral DP reaction. Nucleotide analogues do not rely on viral proteins for their activation and are phosphorylated by cellular kinases to the active diphosphate form. After phosphorylation, the active metabolites compete with the natural nucleotides for incorporation into the cellular and viral DNA. Viral DNA polymerases preferentially incorporate the active form of the nucleotide analogues into viral DNA, leading to selective inhibition of viral DNA synthesis. Pyrophosphate analogues interact with the pyrophosphate-binding site of the DNA polymerase and inhibit the cleavage of the pyrophosphate moiety from deoxynucleotide triphosphates (dNTPs) [25,33,34,35]. In γ-herpesviruses, antiviral resistance to nucleoside analogues can arise at the level of the PK and/or DP (purine analogues) or at the TK and/or DP (pyrimidine analogues). For nucleotide and pyrophosphate analogues, resistance is associated exclusively with changes in the viral DP [27,36,37].
At present, there is limited knowledge regarding human γ-herpesvirus drug resistance. This can be attributed to the lack of in vitro cell culture systems permissive for efficient lytic replication [38] which impedes the isolation and characterization of drug-resistant viral variants. To overcome this hurdle, we used murine γ-herpesvirus-68 (MHV-68) in earlier investigations as a surrogate for the study of human γ-herpesviruses [39,40]. MHV-68, in contrast to EBV and KSHV, can infect and efficiently replicate in a broad range of cell types [41], allowing for selection of drug-resistant mutants [26,27,36,37]. Despite progress in understanding the molecular mechanisms of MHV-68 drug resistance, there is currently no information regarding resistance to a number of nucleoside, nucleotide and pyrophosphate analogues. Moreover, the emergence of drug resistance raises the question of the impact of mutations on viral fitness, as alterations to genes vital to replication can diminish viral fitness. Until recently, herpesvirus viral fitness studies were performed using monoculture infection systems [42,43,44]. However, dual infection competition assays, where two viral variants are grown together and the proportions of the competing viruses are carefully measured over time, are currently the “gold standard” to measure viral fitness. Competition experiments have the advantage that they can estimate the replication capacity of two viral variants under identical conditions. While earlier investigations relied on bulk [45,46] or clonal sequencing [47] to determine the ratio of wild-type to drug-resistant viruses, we developed a next-generation sequencing (NGS) approach to accurately quantify the composition of the viral population in herpesvirus dual infection competition assays [48].
In this study, we investigated the mechanism of MHV-68 resistance to nucleoside (GCV), nucleotide (CDV, HPMP-5azaC, HPMPO-DAPy) and pyrophosphate analogues (PFA) and evaluated the impact of mutations associated with drug resistance on viral replication capacity. The viral fitness of wild-type versus drug-resistant viruses was evaluated using dual infection competition assays in the absence and presence of antivirals. The frequency of the competing viral variants was measured using the Illumina NGS platform (Miseq), which allowed identifying variants in a mixed viral population with a high sensitivity. Two amino acid changes were associated with a mutator phenotype (C297W and C981Y), and the association between the C297W amino acid change and a mutator phenotype virus was validated by using CRISPR/Cas9 genome editing.
2. Materials and Methods
2.1. Cells and Virus
Vero (ATCC-CCL81) and HEK293T (ATCC-CRL-321) cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM, ThermoFisher, Merelbeke, Belgium) supplemented with 10% fetal calf serum (FCS), 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate and 10 mM HEPES at 37 °C in a 5% CO2 humidified atmosphere. MHV-68 (G2.4) was provided by Prof A. Nash, Edinburgh, United Kingdom. MHV-68 infection was performed in Vero cells with 2% FCS DMEM.
2.2. Compounds
The following antivirals were used: acyclovir (ACV, [9-(2-hydroxyethoxymethyl)guanine]), GlaxoSmithKline, Stevenage, UK; ganciclovir (GCV, [9-(1,3-dihydroxy-2-propoxymethyl)guanine), Roche, Basel, Switzerland; foscarnet (PFA, [phosphonoformate sodium salt]), Sigma Chemicals, St. Louis, MO, USA; cidofovir (CDV, [(S)-1-(3-hydroxy-2-phosphonomethoxypropyl)cytosine]), Gilead Sciences, Foster City, CA, USA; brivudin (BVDU, [(E)-5-(2-bromovinyl)-2′-deoxyuridine]), Searle, UK; HDVD, 1-[(2S,4S-2-(hydroxymethyl)-1,3-dioxolan-4-yl]5-vinylpyrimidine-2,4(1H,3H)-dione, Dr. D. Chu (University of Georgia, Athens, GA, USA); KAY-2-41 [1-(2-deoxy-1-methyl-4-thio-β-Dribofuranosyl)thymine and KAH-39-139 [1-(2-deoxy-4-amino-4-thio-β-Dribofuranosyl)thymine], Showa University, Tokyo, Japan; HPMP-5azaC [(1-(S)-(3-hydroxy-2-(phosphonomethoxy)propyl)-5-azacytosine] and HPMPO-DAPy [(R)-2,4-diamino-3-hydroxy-6-[2-(phosphonomethoxy)propoxy])pyrimidine], Dr M. Krecmerova, Institute of Organic Chemistry and Biochemistry, Prague, Czech Republic.
2.3. Selection of Drug-Resistant MHV-68
Selection of MHV-68 mutants resistant to GCV, CDV, HPMP-5azaC, HPMPO-DAPy or PFA was performed by sequential passaging of the virus under increasing concentrations of antiviral. Resistance emerged following 21 (GCV), 15 (CDV), 12 (HPMP-5azaC), 13 (HPMPO-DAPy) and 17 (single mutant) or 22 (double mutant) (PFA) passages under antiviral pressure. The heterogeneous populations of drug-resistant viruses were plaque purified by limited dilution to obtain single viral clones.
2.4. Genotypic Analysis
Each drug-resistant viral clone was genotyped using Sanger sequencing. After DNA extraction (QIAmp DNA kit; Qiagen Benelux BV, Venlo, The Netherlands), the viral TK, PK and DP genes were amplified by PCR (Table S1) (FastStart high-fidelity DNA polymerase; Roche Applied Science, Mannheim, Germany). The PCR products were purified (QIAquick purification kit, Qiagen Benelux BV, Venlo, The Netherlands) and sequenced using the BigDye terminator kit version 3.1 on an ABI 3730 sequencing system (ThermoFisher, Merelbeke, Belgium) (Table S1). The sequencing results were analyzed with SeqScape 2.7 (ThermoFisher, Merelbeke, Belgium).
2.5. 3D Modeling
Based on the already published HSV-1 DP structure (pdb code: 2GV9), a model of MHV-68 DP was built using the protein structure homology-modelling server Swiss Model (https://swissmodel.expasy.org, accessed on 1 April 2021). The altered amino acid positions of the resistant viral clones were marked in red. The figures were constructed using Pymol Delano software, open source version 0.99rc6.
2.6. Drug Susceptibility Profile
MHV-68 wild-type and drug-resistant viral clones were characterized by CPE reduction assays in Vero cells as described earlier [23]. For each antiviral, the required concentration to reduce virus-induced CPE by 50% compared to the untreated control (EC50) was determined. For each clone, at least three independent assays were performed, and the mean EC50 values were calculated. The level of drug resistance was expressed as the ratio EC50 resistant virus/EC50 wild-type virus. MHV-68 viral clones were considered resistant or hypersensitive at ≥2-fold change in EC50.
2.7. Replication Capacity and Relative Fitness
The viral replication capacity of Y383SGCV, Q827RHPMP-5azaC, G302WPFA, K442TPFA and G302W+K442TPFA, C297WHPMPO-DAPy and C981YCDV was determined in the absence and presence of antivirals at 750 PFU/well of either the wild-type virus, a drug-resistant viral clone or a 50:50 ratio of both (375 PFU wild-type virus: 375 PFU DP mutant virus), as described earlier [48]. The replication capacity of viral clone Y383SGCV was evaluated using an 80:20 ratio of Y383SGCV/MHV-68 wild-type (600 PFU Y383SGCV/150 PFU wild-type virus). The composition of the viral population was determined using the Illumina Miseq NGS platform as described earlier [48]. PCR amplification of the viral DP was performed using the following primer pair: For: 5′-CCATATAGGCTACTCTACCCTC-3′, and Rev: 5′-GTAGGTCCAGAGTGGTGTATC-3′.
After NGS and data analysis, the relative fitness was calculated using the formula: (1 + s) = exp [1/t × ln (Mt/Wt × Wt0/Mt0)], where s is the selection coefficient and t is the time (in days). Mt and Mt0 are the fractions of mutant virus initially and at the time of the measurement. Wt and Wt0 are the fractions of wild-type virus initially and at the time of the measurement. An RF = 1 indicates that both viruses grew with equivalent fitness, an RF <1 indicates that the mutant virus grows less efficiently than the wild-type virus and an RF >1 indicates increased fitness of the mutant virus relative to that of the wild-type virus [48,49,50]. The mean RF values from two biological replicate infection competition assays are reported. The impact of drug resistance mutations in the viral DP on viral fitness and the impact of antiviral treatment were calculated using one-way Anova (multiple comparisons to MHV-68 wild-type with the Dunnett test (p ≤ 0.01)).
2.8. Correlation between Antiviral Resistance and Relative Fitness
The strength and direction of the relationship between antiviral drug resistance and relative fitness capacity were calculated using Spearman’s rank-order correlation test. Spearman’s correlation coefficient ρ (also signified by rs) measures the strength and direction of association between two ranked variables (rs between 0.00 and 0.19 (very weak), 0.20 and 0.39 (weak), 0.40 and 0.59 (moderate), 0.60 and 0.79 (strong) and 0.80 and 1.0 (very strong)).
2.9. Investigation of the Mutation Frequency in the Viral PK, TK and DP
To determine the mutation frequency of the mutator phenotype viruses C297WHPMPO-DAPy and C981YCDV, three different viral clones of each mutator were selected and expanded by three rounds of amplification. A DNA extract of each clone was obtained at passage 0 and passage 3, and the emergence of spontaneous mutations in the viral PK, TK and DP was determined by NGS as mentioned earlier [48]. The mutations arising in MHV-68 PK, TK and DP were quantified (A → T, A → G, C → A; C → T, G → T; G → A, C → A, C → T), and the intermutational distance (i.e., the distance between two mutations) was quantified. Lastly, the spontaneous mutations were aligned to HSV-1, HSV-2, VZV and HCMV (Uniprot) to determine if the corresponding amino acids were linked to known polymorphisms or mutations associated with drug resistance.
2.10. CRISPR/Cas9 Genome Editing of MHV-68 DP Amino Acid Position C297
2.10.1. Plasmid Construct
The CRISPR/Cas9 plasmid construct was assembled using the GeneArt CRISPR Nuclease Vector, which contains an orange fluorescent protein reporter (ThermoFisher, Merelbeke, Belgium). CRISPRdirect (https://crispr.dbcls.jp/, accessed on 5 October 2015) was used to select a target-specific sgRNA with a reduced number of off-target sites, using the MHV-68 DP sequence as input (sgRNA sequence: 5′-GTCATTTGATATAGAGTGTT-3′) (Figure S1).
Target-specific oligonucleotides (For: 5′-GTCATTTGATATAGAGTGTTGTTTT-3′, and sgRNA Rev: 5′-AACACTCTATATCAAATGACCGGTG-3′) were synthesized with 3′ overhang ends compatible to clone into the GeneArt Vector. The circularized vector was transformed into One Shot TOP10 chemically competent E. coli bacteria (ThermoFisher, Merelbeke, Belgium). Ampicillin-resistant colonies were cultured overnight for DNA extraction (Wizard Plus SV Minipreps DNA Purification System, Promega, Leiden, The Netherlands). The presence of the correct insert was confirmed by Sanger sequencing, using primers flanking the cloning site of the vector (For: 5′-GAGGGCCTATTTCCCATGAT-3′, and Rev: 5′-ACCATGATTACGCCAAGCTC-3′). Following successful cloning, midipreps of 50 mL were performed using the PureLink.HiPure Plasmid Midiprep Kit (ThermoFisher, Merelbeke, Belgium). Plasmid concentration and purity were assessed with Nanodrop ND-1000 (Isogen Life Science, De Meern, The Netherlands).
2.10.2. Transfection/Infection
After cloning the sgRNA coding sequence into the GeneArt Vector, the purified plasmid and the homologous recombination (HR) template were transfected into HEK293T cells by reverse transfection using Lipofectamine 3000 (ThermoFisher, Merelbeke, Belgium), following the manufacturer’s instructions. Then, 2.5 × 105 transfected HEK293T cells were plated in each well of a 24-well plate. The following asymmetric template was used: AGAGATAGACTGTGGTCTGGGAACATCTGTCATCATCCAGAGCTCTCCTCCTGGCCCCCCT-ACAATATCCTGTCATTTGATATAGAGTGGTTAGGTGAATGTGGGTTTCCCTGTG-CCCTGAAAGAA, where the target nucleotide change (T to G) is indicated in bold and highlighted in gray, and a silence mutation (A, bold) was included in the PAM (underlined). Consequently, the thymine at position 891 of MHV-68 DP was altered to guanine (T891G).
The next day, following transfection, the cells were infected with 25 PFU/well, 50 PFU/well or 100 PFU/well of MHV-68 wild-type. After 2 h, the residual virus was removed, and the cells were incubated with 1 mL DMEM 2%. The supernatants were harvested 24 h, 48 h and 120 h post-infection. The obtained viral stocks were plaque purified by limited dilution. The clones were screened for the presence of the T891G nucleotide change by Sanger sequencing and NGS, as described earlier.
3. Results
3.1. MHV-68 Mutants Selected under Pressure of GCV-, HPMP-5azaC- or PFA-Harbored Mutations in the Viral DNA Polymerase (DP)
Selection with GCV, HPMP-5azaC or PFA generated five MHV-68 drug-resistant viral clones with amino acid changes in the viral DP (without changes in the PK or TK genes). The MHV-68 DP consists of six domains: 3′–5′ exonuclease domain (yellow), thumb (green), 5′–3′ exonuclease domain (light blue), NH2 domain (gray), palm (orange) and finger (dark blue) (Figure 1). Drug resistance selection with GCV and PFA gave rise to mutations in the 3′–5′ exonuclease domain. Selection with GCV led to an Y383SGCV amino acid change, whereas under pressure of PFA, three MHV-68 drug-resistant viral clones were obtained: two clones with single substitutions (G302WPFA and K442TPFA) and one clone bearing both mutations (G302W+K442TPFA). Selection with HPMP-5azaC resulted in the amino acid change Q827RHPMP-5azaC, located in the thumb domain of the viral DP.
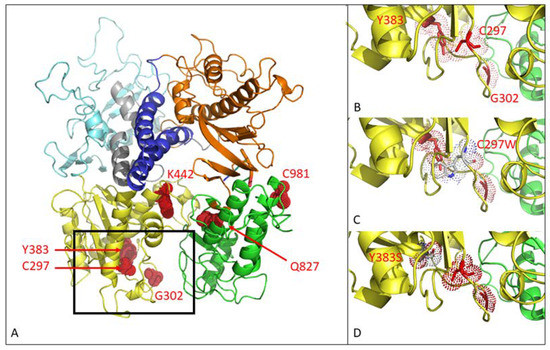
Figure 1.
Three-dimensional modeling of mutations in the MHV-68 viral DNA polymerase. (A) MHV-68 DNA polymerase consists of 6 domains. Mutations C297WHPMPO-DAPy, G302WPFA, Y383SGCV and K442TPFA map to the 3′–5′ exonuclease domain (yellow). Q827RHMP-5azaC and C981YCDV map to the thumb domain (green). There were no mutations in the other domains (5′–3′ exonuclease domain (light blue), NH2 domain (gray), palm (orange), finger (dark blue)). (B) Close view of positions C297, G302 and Y383. (C) Close view of the amino acid change C297W. (D) Close view of the amino acid change Y383S.
The inhibitory effects of a panel of antivirals (ACV, GCV, BVDU, HDVD, KAH-39-139, KAY-2-41, CDV, HPMP-5azaC, HPMPO-DAPy or PFA) against the drug-resistant MHV-68 viral clones were determined (Figure 2A and Table S2). The compound concentration to reduce virus-induced CPE by 50% compared to the untreated control, i.e., EC50, was calculated for the drug-resistant viruses. The viral variants Y383SGCV and Q827RHPMP-5azaC were resistant to GCV (4-fold) and HPMP-5azaC (5- and 56-fold, respectively) and were cross-resistant to KAY-2-41 (3-fold), CDV (9- and 40-fold, respectively) and HPMPO-DAPy (22- and 24-fold, respectively). The viral variant Y383SGCV also demonstrated cross-resistance to HDVD (2-fold) and PFA (3-fold), and Q827RHPMP-5azaC was hypersensitive to PFA (0.4-fold). The viral variants G302WPFA, K442TPFA and G302W+K442TPFA were resistant to PFA (2-, 2- and 3-fold, respectively) and had cross-resistance to GCV (4-, 2- and 3-fold, respectively), CDV (5-, 3- and 2-fold, respectively), HPMP-5azaC (2-fold) and HPMPO-DAPy (9-, 2- and 7-fold, respectively). Viral variants G302WPFA and G302W+K442TPFA were resistant to HDVD (3-fold), and the viral variant G302WPFA was resistant to KAH-39-139 (5-fold), while the other selected clones under PFA remained equally sensitive to this drug. None of the stated mutants showed resistance to ACV or BVDU.
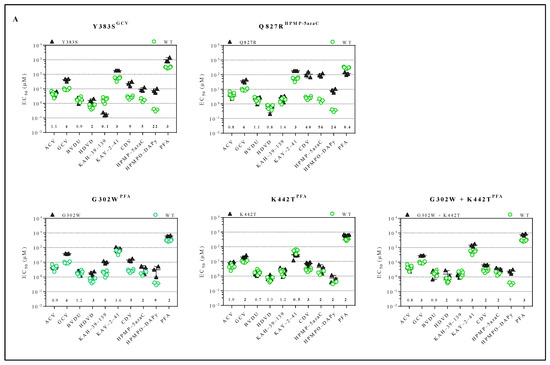
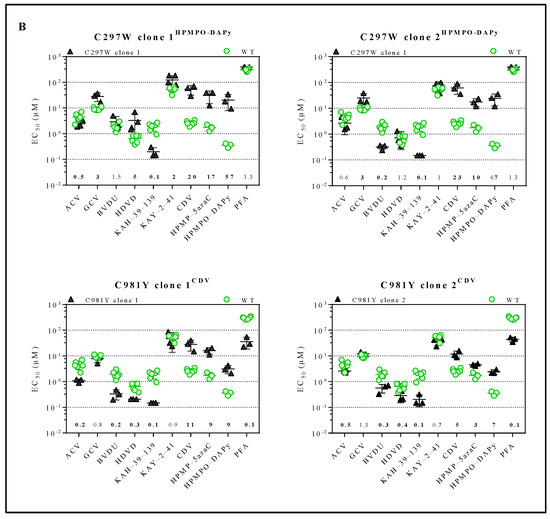
Figure 2.
Drug susceptibility profile of MHV-68 wild-type and drug-resistant clones. (A) Drug susceptibility profile of viral variants Y383SGCV, Q827RHPMP-5azaC, G302WPFA, K442TPFA and G302W+K442TPFA. (B) Drug susceptibility profile of 2 different viral clones of mutator phenotype viruses C297WHPMPO-DAPy and C981YCDV. In each graph, the wild-type virus is marked with a circle (green), and the drug-resistant virus is marked with a triangle (black). The fold resistance, calculated as the EC50 of the drug-resistant virus to the wild-type virus, is marked at the bottom of the graph. MHV-68 viral clones were considered drug-resistant or hypersensitive at ≥2-fold change in EC50 (bold).
3.2. MHV-68 Mutants Selected under Pressure of CDV or HPMPO-DAPy Were Associated with a Mutator Phenotype Virus
In contrast to Y383SGCV and Q827RHPMP-5azaC, G302WPFA, K442TPFA and G302W+K442TPFA genotyping of C297WHPMPO-DAPy (Sanger sequencing) and C981YCDV (NGS) showed multiple spontaneous mutations arising throughout the viral PK, TK and DP genes, suggesting that these substitutions were associated with a mutator phenotype virus (Table S3).
The C297WHPMPO-DAPy amino acid change is in the 3′–5′exonuclease domain (yellow), and C981YCDV is in the thumb domain (green) of the viral DP (Figure 1A). Amino acid changes at positions C297, G302, and Y383 are located closely together in the 3′–5′ exonuclease domain, as depicted in Figure 1B. The amino acid substitution C297W is visualized in Figure 1C, where the change of a cysteine to tryptophan appears to induce stereochemical hindrance, which may lead to an impaired function of the 3′–5′ exonuclease domain, giving rise to a mutator phenotype virus. The Y383S amino acid change is depicted in Figure 1D, where no stereochemical hindrance appears to occur.
For each mutator phenotype virus, the drug susceptibility profile of two viral clones (C297WHPMPO-DAPy clones 1 and 2, and C981YCDV clones 1 and 2) was determined (Figure 2B and Table S2). Interestingly, C297WHPMPO-DAPy clones 1 and 2 had different drug susceptibility profiles. C297WHPMPO-DAPy clones 1 and 2 were both resistant to GCV (3-fold), CDV (20- and 23-fold, respectively), HPMP-5azaC (17- and 10-fold, respectively) and HPMPO-DAPy (57- and 67-fold, respectively) and demonstrated hypersensitivity to KAH-39-139 (0.1-fold). C297WHPMPO-DAPy clone 1 remained equally sensitive to BVDU, while clone 2 was hypersensitive (0.2-fold). C297WHPMPO-DAPy clone 1 was resistant to HDVD (5-fold) and KAY-2-41 (2-fold), while clone 2 remained equally sensitive. C981YCDV clones 1 and 2 had a similar drug susceptibility profile and demonstrated resistance to nucleotide analogues (CDV (11- and 5-fold, respectively), HMP-5azaC (9- and 3-fold, respectively) and HPMPO-DAPy (9- and 7-fold, respectively)). Both viral clones were hypersensitive to ACV (0.3- and 0.5-fold), BVDU (0.2- and 0.3-fold), KAH-39-139 (0.1-fold) and PFA (0.1-fold).
3.3. Without Antiviral Pressure, the Viral Fitness of Q827RHPMP-5azaC, G302WPFA, K442TPFA and G302W+K442TPFA Remained Equal to Wild-Type Virus, While Y383SGCV Had an Increased Viral Fitness
The replication capacity of MHV-68 drug-resistant clones versus the wild-type virus was determined by dual infection competition assays. A mixture containing equal infectious units of wild-type and drug-resistant viruses was cultured for 5 days. The composition of the viral population in the absence of antivirals was determined by NGS at the time of infection and after 5 days of growth. The replication capacity of each viral clone was evaluated by calculating the 1 + s relative fitness (RF) values (Table S4).
Without antiviral pressure, there was no significant increase or decrease in the viral fitness of Q827RHPMP-5azaC (RF = 1.08), G302WPFA (RF = 0.98), K442TPFA (RF = 1.02) or G302W+K442TPFA (RF = 0.97) compared to the untreated wild-type virus. Interestingly, Y383SGCV had an increased fitness compared to the untreated wild-type virus (RF = 1.33, p = 0.009).
3.4. Antiviral Drug Treatment Altered the Viral Fitness of Y383SGCV, Q827RHPMP-5azaC, G302WPFA, K442TPFA and G302W+K442TPFA
The impact of antiviral treatment on the replication capacity was evaluated by competition of the viruses in the presence of various antiviral drugs (Figure 3A and Table S4). Y383SGCV preserved increased viral fitness under the pressure of ACV, GCV, HDVD, CDV and PFA and was equally as fit as the wild-type virus under the pressure of BVDU. Q827RHPMP-5azaC viral fitness increased under the pressure of GCV, CDV and HPMP-5azaC and diminished in the presence of PFA. The three viral clones selected under PFA (G302WPFA, K442TPFA and G302W+K442TPFA) increased in viral fitness under the pressure of GCV, HDVD, CDV and PFA. G302WPFA and G302W+K442TPFA populations diminished under the pressure of ACV, while K442TPFA increased under the pressure of ACV.
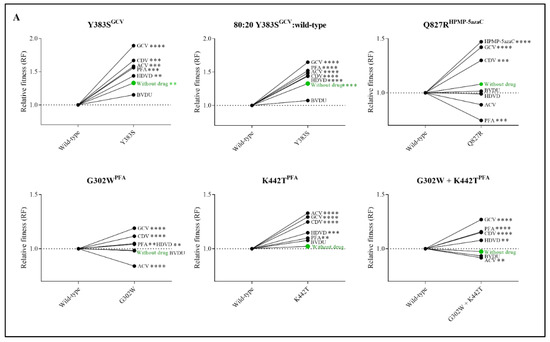
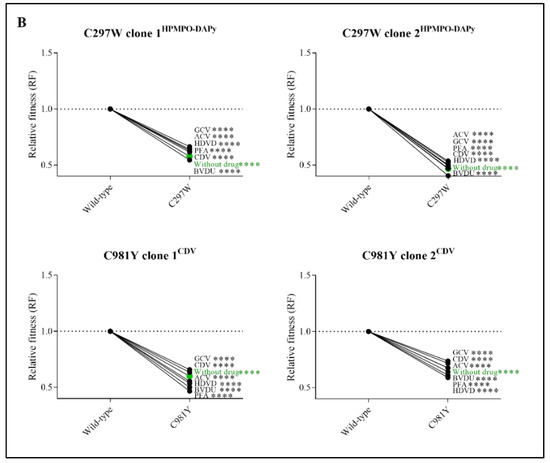
Figure 3.
Relative fitness (RF) of MHV-68 wild-type (WT) and drug-resistant clones (A) or mutator phenotype viruses (B) in growth competition assays in the absence and presence of antiviral drugs. Competition between mutant versus wild-type virus was evaluated without drug (in green) and in the presence of ACV (9 µM), GCV (8 µM), BVDU (1.50 µM), HDVD (0.20 µM), CDV (1.60 µM) or PFA (160 µM). Additionally, the viral mutant Y383SGCV was evaluated with a ratio of 80:20 of Y383SGCV/MHV-68 wild-type. Values represent the means of the viral frequencies of two biological replicates, determined by NGS. Statistical significance was calculated using one-way Anova (multiple comparisons to MHV-68 wild-type virus with the Dunnett test. p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****)).
Due to the increased viral fitness of clone Y383SGCV, viral fitness was further investigated using an 80:20 ratio of Y383SGCV/MHV-68 wild-type (Figure 3A and Table S4). Altering the ratio of mutant versus wild-type viruses did not affect the viral fitness pattern. Without antiviral pressure, Y383SGCV was more fit compared to the wild-type virus (RF = 1.33, p < 0.0001). Additionally, Y383SGCV was more fit under the pressure of antivirals (ACV (RF = 1.49, p < 0.0001), GCV (RF = 1.65, p < 0.0001), HDVD (RF = 1.44, p < 0.0001), CDV (RF = 1.45, p < 0.0001) and PFA (RF = 1.52, p < 0.0001)), with the exception of BVDU, where both viruses grew equally (RF = 1.07, p = 0.17).
3.5. Relative Fitness Strongly Correlated with Drug Resistance Levels for Y383SGCV, Q827RHPMP-5azaC, G302WPFA and G302W+K442TPFA, and Moderately for K442TPFA
To understand the connection between viral fitness and drug resistance, changes in viral fitness were analyzed as a function of the fold resistance using Spearman’s rank-order correlation test (Figure 4A). Q827RHPMP-5azaC, G302WPFA and G302W+K442TPFA had a very strong positive correlation between relative fitness and the degree of drug resistance (rs = 0.96, p < 0.05, rs = 0.94, p < 0.05 and rs = 1.00, p < 0.05, respectively). Y383SGCV (rs = 0.77, p = 0.10) had a strong correlation between relative fitness and drug resistance, and K442TPFA (rs = 0.41, p = 0.43) had a moderate correlation.
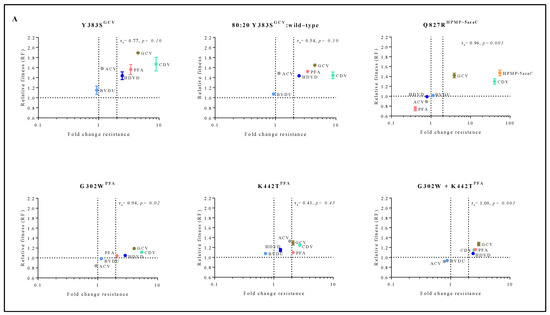
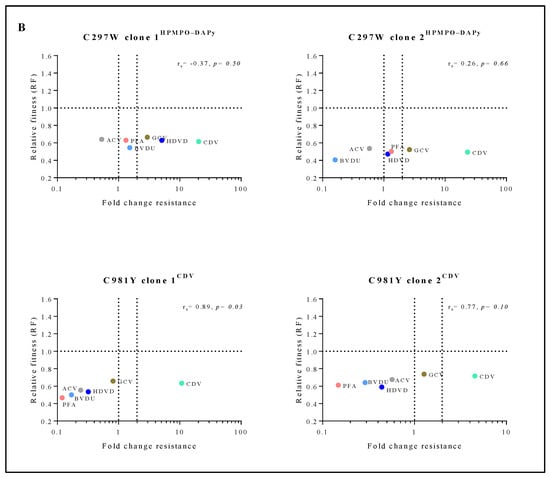
Figure 4.
Correlation between relative fitness and drug resistance levels of MHV-68 viral clones bearing amino acid substitutions in the viral DP (A) or mutator phenotype viruses (B). Spearman’s rank-order correlation test was used to calculate the strength and direction of the relationship between antiviral drug resistance (fold resistance, as shown in Figure 2A,B) and relative fitness (RF, as shown in Figure 3A,B). Spearman’s correlation coefficient (ρ, also signified by rs) measures the strength and direction of association between two ranked variables (rs between 0.00 and 0.19 (very weak), 0.20 and 0.39 (weak), 0.40 and 0.59 (moderate), 0.60 and 0.79 (strong) and 0.80 and 1.0 (very strong)).
3.6. Mutator Phenotype Viruses Had a Severely Impaired Viral Fitness in the Absence and Presence of Antivirals
Competitive fitness studies for mutator phenotype viruses C297WHPMPO-DAPy and C981YCDV were performed using two different viral clones (C297WHPMPO-DAPy clones 1 and 2; C981YCDV clones 1 and 2). Without antiviral pressure, C297WHPMPO-DAPy clones 1 and 2 had a severely impaired replication capacity (RF = 0.58, p < 0.0001 and RF= 0.47, p < 0.0001, respectively). Likewise, C981YCDV clones 1 and 2 had a diminished replication capacity (RF = 0.60, p < 0.0001 and RF = 0.67, p < 0.0001). There was no increase in viral fitness under antiviral pressure (Figure 3B and Table S4).
During dual infection competition experiments, monocultures of both the wild-type virus and resistant virus were included as controls. Monoculture of the C981YCDV clone 2 in the viral fitness experiments led to the emergence of a second distinct viral population (G302W). Although not present in the original stock, the mutation G302W (±7%) arose in the untreated C981YCDV clone 2 (93%) viral population. Under the pressure of PFA, the pro-portion of G302W in the population increased to 45% with a corresponding decrease in C981Y to 48%. Under the pressure of HDVD, G302W increased to 21% of the total population (C981Y 74%). Subsequent cloning of the mixed viral population showed that G302W overgrew the mixed viral population, resulting in a pure G302W population with no detectable C981Y nor spontaneous mutations present, indicating an escape mutant. For the three other mutator phenotype viral clones, no escape viruses were detected.
3.7. Mutator Phenotype Virus C981YCDV, but Not C297WHPMPO-DAPy, Demonstrated a Correlation between Relative Fitness and Antiviral Drug Resistance Levels
The mutator phenotype viruses C981YCDV clones 1 and 2 demonstrated a very strong to strong positive correlation between relative fitness and antiviral resistance (rs = 0.89, p < 0.05 and rs = 0.77, p = 0.10, respectively) (Figure 4B). This is in contrast to C297WHPMPO-DAPy clones 1 and 2 that demonstrated a very weak negative (rs = −0.37, p = 0.50) or positive correlation (rs = 0.26, p = 0.66), respectively.
3.8. Investigation of the Mutation Frequency in the Viral PK, TK and DP
The mutation frequency of three clones of C297WHPMPO-DAPy and three clones of C981YCDV was investigated by NGS. DNA extracts were prepared at passage 0 and after three rounds of amplification, which allowed evaluating the appearance of new mutations. When comparing the amplified virus to the starting stock, the C297WHPMPO-DAPy clones had 34 newly generated mutations over the span of the viral PK, TK and DP, and the C981YCDV clones generated 14 new mutations (Table 1).

Table 1.
Mutation frequency in the viral PK, TK and DP of mutator phenotype viral clones.
C297WHPMPO-DAPy viral clones had a short intermutational distance, mostly ranging from 0 to 500 bp (Figure 5A,B). C981YCDV viral clones had a larger intermutational distance from 100 to 1500 bp (Figure 5C,D). Between ungrown (Figure 5A (C297W) and Figure 5C (C981Y)) and amplified viruses (Figure 5B (C297W) and Figure 5D (C981Y)), no shift in intermutational distance was found. The spontaneous mutations arising in C297WHPMPO-DAPy and C981YCDV viral clones were aligned with HSV-1, HSV-2, VZV and HCMV, and the positions were compared to current information regarding herpesvirus naturally occurring polymorphisms and drug resistance mutations (Tables S5 and S6). Interestingly, various spontaneous mutations were located in conserved regions and were at homologue positions known to confer drug resistance, explaining the variation in drug susceptibility profiles between the two C297WHPMPO-DAPy viral clones (Figure 2B and Table S2).
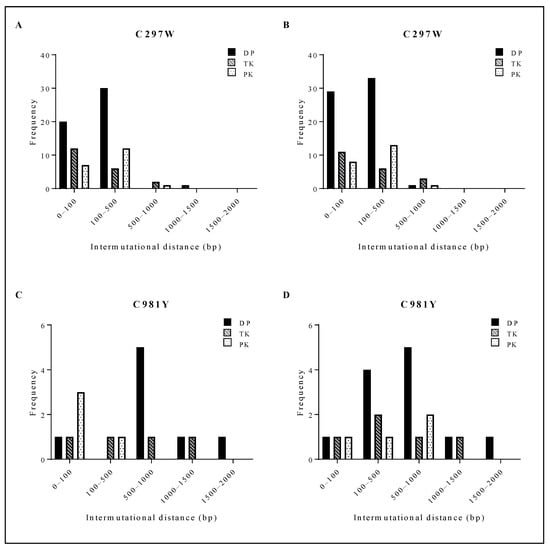
Figure 5.
Distribution of the intermutational distance in the viral PK, TK and DP of C297W and C981Y. For both C297W and C981Y, next-generation sequencing of the viral DP, TK and PK was performed on three viral clones at two different points of expansion (i.e., three clones #0 and three clones #3). (A) Intermutational distance of 3 viral clones bearing C297W amino acid change without amplification. (B) Intermutational distance of 3 viral clones bearing C297W amino acid change after 3 rounds of amplification. (C) Intermutational distance of 3 viral clones bearing C981Y amino acid change without amplification. (D) Intermutational distance of 3 viral clones bearing C981Y amino acid change after 3 rounds of amplification.
3.9. CRISPR/Cas9 Genome Editing Was a Successful Strategy for the Generation of MHV-68 Virus with a Missense Mutation Resulting in a Mutator Phenotype Virus
To formally assess the association of the novel identified mutation C297W with a mutator phenotype, i.e., increased frequency of spontaneous mutations, the generation of a recombinant virus is needed to be sure that this mutation actually confers the mutator phenotype. We used a CRISPR/Cas9-based gene editing approach to generate the C297W amino acid change in MHV-68 (Figure S1). The cysteine at position 297 is extremely conserved among α-, β- and γ-herpesviruses (Figure 6). HEK293T cells were transfected with the CRISPR/Cas9 plasmid, and after 24 h, the cells were infected with MHV-68. Multiple concentrations of virus were used as MHV-68 is able to infect HEK293T cells but is not able to generate visible cytopathic effects (CPE). To aid efficient and precise genome editing, the asymmetric template was designed with the PAM sequence as close as possible to the target site.

Figure 6.
Alignment of MHV-68 DP with human DNA polymerase delta, human DNA polymerase epsilon, HSV-1 DP, HSV-2 DP, VZV DP, HCMV DP, EBV DP, KSHV DP, herpesvirus saimiri DP, rhesus rhadinovirus DP, S. Cerevisiae DNA polymerase delta and S. Cerevisiae DNA polymerase epsilon. The cysteine at position 297 (marked in bold) is in the Exo I domain (green box) of MHV-68 DP, and it is extremely conserved among B family DNA polymerases. It is part of the FDIEC amino acid sequence, in which amino acid changes are frequently associated with the formation of a mutator phenotype. The amino acid positions associated with a mutator are marked in bold red and encompass the following amino acid changes: human DNA polymerase delta: D316H, D316G, C319Y, P327L; human DNA polymerase epsilon: D275A, P286R; HSV-1 DP: D368A, E370A; S. Cerevisiae DNA polymerase delta: D321A, E323A (stated in gray) [51,52,53,54,55,56,57,58,59,60,61,62,63,64,65].
Our genome editing approach using 25 PFU/well resulted in ~17% efficiency as 3 out of 17 tested clones had the desired mutation (Sanger sequencing). The PK, TK and DP of three C297WCRISPR clones and three wild-type clones (obtained after unsuccessful genome editing) were analyzed by NGS. The three C297WCRISPR clones had spontaneous mutations arising through the viral PK, TK and DP, validating the mutator phenotype (Table S7). Wild-type clones 1 and 2 had no mutations in the viral PK or TK.
Nevertheless, they had a minor fraction with insertions/deletions at position 888 in the viral DP, likely acquired through non-homologous end joining (NHEJ) repair, which is an error-prone DNA repair mechanism, of the Cas9 cut site. Wild-type clone 3 had no mutations in the viral PK, TK, or DP.
4. Discussion
In this research, we investigated the resistance of the γ-herpesvirus MHV-68 to several nucleoside (ganciclovir), nucleotide (cidofovir, HPMP-5azaC and HPMPO-DAPy) and pyrophosphate analogues (foscarnet). We evaluated the impact of these mutations on the viral replication capacity by dual infection growth assays and determined the composition of the mixed viral population by NGS. The use of NGS allowed determining the replication capacity of two viral variants under identical conditions with a sensitivity of ≥1%.
The Y383SGCV change is in the exonuclease domain of the viral DP (Exo II) and the viral clone bearing this change had resistance to the purine nucleoside GCV, but not ACV, and cross-resistance to CDV and PFA. Our earlier investigations characterized ACV resistance in MHV-68 and demonstrated a mutation in the activating enzyme (viral PK), but not DP. Similar observations have been made regarding HCMV GCV resistance, where mutations associated with GCV resistance frequently arise in the 3′–5′ exonuclease domain (albeit mutations in the HCMV PK are also reported) [66,67,68]. This could possibly be attributed to the different mechanisms of action of ACV versus GCV. ACV-TP does not possess a hydroxyl group in the 3′ position and is an obligate chain terminator [69], while the presence of a 3′ hydroxyl group in GCV-TP allows for the integration of one additional nucleotide (short-chain terminator) [1].
PFA-resistant clone G302WPFA maps to the exonuclease domain (Exo I), and K442TPFA is adjacent to the δ-C region. The third PFA-resistant viral clone is a double mutant carrying G302W+K442TPFA. G302WPFA, K442TPFA and G302W+K442TPFA viral clones proved to be resistant to PFA, GCV and CDV, a combination that can be found for mutations located in domain III of the viral DP in HCMV, not in the exonuclease domain or δ-C region [70]. However, for HCMV, few substitutions in the exonuclease region have been implicated in clinical PFA resistance (N495K, D515Y) [71,72] or even observed after PFA exposure in cell culture [73].
C297WHPMPO-DAPy (Exo I) and Q827RHPMP-5azaC (thumb) were resistant to HPMPO-DAPy and HPMP-5azaC, respectively, and cross-resistant to GCV and CDV. This in accordance with reports of HCMV resistance where mutations conferring resistance to ganciclovir and cross-resistance to cidofovir can be found in the exonuclease and thumb domains [74]. C981YCDV, which is also located in the thumb domain, conferred resistance to CDV and other nucleotides, but not to GCV.
Resistance mutations normally arise at the active site of the enzyme, thereby affecting substrate binding and the catalysis rates. Frequently, the affected enzyme is essential to the viral function, which leads to a lower viral fitness [75,76,77]. However, there is scarce information regarding the impact of drug resistance mutations on the viral replication capacity in herpesviruses. Studies on HCMV and HSV-1 demonstrated that drug-resistant herpesviruses have an equal or impaired fitness compared to the wild-type virus, with no reports of increased viral fitness [43,78]. Our earlier work focused on the impact of drug resistance mutations in the MHV-68 viral PK or TK on viral replication capacity and demonstrated that, without antiviral pressure, there was a significant reduction in the viral fitness of a PK:GT-deletion frameshift virus and a decrease, albeit not significant, in the viral fitness of a TK:C-insert frameshift virus [48]. These findings are in line with the current understanding of the functions of the γ-herpesvirus PK and TK, where the viral PK plays an essential role in lytic replication and the viral TK is dispensable [79,80,81,82,83]. The viral DP is an essential enzyme during lytic replication. Nevertheless, in our current research, we showed that in vitro, viruses with a drug resistance mutation in the MHV-68 viral DP had an equal viral fitness compared to the wild-type virus in the absence of antiviral pressure (G302WPFA, K442TPFA, G302W+K442TPFA and Q827RHPMP-5azaC) or increased viral fitness as demonstrated with viral clone Y383SGCV (Figure 3A and Table S4). In the presence of antivirals, drug-resistant viruses had a growth advantage over the wild-type virus, and this growth advantage correlated with antiviral resistance (Figure 4A).
The Y383GCV change maps to the active site of the 3′–5′ exonuclease (Exo II). Investigations by Chen et al. demonstrated that mutations associated with GCV resistance in the HCMV exonuclease domain reduced exonuclease activity, making the polymerase less likely to terminate chain elongation. This allowed further polymerase activity and chain elongation after GCV-TP incorporation and generation of full-length gene products [66]. During DNA synthesis, also correctly incorporated nucleotides are frequently excised [84]. Thus, we can hypothesize that the mutation Y383SGCV affects the exonuclease activity, reducing excision of correct nucleotides and/or elongating the viral DNA after antiviral incorporation, consequently leading to a growth advantage of the drug-resistant viral clone over the wild-type virus.
This study identified C297WHPMPO-DAPy and C981YCDV as mutator phenotype viruses. Mutator phenotype viruses can arise through different mechanisms as DNA replication fidelity depends on a combination of nucleotide selection, exonucleolytic proofreading and post-replicative mismatch repair (MMR) [52]. Although frequently described in RNA viruses, limited herpesvirus mutator or antimutator phenotypes have been described in the literature (HSV-1 mutators: Y577H+D581A [54], D368A, E370A, D471A, Y538S, U557S, Y557F, D581A [55,69]; CMV mutator: D413A [85,86]). Mutation C297WHPMPO-DAPy is in the 3′–5′ exonuclease domain, a region that harbors many identified mutator positions (Figure S2). Position C297 is extremely conserved among DNA polymerases, i.e., α-, β- and γ-herpesviruses, human DNA polymerase delta, human DNA polymerase epsilon and S. Cerevisiae DNA polymerase delta. Moreover, an amino acid change at the exact homologous position (C319) of human DNA polymerase δ was proposed to be the cause of the altered mutation frequency in ultra-hypermutated malignant brain tumors (Figure 6) [64]. Both C297W in MHV-68 DP and C319Y in human DP δ are changes to an aromatic amino acid. The 3′–5′ exonuclease domain has a proofreading capacity, i.e., it catalyzes the removal of mismatched base pairs at the primer terminus, producing deoxyribonucleotide monophosphates [87]. Mutations in the 3′–5′ exonuclease domain can then impair the proofreading capacity and result in an increased mutation frequency, i.e., mutator phenotypes [88]. If the mutations are left unrepaired, they can be propagated in the next round of amplification when the mutated strand acts as a template [51]. In eukaryotes, the distorted sequence is subsequently recognized by DNA mismatch repair mechanisms (MMR) that perform excision and re-synthesis of the nascent strand, a process that is lacking in herpesviruses [63].
C981YCDV, on the other hand, is in the thumb region, for which much fewer mutators and antimutators are known. At this moment, two different mechanisms of mutator generation in the thumb region are described. (i) Mutators in the thumb domain are associated with an altered nucleotide selectivity [89], e.g., in Sulfolobus solfataricus DNA polymerase Dpo4, mutation A141D in the thumb domain led to a higher nucleotide incorporation rate, accompanied by an incorrect incorporation of dNTPs, leading to a mutator phenotype virus [90]. (ii) It has been demonstrated that, despite being organized in two different domains, the 3′–5′ exonuclease and thumb domains are structurally and functionally interconnected. More specifically, the putative exonuclease site is in the groove formed between the 3′–5′ exonuclease domain and the tip of the thumb domain [91]. In bacteriophage T4, mutations in the thumb domain (A737V and A777V) were associated with an antimutator phenotype. The mutant strains had an increased hydrolysis of dNTPs per base pair synthesized compared to the wild-type combined with less processivity and less efficient strand displacement synthesis. On the contrary, mutator phenotypes lying in the thumb region hydrolyze less dNTPs per base pair synthesized than the wild-type, indicating the importance of the balance between proofreading and primer extension [52]. It is interesting to note that selection of cidofovir-resistant vaccinia virus also led to a mutator phenotype virus (A684V located in the Pol III catalytic domain) [92].
Monoculture of C981YCDV clone 3 in the viral fitness experiment led to a second viral population, containing a single G302W mutation in the viral DP. In the untreated dual infection experiment, G302W was present at a low frequency but increased profoundly under the pressure of PFA and HDVD. Interestingly, in this project, we identified C981YCDV as hypersensitive to PFA and mutation G302W as conferring resistance to PFA (Figure 2 and Table S2). Mutator populations restore the mutation frequency when the mutation rate exceeds 10−3 (error-induced extinction). This occurs by acquiring a compensating mutation [54], reversion to the wild-type virus (true revertant) [93], formation of an escape mutant [94] or generating antimutator mutations [51]. In our experiments, G302W was an escape mutant that overgrew C981Y with no detectable C981Y nor remaining spontaneous mutations. The disappearance of C981Y indicated that G302W was not a compensating mutation, and the lack of a detectable wild-type virus excluded a true revertant. The absence of remaining spontaneous mutations and intact viral fitness (Figure 3B and Table S4) indicated that G302W was not an antimutator position.
Our results regarding the severely impaired fitness of MHV-68 mutator phenotype viruses correspond to the current state of the art, i.e., an alteration of the mutation frequency of a virus (mutator or antimutator) results in an impaired replication capacity [95,96]. Additionally, we highlighted that for these viruses, antiviral resistance did not result in increased fitness. Despite that both C297WHPMPO-DAPy and C981YCDV are mutator viruses, the mutation frequency differs between them, thereby resulting in different strength mutators. C297W showed a higher mutation frequency (Table 1) and a shorter intermutational distance (Figure 5A,B), compared to C981Y (Figure 5C,D).
The fact that different clones of the mutator phenotype viruses have different sensitivity profiles indicates that some of the mutations arising spontaneously contribute to drug resistance. This would have important consequences in the clinic if such mutator phenotypes arise in human herpesviruses.
In viruses, CRISPR/Cas9 genome editing has thus far been used to disrupt specific viral genes [97,98]; however, to our knowledge, this is the first report of a specific amino acid change in a virus generated by CRISPR/Cas9. We validated the mutator phenotype virus associated with amino acid change C297W in the 3′–5′ exonuclease domain by employing a CRISPR/Cas9 approach. Subsequent NGS analysis of the three generated C297WCRISPR clones confirmed the mutator phenotype of the genome-edited clones as spontaneous mutations were identified in the viral PK, TK and DP (Table S7). There were no detectable off-target effects in the rest of the viral genome, demonstrated by NGS of three wild-type viruses obtained after unsuccessful genome editing.
5. Conclusions
To summarize, our research contributes to the field of antiviral resistance and allows for a comprehensive investigation of the impact of mutations in the DP on viral fitness. While some mutations confer an intrinsic higher replication capacity such as Y383SGCV, others have a growth advantage over the wild-type virus in the presence of antivirals (G302WPFA, K442TPFA, G302W+K442TPFA and Q827RHPMP-5azaC). Our investigations led us to the identification of two mutations associated with a mutator phenotype virus, i.e., C297WHPMPO-DAPy, located in the 3′–5′ exonuclease domain, and C981YCDV, located in the thumb domain. Both mutator phenotype viruses had a severely impaired replication capacity, independent from drug resistance patterns. Lastly, we successfully confirmed C297W to be associated with a mutator phenotype virus using CRISPR/Cas9 genome editing.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/v13060985/s1, Figure S1: Steps followed in the generation of C297W DNA polymerase gene-edited virus, Figure S2: Alignment of MHV-68 DP with human DNA polymerase delta, human DNA polymerase epsilon, HSV-1 DP, HSV-2 DP, VZV DP, CMV DP, EBV DP, KSHV DP, herpesvirus saimiri DP, rhesus rhadinovirus DP, S. Cerevisiae DNA polymerase delta and S. Cerevisiae DNA polymerase epsilon, Table S1: Primers used for PCR amplification and Sanger sequencing of the viral PK, TK and DP, Table S2: The inhibitory effects of a range of antivirals on the replication of MHV-68 wild-type and drug-resistant viruses in Vero cells, Table S3: Spontaneous mutations appearing in the viral PK, TK or DP of different viral clones of C297W and C981Y that led to the hypothesis that amino acid changes C297W and C981Y are associated with mutator phenotype viruses, Table S4: Relative fitness (RF) of MHV-68 wild-type (WT) and drug-resistant clones (4A) or mutator phenotype viruses (4B) in growth competition assays in the absence and presence of antiviral drugs, Table S5: Alignment of C297W spontaneous mutations with HSV-1, HSV-2, VZV and HCMV in the viral PK, TK and DP in the mutation frequency experiment, Table S6: Alignment of C981Y spontaneous mutations with HSV-1, HSV-2, VZV and HCMV in the viral PK, TK and DP in the mutation frequency experiment, Table S7: Alignment of C297WCrispr spontaneous mutations with HSV-1, HSV-2, VZV and HCMV in the viral PK, TK and DP.
Author Contributions
Conceptualization, E.T., R.S. and G.A.; methodology, E.T., A.T., S.G. and G.A.; software, E.T., A.T., D.T. and S.G.; validation, E.T., A.T., S.G. and G.A.; formal analysis, E.T. and G.A.; investigation, E.T., D.T., R.S. and G.A.; resources, E.T., R.S. and G.A.; data curation, E.T. and G.A.; writing—original draft preparation, E.T.; writing—review and editing, E.T., A.T., S.G., D.T., R.S. and G.A.; visualization, E.T., D.T. and G.A.; supervision, R.S. and G.A.; project administration, E.T.; funding acquisition, E.T., R.S. and G.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a Strategic Basic Research Grant from FWO, grant number 1S04516N.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. All data generated or analyzed during this study are included in this published article (and its Supplementary Information Files).
Acknowledgments
We thank Ellen De Waegenaere and Arif Sahin for their excellent technical support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Topalis, D.; Gillemot, S.; Snoeck, R.; Andrei, G. Distribution and effects of amino acid changes in drug-resistant alpha and beta herpesviruses DNA polymerase. Nucleic Acids Res. 2016, 44, 9530–9554. [Google Scholar] [CrossRef] [PubMed]
- Renner, D.W.; Szpara, M.L. Impacts of Genome-Wide Analyses on Our Understanding of Human Herpesvirus Diversity and Evolution. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Cesarman, E. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett. 2011, 305, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Jha, H.C.; Banerjee, S.; Robertson, E.S. The Role of Gammaherpesviruses in Cancer Pathogenesis. Pathogens 2016, 5, 18. [Google Scholar] [CrossRef]
- El-Sharkawy, A.; Al Zaidan, L.; Malki, A. Epstein-Barr Virus-Associated Malignancies: Roles of Viral Oncoproteins in Carcinogenesis. Front. Oncol. 2018, 8, 265. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)—An update. Curr. Opin. Virol. 2013, 3, 238–244. [Google Scholar] [CrossRef]
- Manners, O.; Murphy, J.C.; Coleman, A.; Hughes, D.J.; Whitehouse, A. Contribution of the KSHV and EBV lytic cycles to tumourigenesis. Curr. Opin. Virol. 2018, 32, 60–70. [Google Scholar] [CrossRef]
- Munz, C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef]
- Hong, G.K.; Gulley, M.L.; Feng, W.H.; Delecluse, H.J.; Holley-Guthrie, E.; Kenney, S.C. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J. Virol. 2005, 79, 13993–14003. [Google Scholar] [CrossRef]
- Cohen, M.; Vistarop, A.G.; Huaman, F.; Narbaitz, M.; Metrebian, F.; De Matteo, E.; Preciado, M.V.; Chabay, P.A. Epstein-Barr virus lytic cycle involvement in diffuse large B cell lymphoma. Hematol. Oncol. 2018, 36, 98–103. [Google Scholar] [CrossRef]
- Jones, R.J.; Seaman, W.T.; Feng, W.H.; Barlow, E.; Dickerson, S.; Delecluse, H.J.; Kenney, S.C. Roles of lytic viral infection and IL-6 in early versus late passage lymphoblastoid cell lines and EBV-associated lymphoproliferative disease. Int. J. Cancer 2007, 121, 1274–1281. [Google Scholar] [CrossRef]
- Beatty, P.R.; Krams, S.M.; Martinez, O.M. Involvement of IL-10 in the autonomous growth of EBV-transformed B cell lines. J. Immunol. 1997, 158, 4045–4051. [Google Scholar] [PubMed]
- Jones, K.D.; Aoki, Y.; Chang, Y.; Moore, P.S.; Yarchoan, R.; Tosato, G. Involvement of interleukin-10 (IL-10) and viral IL-6 in the spontaneous growth of Kaposi’s sarcoma herpesvirus-associated infected primary effusion lymphoma cells. Blood 1999, 94, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, P.; Sakakibara, S.; Tosato, G. Contribution of viral and cellular cytokines to Kaposi’s sarcoma-associated herpesvirus pathogenesis. J. Leukoc Biol. 2008, 84, 994–1000. [Google Scholar] [CrossRef][Green Version]
- Hong, G.K.; Kumar, P.; Wang, L.; Damania, B.; Gulley, M.L.; Delecluse, H.J.; Polverini, P.J.; Kenney, S.C. Epstein-Barr virus lytic infection is required for efficient production of the angiogenesis factor vascular endothelial growth factor in lymphoblastoid cell lines. J. Virol. 2005, 79, 13984–13992. [Google Scholar] [CrossRef]
- Quinlivan, E.B.; Zhang, C.; Stewart, P.W.; Komoltri, C.; Davis, M.G.; Wehbie, R.S. Elevated virus loads of Kaposi’s sarcoma-associated human herpesvirus 8 predict Kaposi’s sarcoma disease progression, but elevated levels of human immunodeficiency virus type 1 do not. J. Infect. Dis. 2002, 185, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Coen, N.; Duraffour, S.; Snoeck, R.; Andrei, G. KSHV targeted therapy: An update on inhibitors of viral lytic replication. Viruses 2014, 6, 4731–4759. [Google Scholar] [CrossRef]
- Colombini, E.; Guzzo, I.; Morolli, F.; Longo, G.; Russo, C.; Lombardi, A.; Merli, P.; Barzon, L.; Murer, L.; Piga, S.; et al. Viral load of EBV DNAemia is a predictor of EBV-related post-transplant lymphoproliferative disorders in pediatric renal transplant recipients. Pediatr. Nephrol. 2017, 32, 1433–1442. [Google Scholar] [CrossRef]
- Fellner, M.D.; Durand, K.A.; Solernou, V.; Bosaleh, A.; Balbarrey, Z.; Garcia de Davila, M.T.; Rodriguez, M.; Irazu, L.; Alonio, L.V.; Picconi, M.A. Epstein-Barr virus load in transplant patients: Early detection of post-transplant lymphoproliferative disorders. Rev. Argent. Microbiol. 2016, 48, 110–118. [Google Scholar] [CrossRef][Green Version]
- Malouf, M.A.; Chhajed, P.N.; Hopkins, P.; Plit, M.; Turner, J.; Glanville, A.R. Anti-viral prophylaxis reduces the incidence of lymphoproliferative disease in lung transplant recipients. J. Heart Lung Transplant. 2002, 21, 547–554. [Google Scholar] [CrossRef]
- AlDabbagh, M.A.; Gitman, M.R.; Kumar, D.; Humar, A.; Rotstein, C.; Husain, S. The Role of Antiviral Prophylaxis for the Prevention of Epstein-Barr Virus-Associated Posttransplant Lymphoproliferative Disease in Solid Organ Transplant Recipients: A Systematic Review. Am. J. Transplant. 2017, 17, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Pagano, J.S.; Whitehurst, C.B.; Andrei, G. Antiviral Drugs for EBV. Cancers 2018, 10, 197. [Google Scholar] [CrossRef]
- Andrei, G.; Trompet, E.; Snoeck, R. Novel Therapeutics for Epstein(-)Barr Virus. Molecules 2019, 24, 997. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.R. Epstein-Barr virus (EBV) reactivation and therapeutic inhibitors. J. Clin. Pathol. 2019, 72, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Crumpacker, C.S. Mechanism of action of foscarnet against viral polymerases. Am. J. Med. 1992, 92, 3S–7S. [Google Scholar] [CrossRef]
- Coen, N.; Duraffour, S.; Naesens, L.; Krecmerova, M.; Van den, O.J.; Snoeck, R.; Andrei, G. Evaluation of novel acyclic nucleoside phosphonates against human and animal gammaherpesviruses revealed an altered metabolism of cyclic prodrugs upon Epstein-Barr virus reactivation in P3HR-1 cells. J. Virol. 2013, 87, 12422–12432. [Google Scholar] [CrossRef]
- Coen, N.; Duraffour, S.; Topalis, D.; Snoeck, R.; Andrei, G. Spectrum of activity and mechanisms of resistance of various nucleoside derivatives against gammaherpesviruses. Antimicrob. Agents Chemother. 2014, 58, 7312–7323. [Google Scholar] [CrossRef]
- Lin, J.C.; De Clercq, E.; Pagano, J.S. Inhibitory effects of acyclic nucleoside phosphonate analogs, including (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine, on Epstein-Barr virus replication. Antimicrob. Agents Chemother. 1991, 35, 2440–2443. [Google Scholar] [CrossRef][Green Version]
- Datta, A.K.; Colby, B.M.; Shaw, J.E.; Pagano, J.S. Acyclovir inhibition of Epstein-Barr virus replication. Proc. Natl. Acad. Sci. USA 1980, 77, 5163–5166. [Google Scholar] [CrossRef]
- Davis, C.L. The antiviral prophylaxis of post-transplant lymphoproliferative disorder. Springer Semin. Immunopathol. 1998, 20, 437–453. [Google Scholar] [CrossRef]
- Casper, C.; Wald, A. The use of antiviral drugs in the prevention and treatment of Kaposi sarcoma, multicentric Castleman disease and primary effusion lymphoma. Curr. Top. Microbiol. Immunol. 2007, 312, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.; Hwang, Y.Y.; Chan, T.S.; Pang, A.W.; Leung, A.Y.; Tse, E.; Kwong, Y.L. Valganciclovir suppressed Epstein Barr virus reactivation during immunosuppression with alemtuzumab. J. Clin. Virol. 2014, 59, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, E.A.; Chillemi, A.C.; Sage, D.R.; Fingeroth, J.D. The Epstein-Barr virus thymidine kinase does not phosphorylate ganciclovir or acyclovir and demonstrates a narrow substrate specificity compared to the herpes simplex virus type 1 thymidine kinase. Antimicrob. Agents Chemother. 1998, 42, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Deville-Bonne, D.; El Amri, C.; Meyer, P.; Chen, Y.; Agrofoglio, L.A.; Janin, J. Human and viral nucleoside/nucleotide kinases involved in antiviral drug activation: Structural and catalytic properties. Antivir. Res. 2010, 86, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Hagemeier, S.R.; Fingeroth, J.D.; Gershburg, E.; Pagano, J.S.; Kenney, S.C. The Epstein-Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J. Virol. 2010, 84, 4534–4542. [Google Scholar] [CrossRef]
- Coen, N.; Singh, U.; Vuyyuru, V.; Van den Oord, J.J.; Balzarini, J.; Duraffour, S.; Snoeck, R.; Cheng, Y.C.; Chu, C.K.; Andrei, G. Activity and mechanism of action of HDVD, a novel pyrimidine nucleoside derivative with high levels of selectivity and potency against gammaherpesviruses. J. Virol. 2013, 87, 3839–3851. [Google Scholar] [CrossRef]
- Coen, N.; Duraffour, S.; Haraguchi, K.; Balzarini, J.; van den Oord, J.J.; Snoeck, R.; Andrei, G. Antiherpesvirus activities of two novel 4’-thiothymidine derivatives, KAY-2-41 and KAH-39-149, are dependent on viral and cellular thymidine kinases. Antimicrob. Agents Chemother. 2014, 58, 4328–4340. [Google Scholar] [CrossRef]
- Kenney, S.C. Reactivation and lytic replication of EBV. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Fujiwara, S. Animal Models of Human Gammaherpesvirus Infections. Adv. Exp. Med. Biol 2018, 1045, 413–436. [Google Scholar] [CrossRef]
- Rajcani, J.; Kudelova, M. Murine herpesvirus pathogenesis: A model for the analysis of molecular mechanisms of human gamma herpesvirus infections. Acta Microbiol. Immunol. Hung. 2005, 52, 41–71. [Google Scholar] [CrossRef]
- Dong, S.; Forrest, J.C.; Liang, X. Murine Gammaherpesvirus 68: A Small Animal Model for Gammaherpesvirus-Associated Diseases. Adv. Exp. Med. Biol. 2017, 1018, 225–236. [Google Scholar] [CrossRef]
- Martin, M.; Goyette, N.; Boivin, G. Contrasting effects on ganciclovir susceptibility and replicative capacity of two mutations at codon 466 of the human cytomegalovirus UL97 gene. J. Clin. Virol. 2010, 49, 296–298. [Google Scholar] [CrossRef]
- Chevillotte, M.; Schubert, A.; Mertens, T.; von Einem, J. Fluorescence-based assay for phenotypic characterization of human cytomegalovirus polymerase mutations regarding drug susceptibility and viral replicative fitness. Antimicrob. Agents Chemother. 2009, 53, 3752–3761. [Google Scholar] [CrossRef][Green Version]
- Pesola, J.M.; Coen, D.M. In vivo fitness and virulence of a drug-resistant herpes simplex virus 1 mutant. J Gen. Virol. 2007, 88, 1410–1414. [Google Scholar] [CrossRef]
- Sharma, P.L.; Crumpacker, C.S. Attenuated replication of human immunodeficiency virus type 1 with a didanosine-selected reverse transcriptase mutation. J. Virol. 1997, 71, 8846–8851. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lerma, J.G.; MacInnes, H.; Bennett, D.; Weinstock, H.; Heneine, W. Transmitted human immunodeficiency virus type 1 carrying the D67N or K219Q/E mutation evolves rapidly to zidovudine resistance in vitro and shows a high replicative fitness in the presence of zidovudine. J. Virol. 2004, 78, 7545–7552. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.A.; Thompson, M.G.; Paintsil, E.; Ricketts, M.; Gedzior, J.; Alexander, L. Competitive fitness of nevirapine-resistant human immunodeficiency virus type 1 mutants. J. Virol. 2004, 78, 603–611. [Google Scholar] [CrossRef]
- Trompet, E.; Topalis, D.; Gillemot, S.; Snoeck, R.; Andrei, G. Viral fitness of MHV-68 viruses harboring drug resistance mutations in the protein kinase or thymidine kinase. Antiviral. Res. 2020, 182, 104901. [Google Scholar] [CrossRef]
- Abram, M.E.; Hluhanich, R.M.; Goodman, D.D.; Andreatta, K.N.; Margot, N.A.; Ye, L.; Niedziela-Majka, A.; Barnes, T.L.; Novikov, N.; Chen, X.; et al. Impact of primary elvitegravir resistance-associated mutations in HIV-1 integrase on drug susceptibility and viral replication fitness. Antimicrob. Agents Chemother. 2013, 57, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Andreatta, K.N.; Goodman, D.D.; Miller, M.D.; White, K.L. Reduced viral fitness and lack of cross-class resistance with integrase strand transfer inhibitor and nucleoside reverse transcriptase inhibitor resistance mutations. Antimicrob. Agents Chemother. 2015, 59, 3441–3449. [Google Scholar] [CrossRef][Green Version]
- Herr, A.J.; Ogawa, M.; Lawrence, N.A.; Williams, L.N.; Eggington, J.M.; Singh, M.; Smith, R.A.; Preston, B.D. Mutator suppression and escape from replication error-induced extinction in yeast. PLoS Genet. 2011, 7, e1002282. [Google Scholar] [CrossRef]
- Herr, A.J.; Williams, L.N.; Preston, B.D. Antimutator variants of DNA polymerases. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 548–570. [Google Scholar] [CrossRef][Green Version]
- Briggs, S.; Tomlinson, I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J. Pathol. 2013, 230, 148–153. [Google Scholar] [CrossRef]
- Sanjuan, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, F.J.; Knopf, C.W. Herpes simplex virus type 1 DNA polymerase. Mutational analysis of the 3’-5’-exonuclease domain. J. Biol. Chem. 1996, 271, 29245–29254. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Tomlinson, I. Replicative DNA polymerase mutations in cancer. Curr Opin Genet Dev. 2014, 24, 107–113. [Google Scholar] [CrossRef]
- Jin, Y.H.; Garg, P.; Stith, C.M.; Al-Refai, H.; Sterling, J.F.; Murray, L.J.; Kunkel, T.A.; Resnick, M.A.; Burgers, P.M.; Gordenin, D.A. The multiple biological roles of the 3’-->5’ exonuclease of Saccharomyces cerevisiae DNA polymerase delta require switching between the polymerase and exonuclease domains. Mol. Cell Biol. 2005, 25, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, H.; Scollon, S.; Reuther, J.; Voicu, H.; Rednam, S.P.; Lin, F.Y.; Fisher, K.E.; Chintagumpala, M.; Adesina, A.M.; Parsons, D.W.; et al. Germline POLE mutation in a child with hypermutated medulloblastoma and features of constitutional mismatch repair deficiency. Cold Spring Harb. Mol. Case Stud. 2019, 5. [Google Scholar] [CrossRef]
- Bellido, F.; Pineda, M.; Aiza, G.; Valdes-Mas, R.; Navarro, M.; Puente, D.A.; Pons, T.; Gonzalez, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016, 18, 325–332. [Google Scholar] [CrossRef]
- Shinbrot, E.; Henninger, E.E.; Weinhold, N.; Covington, K.R.; Goksenin, A.Y.; Schultz, N.; Chao, H.; Doddapaneni, H.; Muzny, D.M.; Gibbs, R.A.; et al. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014, 24, 1740–1750. [Google Scholar] [CrossRef] [PubMed]
- Rayner, E.; van Gool, I.C.; Palles, C.; Kearsey, S.E.; Bosse, T.; Tomlinson, I.; Church, D.N. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer 2016, 16, 71–81. [Google Scholar] [CrossRef]
- Dennis, D.G.; McKay-Fleisch, J.; Eitzen, K.; Dowsett, I.; Kennedy, S.R.; Herr, A.J. Normally lethal amino acid substitutions suppress an ultramutator DNA Polymerase delta variant. Sci. Rep. 2017, 7, 46535. [Google Scholar] [CrossRef] [PubMed]
- Shlien, A.; Campbell, B.B.; de Borja, R.; Alexandrov, L.B.; Merico, D.; Wedge, D.; Van Loo, P.; Tarpey, P.S.; Coupland, P.; Behjati, S.; et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015, 47, 257–262. [Google Scholar] [CrossRef]
- Leon-Castillo, A.; Britton, H.; McConechy, M.K.; McAlpine, J.N.; Nout, R.; Kommoss, S.; Brucker, S.Y.; Carlson, J.W.; Epstein, E.; Rau, T.T.; et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J. Pathol. 2020, 250, 323–335. [Google Scholar] [CrossRef]
- Chen, H.; Beardsley, G.P.; Coen, D.M. Mechanism of ganciclovir-induced chain termination revealed by resistant viral polymerase mutants with reduced exonuclease activity. Proc. Natl. Acad. Sci. USA 2014, 111, 17462–17467. [Google Scholar] [CrossRef] [PubMed]
- Lurain, N.S.; Chou, S. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 2010, 23, 689–712. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Ercolani, R.J.; Lanier, E.R. Novel Cytomegalovirus UL54 DNA Polymerase Gene Mutations Selected In Vitro That Confer Brincidofovir Resistance. Antimicrob. Agents Chemother. 2016, 60, 3845–3848. [Google Scholar] [CrossRef]
- James, S.H.; Prichard, M.N. Current and future therapies for herpes simplex virus infections: Mechanism of action and drug resistance. Curr. Opin. Virol. 2014, 8, 54–61. [Google Scholar] [CrossRef]
- Hakki, M.; Chou, S. The biology of cytomegalovirus drug resistance. Curr. Opin. Infect. Dis. 2011, 24, 605–611. [Google Scholar] [CrossRef]
- Andouard, D.; Mazeron, M.C.; Ligat, G.; Couvreux, A.; Pouteil-Noble, C.; Cahen, R.; Yasdanpanah, Y.; Deering, M.; Viget, N.; Alain, S.; et al. Contrasting effect of new HCMV pUL54 mutations on antiviral drug susceptibility: Benefits and limits of 3D analysis. Antiviral. Res. 2016, 129, 115–119. [Google Scholar] [CrossRef]
- Ducancelle, A.; Champier, G.; Alain, S.; Petit, F.; Le Pors, M.J.; Mazeron, M.C. A novel mutation in the UL54 gene of human cytomegalovirus isolates that confers resistance to foscarnet. Antivir. Ther. 2006, 11, 537–540. [Google Scholar] [PubMed]
- Chou, S. Foscarnet resistance mutations mapping to atypical domains of the cytomegalovirus DNA polymerase gene. Antiviral. Res. 2017, 138, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Chou, S. Approach to drug-resistant cytomegalovirus in transplant recipients. Curr. Opin. Infect. Dis. 2015, 28, 293–299. [Google Scholar] [CrossRef]
- Mason, S.; Devincenzo, J.P.; Toovey, S.; Wu, J.Z.; Whitley, R.J. Comparison of antiviral resistance across acute and chronic viral infections. Antiviral. Res. 2018, 158, 103–112. [Google Scholar] [CrossRef]
- Nijhuis, M.; van Maarseveen, N.M.; Boucher, C.A. Antiviral resistance and impact on viral replication capacity: Evolution of viruses under antiviral pressure occurs in three phases. Handb. Exp. Pharmacol. 2009, 299–320. [Google Scholar] [CrossRef]
- Tu, V.; Abed, Y.; Fage, C.; Baz, M.; Boivin, G. Impact of R152K and R368K neuraminidase catalytic substitutions on in vitro properties and virulence of recombinant A(H1N1)pdm09 viruses. Antiviral. Res. 2018, 154, 110–115. [Google Scholar] [CrossRef]
- Martin, M.; Azzi, A.; Lin, S.X.; Boivin, G. Opposite effect of two cytomegalovirus DNA polymerase mutations on replicative capacity and polymerase activity. Antivir. Ther. 2010, 15, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Lee, C.P.; Huang, Y.H.; Yang, P.W.; Wang, J.T.; Chen, M.R. Epstein-Barr virus protein kinase BGLF4 targets the nucleus through interaction with nucleoporins. J. Virol. 2012, 86, 8072–8085. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.P.; Huang, Y.H.; Lin, S.F.; Chang, Y.; Chang, Y.H.; Takada, K.; Chen, M.R. Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J. Virol. 2008, 82, 11913–11926. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.P.; Chen, J.Y.; Wang, J.T.; Kimura, K.; Takemoto, A.; Lu, C.C.; Chen, M.R. Epstein-Barr virus BGLF4 kinase induces premature chromosome condensation through activation of condensin and topoisomerase II. J. Virol. 2007, 81, 5166–5180. [Google Scholar] [CrossRef]
- Nakayama, S.; Murata, T.; Murayama, K.; Yasui, Y.; Sato, Y.; Kudoh, A.; Iwahori, S.; Isomura, H.; Kanda, T.; Tsurumi, T. Epstein-Barr virus polymerase processivity factor enhances BALF2 promoter transcription as a coactivator for the BZLF1 immediate-early protein. J. Biol. Chem. 2009, 284, 21557–21568. [Google Scholar] [CrossRef]
- Zhu, J.; Liao, G.; Shan, L.; Zhang, J.; Chen, M.R.; Hayward, G.S.; Hayward, S.D.; Desai, P.; Zhu, H. Protein array identification of substrates of the Epstein-Barr virus protein kinase BGLF4. J. Virol. 2009, 83, 5219–5231. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Pandey, M.; Nandakumar, D.; Raney, K.D.; Yin, Y.W.; Patel, S.S. Excessive excision of correct nucleotides during DNA synthesis explained by replication hurdles. EMBO J. 2020, 39, e103367. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Marousek, G.; Bowlin, T.L. Cyclopropavir susceptibility of cytomegalovirus DNA polymerase mutants selected after antiviral drug exposure. Antimicrob. Agents Chemother. 2012, 56, 197–201. [Google Scholar] [CrossRef]
- Chou, S.; Marousek, G.I. Accelerated evolution of maribavir resistance in a cytomegalovirus exonuclease domain II mutant. J. Virol. 2008, 82, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Khare, V.; Eckert, K.A. The proofreading 3’–>5’ exonuclease activity of DNA polymerases: A kinetic barrier to translesion DNA synthesis. Mutat. Res. 2002, 510, 45–54. [Google Scholar] [CrossRef]
- Mansky, L.M.; Cunningham, K.S. Virus mutators and antimutators: Roles in evolution, pathogenesis and emergence. Trends Genet. 2000, 16, 512–517. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Bebenek, K. DNA replication fidelity. Annu. Rev. Biochem. 2000, 69, 497–529. [Google Scholar] [CrossRef]
- Wang, L.; Liang, C.; Wu, J.; Liu, L.; Tyo, K.E.J. Increased Processivity, Misincorporation, and Nucleotide Incorporation Efficiency in Sulfolobus solfataricus Dpo4 Thumb Domain Mutants. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef]
- Tian, W.; Hwang, Y.T.; Lu, Q.; Hwang, C.B. Finger domain mutation affects enzyme activity, DNA replication efficiency, and fidelity of an exonuclease-deficient DNA polymerase of herpes simplex virus type 1. J. Virol. 2009, 83, 7194–7201. [Google Scholar] [CrossRef]
- Andrei, G.; Gammon, D.B.; Fiten, P.; De Clercq, E.; Opdenakker, G.; Snoeck, R.; Evans, D.H. Cidofovir resistance in vaccinia virus is linked to diminished virulence in mice. J. Virol. 2006, 80, 9391–9401. [Google Scholar] [CrossRef]
- Pult, I.; Abbott, N.; Zhang, Y.Y.; Summers, J. Frequency of spontaneous mutations in an avian hepadnavirus infection. J. Virol. 2001, 75, 9623–9632. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Regoes, R.R.; Hamblin, S.; Tanaka, M.M. Viral mutation rates: Modelling the roles of within-host viral dynamics and the trade-off between replication fidelity and speed. Proc. Biol. Sci. 2013, 280, 20122047. [Google Scholar] [CrossRef] [PubMed]
- Rozen-Gagnon, K.; Stapleford, K.A.; Mongelli, V.; Blanc, H.; Failloux, A.B.; Saleh, M.C.; Vignuzzi, M. Alphavirus mutator variants present host-specific defects and attenuation in mammalian and insect models. PLoS Pathog. 2014, 10, e1003877. [Google Scholar] [CrossRef]
- Gnadig, N.F.; Beaucourt, S.; Campagnola, G.; Borderia, A.V.; Sanz-Ramos, M.; Gong, P.; Blanc, H.; Peersen, O.B.; Vignuzzi, M. Coxsackievirus B3 mutator strains are attenuated in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, E2294–E2303. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. CRISPR/Cas9-Based Antiviral Strategy: Current Status and the Potential Challenge. Molecules 2019, 24, 1349. [Google Scholar] [CrossRef]
- De Silva Feelixge, H.S.; Stone, D.; Roychoudhury, P.; Aubert, M.; Jerome, K.R. CRISPR/Cas9 and Genome Editing for Viral Disease-Is Resistance Futile? ACS Infect. Dis. 2018, 4, 871–880. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).